

Abstract
In neurons exposed to glutamate, Ca2+ influx triggers intracellular Zn2+ release via an as yet unclear mechanism. Since glutamate induces a Ca2+-dependent cytosolic acidification, the present work tested the relationships among intracellular Ca2+ concentration ([Ca2+]i), intracellular pH (pHi), and [Zn2+]i. Cultured hippocampal neurons were exposed to glutamate and glycine (Glu/Gly), while [Zn2+]i, [Ca2+]i and pHi were monitored using FluoZin-3, Fura2-FF, and 2′,7′-bis-(2-carboxyethyl)-5(6)-carboxyfluorescein, respectively. Glu/Gly applications decreased pHi to 6.1 and induced intracellular Zn2+ release in a Ca2+-dependent manner, as expected. The pHi drop reduced the affinity of FluoZin-3 and Fura-2-FF for Zn2+. The rate of Glu/Gly-induced [Zn2+]i increase was not correlated with the rate of [Ca2+]i increase. Instead, the extent of [Zn2+]i elevations corresponded well to the rate of pHi drop. Namely, [Zn2+]i increased more in more highly acidified neurons. Inhibiting the mechanisms responsible for the Ca2+-dependent pHi drop (plasmalemmal Ca2+ pump and mitochondria) counteracted the Glu/Gly-induced intracellular Zn2+ release. Alkaline pH (8.5) suppressed Glu/Gly-induced intracellular Zn2+ release whereas acidic pH (6.0) enhanced it. A pHi drop to 6.0 (without any Ca2+ influx or glutamate receptor activation) led to intracellular Zn2+ release; the released Zn2+ (free Zn2+ plus Zn2+ bound to Fura-2FF and FluoZin-3) reached 1 μM.
Keywords: Fura-2FF, FluoZin-3, BCECF, TPEN, 2-deoxy-D-glucose, vanadate, rotenone, oligomycin
Introduction
Destabilization of zinc homeostasis has been implicated in the degeneration of ischemic neurons (Koh et al. 1996). Recent data indicate that neurons containing zinc and glutamate in their synaptic vesicles are primarily responsible for extra- and intracellular [Zn2+] ([Zn2+]i) elevations taking place during spreading depression monitored in hippocampal slices (Carter et al. 2011). Similar phenomena may take place during repetitive spreading depression events, such as peri-infarct depolarizations which contribute to neurodegeneration in animal models of stroke (Mies et al. 1993). However, vesicular Zn2+ release is not solely responsible for the increases in [Zn2+]i because elevated Zn2+ levels following injury were also observed in thalamic neurons which are not innervated by fibers containing zinc and glutamate in synaptic vesicles (Land and Aizenman 2005) and also in neurons of ZnT3 knockout mice that lack zinc in synaptic vesicles (Lee et al. 2000). In such instances, [Zn2+]i elevates due to a Zn2+ release from intracellular stores. A number of investigators reported a Ca2+-dependent Zn2+ release from intracellular stores in glutamate-treated neurons (Sensi et al. 2003; Dineley et al. 2008; Dittmer et al. 2009; Kiedrowski 2011). The mechanism of this Ca2+-dependent Zn2+ release remains unclear. Oxidative agents are known to induce Zn2+ release from intracellular stores (Aizenman et al. 2000) and Dineley et al. (2008) suggested that intracellular zinc is released in glutamate-treated neurons due to a Ca2+-dependent production of reactive oxygen species. Intracellular pH (pHi) fluctuations also strongly affect [Zn2+]i levels following activation of glutamate receptors (Frazzini et al. 2007; Kiedrowski 2011). The present work follows up on the latter findings and tests the idea that glutamate-induced and Ca2+-dependent cytosolic acidification, which is well recognized (Hartley and Dubinsky 1993; Irwin et al. 1994; Koch and Barish 1994; Wang et al. 1994), may serve as a trigger to induce intracellular Zn2+ release. The new data support this idea.
Materials and Methods
Primary cultures of mouse hippocampal neurons
A SPOT™ kit from the University of Illinois at Chicago Research Resources Center (http://www.rrc.uic.edu/portal/SPOT_Culture_Kit) was used to prepare primary cultures of hippocampal neurons obtained from c57/bl/6 mice at embryonic day 16. The kit production was approved by the Institutional Animal Care and Use Committee. Cells from a single kit were plated on about 30 glass coverslips, as previously described (Kiedrowski, 2011). The cells were used for experiments after 13 days in vitro.
Fluorescence imaging and superfusion equipment
Hoffman modulation contrast images were taken using a Zeiss LD A-Plan 20X/0.3 HMC objective (Carl Zeiss Mikroskopie, Jena, Germany). Fluorescence was monitored using a Zeiss Fluar 20x, NA 0.75 objective and an Attofluor digital imaging system (Atto Instruments, Rockville, MD) connected to Zeiss Axiovert 100 microscope. Superfusion media were delivered directly onto the cells via an 8-channel manifold (MPRE-8, Cell MicroControls, Norfolk, VA) with a computer-controlled flow using an 8-channel valve switch (cFlow8, Cell MicroControls). Cells were superfused at a rate of 0.5 ml/min at 37°C. Temperature was maintained at 37°C using a bipolar temperature controller (TC2BIP, Cell MicroControls).
Monitoring Fura2-FF and FluoZin-3 fluorescence in neurons
Coverslips with SPOT™ cultures were placed in custom-made 50 μl imaging chambers, where the cells were loaded for 5 minutes at 37°C with 0.1 μM FluoZin-3 AM and 0.1 μM Fura-2FF AM added to the medium in which the cells were cultured. Immediately after the loading, the imaging chamber was superfused with Locke’s buffer [(in mM) NaCl (157.6), KCl (2.0), KHCO3 (3.6), MgCl2 (1.0), CaCl2 (1.3), HEPES (10), glucose (5), and pH 7.4 adjusted with Tris], which was delivered directly onto the cells via an 8-channel manifold at 37 °C. For time-lapse imaging, cells were exposed every 5 seconds to a sequence of 340, 380, 488 and 356 nm excitation and the images of fluorescence emitted at >520 nm (F340, F380, F488, and F356) were saved on a computer hard-drive for off-line analysis. At the end of each experiment, the maximal Fura-2FF F340/F380 ratio (Rmax) and the maximal FluoZin-3 F488 signal (Fmax) were measured after saturating the indicators in situ with Ca2+ and Zn2+, respectively. Rmax was determined while the cells were superfused with Locke’s buffer supplemented with 10 μM ionomycin, 10 μM TPEN, and 10 mM CaCl2. To measure Fmax, Locke’s buffer was supplemented with 20 μM 1-hydroxypyridine-2-thione zinc salt (pyrithione) and 100 μM ZnCl2. In some experiments, the effect of pHi change on Fmax was tested. In these experiments, the Fmax-testing medium was supplemented with 5 μM gramicidin to accelerate the adjustment of intracellular to extracellular pH and (2-[N-morpholino]ethanesulfonic acid instead of HEPES was used when pH was 6.0. Unless stated otherwise, the FluoZin-3 data were expressed as a percentage of Fmax to normalize for artifacts associated with uneven loading of the cells with the indicator (Kiedrowski 2011). Fura-2FF data were expressed in terms of a percentage of Rmax to normalize the data from experiments in which absolute F340/F380 ratio values varied due to technical issues (differences in camera gains or neutral density filters). Fura-2FF isosbestic point (F356) fluorescence data were used to assess intracellular Fura-2FF concentrations reached in various cells and to monitor indicator leakage and/or bleaching during experiments. Unhealthy cells showing the Fura-2FF F340/F380 ratio elevated to Rmax under the basal conditions were occasionally observed; such cells were excluded from analysis. In a few cells, the FluoZin-3 Fmax signal could not be effectively measured because it exceeded the dynamic range of the camera; FluoZin-3 and Fura-2FF data from such cells were excluded from analysis. Data from all other neurons (regardless of the pattern of FluoZin-3 or Fura-2FF signal changes) were analyzed and included in figures showing average responses.
Approximation of intracellular Fura-2FF concentrations
Capillaries (20 μm internal diameter; VitroCom, Mountain Lakes, NJ) were filled with various concentrations of Fura-2FF pentapotassium salt dissolved in 100 mM KNO3, 50 mM PIPES, 10 mM EGTA, pH 7.2. The isosbestic point fluorescence (F356; 356 nm excitation, 515 nm emission) from the capillaries was measured using the Attofluor imaging system with the same optics and camera settings as those used to monitor the Fura-2FF F356 fluorescence emitted from hippocampal neurons. The Fura-2FF concentration in the capillaries that yielded the same F356 as the average F356 measured in 173 Fura-2FF-loaded neurons was taken as an approximation of the intracellular Fura-2FF concentration. It needs to be stressed that the accuracy of this approach is uncertain. Several factors in addition to Fura-2FF concentration affect intracellular F356. Intracellular viscosity is known to enhance fluorescence intensity (Poenie 1990) and may also slightly shift the isosbestic point (Busa 1992). The viscosity effect would enhance F356 fluorescence emitted from cultured neurons. On the other hand, fluorescence from neurons would be lower than that emitted from capillaries because the pathlength through cultured neurons, 5 to 14 μm, is shorter than the internal diameter of the capillaries.
Approximation of intracellular FluoZin-3 concentrations
This was done as described above for Fura-2FF except that the capillaries were filled with various concentrations of FluoZin-3 tetrapotassium salt dissolved in 100 mM KNO3, 50 mM PIPES, 10 mM EGTA, 10 mM ZnSO4 (10 μM free Zn2+), pH 7.2. Under such conditions, FluoZin-3 is saturated with Zn2+ and its fluorescence (F488; 488 nm excitation and 515 nm emission) reflects the concentration of the indicator. The FluoZin-3 concentration in capillaries yielding the F488 value equal to the average Fmax measured in neurons loaded with FluoZin-3 was taken as an approximation of FluoZin-3 concentration in the neurons.
Measuring FluoZin-3 and Fura-2FF apparent zinc Kd at a pH of 7.2 and 6.1
Fluorescence of media supplemented with 10 nM FluoZin-3 tetrapotassium salt or 10 nM Fura-2FF pentapotassium salt and containing free Zn2+ concentrations ranging from 0 to 10 μM was measured at 25°C using a PTI QuantaMaster spectrofluorometer (Photon Technology International, Inc., Birmingham, NJ). The media were prepared using 100 mM KNO3, 50 mM PIPES, 10 mM EGTA and 0 – 10 mM ZnSO4 at the pH of 7.2 or 6.1 (adjusted using KOH). Free Zn2+ concentration was calculated using the WEBMAXC STANDARD program (Patton 2009). The fluorescence of FluoZin-3 was excited at 488 nm (F488) and that of Fura-2FF at 346 nm (F346); the emission was measured in both cases at 515 nm. F346 was chosen to excite Fura-2FF because at this weavelength the fluorescence of Fura-2FF-Zn complex reaches maximum (Kiedrowski, 2011). FluoZin-3 data were fitted to a 2 parameter equation F488 = Fmax × [Zn]/(Kd + [Zn]); Fura-2FF data were fitted to a 3 parameter equation F346 = y0 + Fmax × [Zn]/(Kd + [Zn]), where Fmax is the maximal F488 or F346, Kd is the apparent zinc dissociation constant, and y0 is the F346 at [Zn] = 0. To fit the data, SigmaPlot 10 software (Systat Software Inc., Richmond, CA) was used.
Approximation of the concentrations of released Zn2+
In a cell loaded with Fura-2FF and FluoZin-3, a certain amount of Zn2+ binds to Fura-2FF and FluoZin-3. The concentration of released Zn2+ ([Zn released]) sensed by the fluorescent indicators can be defined as:
| [1] |
where [Zn2+]i is the free Zn2+ concentration in the cytosol, [Fura-2FF-Zn] is the concentration of Fura-2FF-Zn complex and [FluoZin-3-Zn] is the concentration of FluoZin-3-Zn complex.
To approximate [Znreleased], [Fura-2FF-Zn] and [FluoZin-3-Zn] were calculated as described below and [Zn2+]i was calibrated in situ using the equation:
| [2] |
where Fmin is the minimal F488 measured after exposing the cells to 10 μM TPEN, Fmax is the maximal F488 measured as described above, and Kd is the apparent zinc dissociation of FluoZin-3 measured at the pH of 6.1.
[Fura-2FF-Zn] and [FluoZin-3-Zn] were calculated as described by Dineley et al. (2002):
| [3] |
where [dye-Zn] represents intracellular [Fura-2FF-Zn] or [FluoZin-3-Zn], [Znt] represents the total [Zn2+], [dye] represents the total intracellular [Fura-2FF] or [FluoZin-3]. [dye-Zn] was calculated separately for Fura-2FF and FluoZin-3 by testing various [Znt] values in eq. 3 and selecting the [Znt] at which [Znt] − [dye-Zn] = [Zn2+]i. To calculate [Zn released], [dye-Zn] values calculated using eq. 3 and [Zn2+]i calculated using eq. 2 were entered to eq. 1.
Monitoring intracellular pH (pHi) in neurons
The assay was conducted as described in detail in Kiedrowski (2011) except that in the experiments in which cells were co-loaded with Fura-2FF and BCECF, the loading BCECF-AM concentration was reduced from 1 μM to 5 nM, such that the intracellular BCECF fluorescence observed after 340 and 380 nm excitation was close to background levels (to minimize Fura-2FF signal contamination by BCECF fluorescence). To monitor BCECF fluorescence, the neurons were exposed every 5 seconds to 488 and 440 nm excitation and the images of fluorescence emitted at >520 nm (F488 and F440) were saved on a computer hard-drive for off-line analysis. In selected experiments, F488/F440 ratios were converted to pHi values based on in situ calibration performed at the end of the experiments as described in Kiedrowski (2011).
Experimental media
Glutamate receptors were activated with 100 μM glutamate and 10 μM glycine (Gly/Glu) dissolved in a nominally Mg2+-free Locke’s buffer in which NaCl was substituted with a chloride salt of N-methyl-D-glucamine, as it was shown earlier that Glu/Gly-induced [Zn2+]i elevations are greatly enhanced under such conditions (Kiedrowski 2011). To remove Ca2+ and Zn2+ from the buffer, CaCl2 was omitted and 100 μM EGTA was added. To remove Zn2+ but not Ca2+, the buffer was supplemented with 1 mM CaEDTA. The latter was added simultaneously with Glu/Gly. Exposure to Glu/Gly plus CaEDTA lasted no longer than 5 – 10 min, which was not long enough to deplete intracellular Zn2+ by the extracellular chelator. In fact, the [Zn2+]i elevations were not affected by CaEDTA (Kiedrowski 2011).
Testing Zn2+ chelation by reagents
F488 fluorescence (488 nm excitation, 515 nm emission) of Locke’s buffer supplemented with 100 nM FluoZin-3 tetrapotassium was measured at 25°C using a PTI QuantaMaster spectrofluorometer prior to and after adding 10 nM ZnSO4 using a volumetric 50 mM ZnSO4 standard (Fluka, Germany). Then the tested concentration of a reagent (vanadate, rotenone, oligomycin, or 2DG) was added and F488 was measured again. This was followed by adding 100 μM TPEN to chelate Zn2+ and measure Fmin. Finally, Fmax was measured after adding 200 μM ZnSO4 to the same cuvette.
Statistical analysis
SigmaStat 3.5 software (Systat Software Inc., Richmond, CA) was used to determine the statistical significance of the data. SigmaPlot 10 software (Systat Software Inc., Richmond, CA) was used to fit the relationship between the rate of Fura-2FF and FluoZin-3 signal increase to a logarithmic equation:
| [4] |
The rates of Fura-2FF and FluoZin-3 signal increase were calculated by linear regression using the linear portion of the data.
Reagents
Fura-2FF AM and Fura-2FF pentapotassium salt was obtained from Teflabs (Austin, TX, USA). Neurobasal medium, B-27 supplement, BCECF AM, FluoZin-3 AM, and FluoZin-3 tetrapotassium salt were from Invitrogen (Carlsbad, CA, USA). All other reagents were from Sigma-Aldrich (St Louis, MO, USA) unless otherwise stated.
Results
The largest and the most rapid [Ca2+]i elevations are negatively correlated with [Zn2+]i increases
Fura-2FF and FluoZin-3 fluorescence were simultaneously monitored in primary cultures of hippocampal neurons exposed to 100 μM glutamate and 10 μM glycine (Glu/Gly). To ensure that the fluorescence changes were related to intracellular Zn2+ release, extracellular Zn2+ was chelated using 1 mM CaEDTA, a plasma membrane-impermeable Zn2+ chelator. After FluoZin-3 and/or Fura-2FF signals were elevated by Glu/Gly applications, the impact of 10 μM TPEN, a plasma membrane-permeable Zn2+ chelator, was tested on these signals. Experiments were performed under Na+-free conditions, as a recent study established that under such conditions, Glu/Gly-induced [Zn2+]i elevations are greatly enhanced (Kiedrowski 2011). Fig. 1a shows data from a typical experiment. Consistent with earlier reports (Devinney et al. 2005; Dineley et al. 2008; Kiedrowski 2011), Glu/Gly applications elevated both Fura-2FF and FluoZin-3 signals and a subsequent application of TPEN eliminated the FluoZin-3 signal in all the cells but failed to decrease the Fura-2FF signal in a majority of the cells, indicating that these signals represent [Zn2+]i and [Ca2+]i elevations, respectively. Surprisingly, in neurons showing the most rapid Glu/Gly-induced [Ca2+]i elevations reaching Fura-2FF-saturating levels, the [Zn2+]i increases were the smallest (Fig. 1a, blue traces). In neurons with intermediate rates of a [Ca2+]i increase, the rates of [Zn2+]i elevations were the highest (Fig. 1 a, red traces) followed by neurons with slower rates of Fura-2FF signal increase (Fig. 1a, green traces). In the latter neurons upon TPEN application, the Fura-2FF signal decreased and remained low or increased after the initial drop (Fig. 1a, arrow), whereas the FluoZin-3 signal dropped to basal or lower levels. This outcome can be interpreted as follows. When [Ca2+]i and [Zn2+]i slowly increase (Fig. 1a, green traces), Fura-2FF primarily binds Zn2+ because the affinity of the indicator for Zn2+ is much higher than that for Ca2+ (Hyrc et al. 2000). When TPEN is applied (arrow in Fig. 1a), it strips Zn2+ from Fura-2FF. At this point, Fura-2FF begins to bind Ca2+ and depending on the amount of ambient cytosolic Ca2+, the Fura-2FF signal may decrease or increase or remain unchanged. The F340/F380 ratio of the Zn2+-saturated Fura-2FF is about 30% of the Ca2+-saturated Fura-2FF (Kiedrowski 2011), which creates a major ambiguity in Fura-2FF signal interpretation. An increase of F340/F380 ratios after TPEN addition most likely means that [Ca2+]i was high prior to the TPEN addition, but the high [Ca2+]i could only be detected after the Zn2+ that had been bound to Fura-2FF was stripped by TPEN. A F340/F380 ratio reaching 30% of Rmax, may indicate that Fura-2FF is saturated with Zn2+ (Kiedrowski 2011), or it may indicate that [Zn2+]i rests at picomolar levels but [Ca2+]i is approaching levels close to the Fura-2FF Kd for Ca2+. Simultaneous monitoring of Fura-2FF and FluoZin-3 signals in combination with an eventual TPEN application allows one to identify at which point the Fura-2FF signal exclusively reports [Ca2+]i. However, certain problems, for example whether Zn2+ affects the mechanisms of Ca2+ homeostasis, cannot be addressed using this method.
Figure 1.

The relationship between the rates of [Ca2+]i and [Zn2+]i increases. (a) Simultaneously monitored Fura-2FF and FluoZin-3 signals in 28 hippocampal neurons exposed to 100 μM glutamate and 10 μM glycine (Glu/Gly) under Na+-free conditions (Na+ was removed at the time Glu/Gly was added) with Na+ substituted with N-methyl-D-glucamine. Extracellular Zn2+ was chelated using 1 mM CaEDTA. Where indicated, 10 μM TPEN was added to chelate intracellular Zn2+. The colors, blue, red, and green, identify neurons with high, intermediate, and slow rates of Fura-2FF signal increase, respectively. (b1–b4) Data from three exemplar neurons A, B, and C from the population of 28 neurons shown in panel (a). (b1) A Hoffman modulation contrast image (left) and a FluoZin-3 Fmax image (right) of the neurons. Bar = 50 μm. (b2, b3) Fura-2FF and FluoZin-3 signals expressed as a percentage of Rmax and Fmax signal, respectively. The Rmax and Fmax signals were measured at the end of the experiment (not shown) as described in Methods. (b4) Fura-2FF F356 signal (isosbestic point) expressed as a percentage of F356 signal measured prior to Glu/Gly application. (c) The rate of FluoZin-3 signal increase does not correlate with the Fura-2FF isosbestic point (F356) fluorescence (left), the maximal FluoZin-3 signal (Fmax) (middle), or the sum of F356 and Fmax (right). The data in (a), (b1–b4) and (c) are from the same experiment, which was repeated 5 times with similar results.
For a better clarity, the data from three representative neurons marked A, B, and C, are singled out in Fig. 1b1–b4. Fig. 1b1 shows a Hoffman modulation contrast and a FluoZin-3 Fmax image of the cells. Despite major differences in the patterns of [Ca2+]i (Fig. 1b2) and [Zn2+]i (Fig. 1b3) changes in these three neurons, the patterns of the isosbestic point fluorescence (F356) of Fura-2FF were similar (Fig. 1b4), indicating that cells A, B, C were similarly affected by phenomena such as cell swelling or Fura-2FF leakage or bleaching.
Differences in rates of [Zn2+]i increase among the cells (Fig. 1a, right panel) may result from differences in Fura-2FF or FluoZin-3 concentrations in these cells. For example, slower rates of [Zn2+]i increases (blue and green FluoZin-3 traces in Fig. 1a) could result from a greater loading of these cells with the indicators. To test whether this is the case, the relative concentrations of Fura-2FF and FluoZin-3 were determined by measuring the F356 (isosbestic point) fluorescence of Fura-2FF, and Fmax of FluoZin-3 in each cell. In Fig. 1c, the rate of [Zn2+]i increase in 28 neurons is plotted as a function of F356 (an index of Fura-2FF concentration), Fmax (an index of FluoZin-3 concentration), and F356 + Fmax. As can be seen, there is no correlation between the rate of [Zn2+]i increase and F356, or Fmax, or both, which indicates that the differences in rates of [Zn2+]i increase among the cells did not result from differences in Fura-2FF or FluoZin-3 concentrations in these cells.
[Zn2+]i elevations are associated with a drop in pHi
The Glu/Gly-induced [Ca2+]i and [Zn2+]i elevations were both prevented by application of an NMDA channel blocker MK-801 (Fig. 2a), which indicates that they resulted from activation of NMDA receptors. As expected (Sensi et al. 2003; Dineley et al. 2008; Dittmer et al. 2009), Glu/Gly-induced [Zn2+]i elevations were Ca2+-dependent; no FluoZin-3 or Fura-2FF signal increase was observed when Glu/Gly was applied under Ca2+-free conditions but as soon as Ca2+ was added to the medium, both signals increased (Fig. 2b). To relate FluoZin-3 and Fura-2FF signal changes to pHi changes, pHi was monitored in an analogous experiment. When hippocampal neurons were exposed to Glu/Gly in the absence of Ca2+, pHi dropped from 7.5 ± 0.03 to 6.7 ± 0.08. A further pHi drop was observed when Ca2+ was added (Fig. 2c). Applications of Glu/Gly in the presence of Ca2+ led to a pHi drop to 6.1 ± 0.03 within 5 minutes. The pHi drop to 6.7 observed when Glu/Gly was applied in the absence of Ca2+ (Fig. 2c) was not caused by Glu/Gly but by the lack of Na+ in the medium in which Glu/Gly was applied; a similar pHi drop was observed in neurons exposed to a Na+-free Locke’s buffer without Glu/Gly (Fig. 2c, inset). It appears that the pHi drop from 6.7 to 6.1 triggers an intracellular Zn2+ release but the pHi drop from 7.5 to 6.7 is insufficient to induce this effect. The drop in the BCECF signal was not affected by Zn2+ chelation with TPEN; Zn2+ influx caused by application of 100 μM ZnCl2 plus 20 μM pyrithione did not prevent BCECF from reporting pHi changes (Fig. 2d).
Figure 2.

Glutamate-induced and Ca2+-dependent [Zn2+]i elevations depend on activation of NMDA receptors and are associated with a drop in pHi. (a) Blocking NMDA channels with 10 μM MK-801 prevents the Glu/Gly-induced increase of Fura-2FF and FluoZin-3 signal. The data are averages ± SE from 29 (right) and 22 (left) neurons. The experiments were repeated 5 times with similar results. In these and all other experiments, Glu/Gly was applied under Na+-free conditions (Na+ was removed at the same time Glu/Gly was added). (b) In the absence of Ca2+ (−Ca), Glu/Gly application failed to increase Fura-2FF and FluoZin-3 signals. Only after 1.3 mM CaCl2 was added (+Ca) did both signals increase. (c) Glu/Gly application decreased pHi in a Ca2+-dependent manner. The cells were exposed to Glu/Gly under analogous experimental conditions as in (b). Note that the Ca2+-dependent FluoZin-3 and Fura-2FF signal increase shown in (b) is associated with a Ca2+-dependent pHi drop. The initial pHi drop, observed in the absence of Ca2+ (−Ca), is caused not by Glu/Gly but by removal of Na+. As shown in the inset, an analogous pHi drop is observed when Na+-free Locke’s buffer (without Glu/Gly) is applied. The data are averages ± SE from 28 (b), 11 (c) and 14 (inset) neurons monitored in a single experiment. All experiments were repeated 4 – 5 times with similar results. (d) Zn2+ chelation with 10 μM TPEN does not affect the Glu/Gly-induced drop in the intracellular BCECF signal. At the end of the experiment, Zn2+ influx and pHi changes were imposed by an application of 100 μM ZnCl2, 20 μM pyrithione, and 5 μM gramicidin at a pH of 6.0 (Zn pH 6.0) and a pH of 7.4 (Zn pH 7.4), as indicated. The data are averages ± SE from 31 neurons monitored in a single experiment that was repeated 3 times with similar results.
Rapid deregulation of Ca2+ homeostasis leads to less pronounced intracellular acidification and smaller [Zn2+]i increases
After plotting the rate of a FluoZin-3 signal increase as the function of Fura-2FF signal increase and fitting the data to equation 4 (for details see Methods), it became evident that the [Ca2+]i elevations with rates up to 10% of Fura-2FF Rmax x min−1 were positively correlated with the rates of [Zn2+]i elevations. However, in neurons with higher rates of [Ca2+]i increases, the correlation becomes negative (Fig. 3a).
Figure 3.
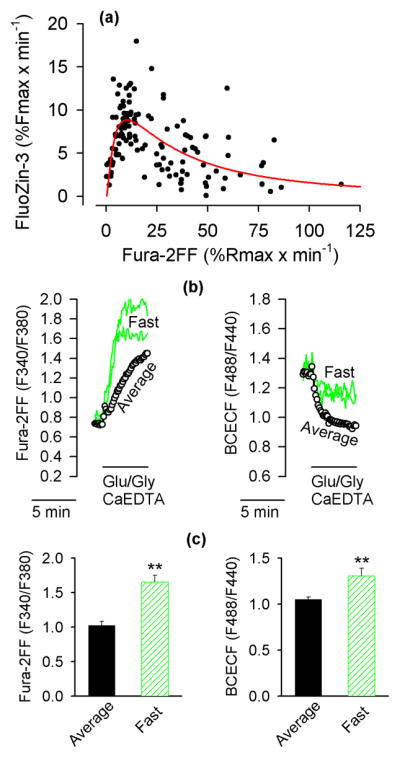
The neurons with the fastest rates of [Ca2+]i increase acidify less and release less Zn2+ from intracellular stores. (a) The relationship between the rate of Fura-2FF signal increases and the rate FluoZin-3 signal increases. The data are from 128 individual neurons monitored in 6 experiments analogous to the one shown in Fig. 1a. The red line indicates the closest fit of the data to equation 4. Note that the rate on FluoZin-3 signal increase is positively correlated with the rate of Fura-2FF signal increase only up to about 10% of Rmax x min−1. At faster rates of Fura-2FF signal increases, the correlation becomes negative. (b) The neurons with the fastest rates of Fura-2FF signal increases acidify less than other neurons. Intracellular Fura-2FF and BCECF signals were simultaneously monitored in neurons exposed to Glu/Gly under the experimental conditions identical to those described in Fig. 1a. The neurons with the fastest rate of Fura-2FF signal increases were singled out (Fast; green traces); the data from all other neurons were averaged (Average, black open circles). Note that the neurons with the fastest rate of Fura-2FF signal increases (left panel) show a less pronounced drop in the BCECF signal (right panel). (c) Fura-2FF and BCECF signals measured 5 min after the onset of Glu/Gly application. The data are means ± SE from 4 experiments analogous to the one shown in (b). ** P < 0.01, Mann-Whitney Rank Sum Test.
Since Glu/Gly-induced [Zn2+]i elevations appear to be associated with a pHi drop (Fig. 2b,c), an experiment was designed to measure pHi in neurons showing various rates of [Ca2+]i increases. In this experiment, Fura-FF and BCECF fluorescence were simultaneously monitored. The data from neurons with the most rapid rates of Fura-2FF signal increase were singled out and compared to the average data from all other neurons in the same field. It was found that the pHi drop was smallest in the neurons with the fastest rates of [Ca2+]i increase (Fig. 3b,c).
The idea that neurons showing particularly fast rates of [Ca2+]i increase but small [Zn2+]i increases (blue traces in Fig. 1a) may have been less acidified than other neurons was tested further. To this end, neuronal energy reserves were compromised by substituting glucose with 2-deoxy-D-glucose (2DG), a glucose analog that is phosphorylated but not further metabolized. It was expected that Glu/Gly-induced [Ca2+]i elevations in such neurons would be accelerated (Vergun et al. 2003). As shown in Fig. 4a (green), in the presence of 2DG the rate of Glu/Gly-induced [Ca2+]i increase was significantly accelerated and this increase was associated with a slower [Zn2+]i increase (Fig. 4b) and less pronounced intracellular acidification (Fig. 4c).
Figure 4.

Effects of substitution of glucose with 2-deoxy-D-glucose (2DG) on [Ca2+]i, [Zn2+]i and pHi in Glu/Gly-treated neurons. Fura-2FF- and FluoZin-3-loaded neurons (a and b) or BCECF-loaded neurons (c) were exposed to Glu/Gly as described in Fig. 1 in the presence of glucose (black traces, filled symbols, black bars) or with glucose substituted with 2DG (green traces, open symbols, hatched bars). The arrow indicates when glucose was replaced by 2DG. The data in the left hand panels are averages ± SE from 13 – 22 neurons monitored in a single experiment; the averages ± SE from 4 such experiments are shown in the right hand panel. ** p < 0.01 (Student’s t-test).
Two major mechanisms contribute to intracellular acidification induced by Ca2+ influx; first, Ca2+ entering the cells is extruded back to the extracellular space by a plasmalemmal Ca2+ pump that works as an ATP-fueled Ca2+/H+ exchanger (Trapp et al. 1996; Wu et al. 1999); second, mitochondria, while they accumulate Ca2+, extrude more protons and the cytosol gets acidified while the matrix becomes more alkaline (Wang et al. 1994; Abad et al. 2004). However, the extent to which mitochondria contribute to the Ca2+-dependent pHi drop vary significantly among neurons (Abad et al. 2004). Therefore, to test the impact of a Ca2+-dependent pHi drop on [Zn2+]i increases, both mechanisms of Ca2+-dependent pHi drop (the plasmalemmal Ca2+ pump and mitochondrial Ca2+ accumulation) were compromised simultaneously. To inhibit the plasmalemmal Ca2+ pump, 1 mM vanadate (Tiffert and Lew 2001) was used, and to prevent Ca2+ accumulation in the mitochondria, 2 μM rotenone, an inhibitor of complex I of the respiratory chain, and 3 μg/ml oligomycin, an inhibitor of mitochondrial ATP synthase were applied. Under such conditions, mitochondria promptly depolarize and cannot accumulate Ca2+ (Budd and Nicholls 1996a, b). Fig. 5a shows data from an experiment in which vanadate, rotenone, and oligomycin (VRO) were applied two minutes prior to Glu/Gly. Upon VRO addition, [Ca2+]i began to increase in many neurons and a subsequent application of Glu/Gly led to a rapid [Ca2+]i elevation, leading to Fura-2FF saturation at a rate significantly faster than that observed in control cells exposed to Glu/Gly without VRO (Fig. 5a). As shown in Fig 5b, VRO treatment compromised the ability of Glu/Gly to induce an intracellular Zn2+ release. As expected, the Glu/Gly-induced cytosolic acidification was significantly reduced when Glu/Gly was applied in the presence of VRO (Fig. 5c).
Figure 5.

Effects of vanadate, rotenone, and oligomycin (VRO) on [Ca2+]i, [Zn2+]i and pHi in Glu/Gly-treated neurons. Fura-2FF- and FluoZin-3-loaded neurons (a and b) or BCECF-loaded neurons (c) neurons were exposed to Glu/Gly in the presence of 1 mM vanadate (to inhibit the plasmalemmal Ca2+ pump) and 2 μM rotenone plus 3 μg/ml oligomycin (to depolarize mitochondria). The arrow indicates when VRO was added. The data in the left hand panels are averages ± SE from 21–31 neurons monitored in a single experiment; the averages ± SE from 5 such experiments are shown in the right hand panel. Black traces, filled symbols, and black bars show data from control neurons. Green traces, open symbols, and hatched bars show data from VRO-treated neurons. ** p < 0.01 (Student’s t-test).
Alkaline pH counteracts and acidic pH promotes a Glu/Gly-induced intracellular Zn2+ release
To further test the role of pHi decrease in Glu/Gly-induced intracellular Zn2+ release, Glu/Gly-induced intracellular acidification was counteracted by increasing the extracellular pH to 8.5. At such an extracellular pH, the Glu/Gly-induced pHi drop (Fig. 6a) and [Zn2+]i increases were suppressed (compare Fig. 6b versus 6c). However, when a rapid drop of pHi to 6.0 was imposed by applying 5 μM gramicidin and simultaneously reducing extracellular pH to 6.0, the FluoZin-3 signal promptly increased (Fig. 6c). This increase represented a Zn2+ release from the intracellular stores because it took place while 1 mM CaEDTA was present in the extracellular solution. The FluoZin-3 signal increase was rapidly obliterated when 10 μM TPEN was added to chelate intracellular Zn2+ (Fig. 6c).
Figure 6.
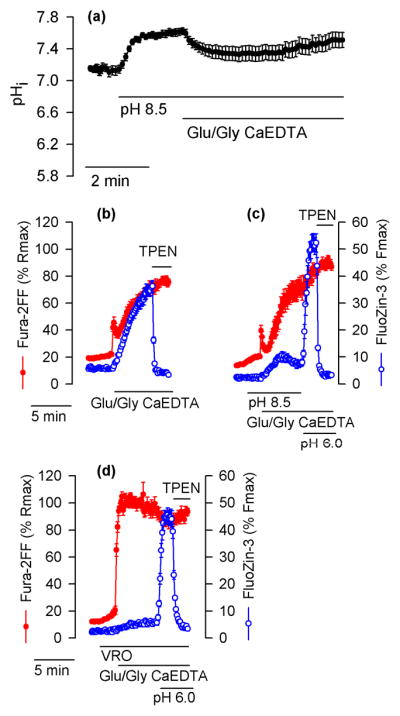
Alkaline pH inhibits and acidic pH promotes intracellular Zn2+ release in Glu/Gly-treated neurons. (a) The effect of alkaline pH on pHi in cultured hippocampal neurons exposed to Glu/Gly in the presence of 1 mM CaEDTA. The experiment was conducted as those shown in Figs. 4c and 5c (control, black traces) except that where indicated extracelluar pH was increased from 7.4 to 8.5. (b) Fura-2FF and FluoZin-3 signals monitored in a control experiment performed at a pH of 7.4. (c) Analogous experiments with Glu/Gly applied at the pH of 8.5 followed by application of a pH of 6.0. To ensure a prompt drop of pHi to 6.0, 5 μM gramicidin was applied at the time the extracellular pH was switched form 8.5 to 6.0. Where indicated, 10 μM TPEN was added to chelate Zn2+. (d) Similar experiment to the one shown in (c) but performed in the presence of VRO. The data are averages ± SE from 16 (a), 28 (b), 17 (c), and 22 (d) neurons monitored in a single experiment. All experiments were repeated with similar results 4 – 5 times.
Increasing extracellular pH to 8.5 not only counteracts the Glu/Gly-induced pHi drop (Fig. 6a) but also inhibits Ca2+ extrusion by the plasmalemmal Ca2+ pump and consequently accelerates [Ca2+]i elevations in glutamate-treated neurons (Khodorov et al. 1995). The faster rate of [Ca2+]i increases in neurons with an inhibited Ca2+ pump and depolarized mitochondria (VRO, Fig. 5a) compared to neurons with an inhibited Ca2+ pump but functional mitochondria (pH 8.5, Fig. 6c) emphasizes the impact of the mitochondrial Ca2+ sequestration on [Ca2+]i clearance.
Inhibition of mitochondrial ATP production by rotenone plus oligomycin is expected to accelerate ATP depletion in Glu/Gly-treated neurons. Therefore, one might be concerned that the VRO effect on intracellular Zn2+ release shown in Fig. 5b is caused by a greater ATP depletion rather than the smaller pHi drop (Fig. 5c). Should this be the case, [Zn2+]i in VRO-treated neurons should not be affected by a pHi drop. However, as shown in Fig. 6d, an experimental maneuver resulting in a pHi drop to 6.0 in VRO-treated neurons led to a prompt [Zn2+]i increase similar to that observed in neurons not treated with VRO (Fig. 6c).
The ability of 2DG, vanadate, rotenone, and/or oligomycin to counteract Glu/Gly-induced [Zn2+]i elevations (Figs. 4b and 5b) could be an artifact should these agents be able to chelate Zn2+. Using the approach described in Methods, it was found that none of these agents (at the concentrations used in this study) acted as a Zn2+ chelator.
A pHi drop alone induces intracellular Zn2+ release
All experiments described thus far were performed under Na+-free conditions. One may be concerned about the physiological significance of such data. In fact, in the presence of Na+, in cultured cortical neurons exposed to the pH of 6.0 for 10 min, Sensi et al. (2003) failed to observe any [Zn2+]i elevations. In cultured cerebellar granule cells, Isaev et al. (2010) did observe [Zn2+]i elevations under such conditions but the exposures lasted as long as 30 min. To test whether a pHi drop to 6.0 performed in the presence of Na+ leads to an intracellular Zn2+ release in hippocampal neurons, the neurons were loaded with Fura-2FF and FluoZin-3 and incubated in Locke’s buffer containing 158 mM Na+. The buffer was nominally Ca2+- and Zn2+-free (it contained 100 μM EGTA to chelate Zn2+ and Ca2+ contamination) and was supplemented with 5 μM gramicidin to accelerate the adjustment of pHi to the extracellular pH (Kiedrowski 2011). As shown in Fig. 7a, the gramicidin addition per se did not affect the Fura-2FF or FluoZin-3 signal. However, when pH 6.0 was applied three minutes later, both signals promptly increased and, when 10 μM TPEN was added to chelate the intracellular Zn2+, the FluoZin-3 signal dropped to basal levels in 100% of neurons; the Fura-2FF signal dropped in 84% of neurons (104 of 124 neurons; 6 experiments). Since neither Zn2+ nor Ca2+ was present in the extracellular medium, the data can be interpreted to indicate that the pHi drop alone (without any Ca2+ influx) was sufficient to induce an intracellular Zn2+ and Ca2+ release, although the latter was overtly apparent in only 16% of neurons.
Figure 7.

A pHi drop (without Ca2+ influx) suffices to induce intracellular Zn2+ release. (a) Fura-2FF- and FluoZin-3-loaded neurons incubated in a Ca2+-free and Zn2+-free Locke’s buffer containing 158 mM Na+ and supplemented with 100 μM EGTA (EGTA). Where indicated, 5 μM gramicidin (Gramic) was applied to permeabilize the plasma membrane to H+, Na+ and K+; to induce pHi drop, extracellular pH was decreased to 6.0; TPEN (10 μM) was added to chelate Zn2+. Left and right panels show Fura-2FF and FluoZin-3 data, respectively, simultaneously monitored in 32 neurons. In three neurons (red) unlike in other neurons (green), the Fura-2FF signal failed to drop upon TPEN application. (b) Average FluoZin-3 ± SE data from all cells shown in (a). At the end of this experiment, the maximal FluoZin-3 signal (Fmax) was measured at a pH of 6.0 (Zn pH 6.0) and a pH of 7.4 (Zn pH 7.4). Note that the pH change failed to affect Fmax. The values listed in the box are discussed in the text. (c) Analogous experiment to the one shown in (b) but without gramicidin. The data are averages ± SE from 24 neurons monitored in a single experiment. All experiments were repeated 5 times with similar results.
Fig. 7b shows average FluoZin-3 data from the same experiment that is shown in Fig. 7a. Fig. 7c shows average FluoZin-3 data from an analogous experiment but performed in the absence of gramicidin. As can be seen, the [Zn2+]i increase observed after applying pH 6.0 in the absence of gramicidin was much smaller than that observed in the presence or gramicidin. At the end of both experiments, it was tested whether the FluoZin-3 Fmax is affected by a pH change from 6.0 to 7.4. To this end, 100 μM ZnCl2, 20 μM pyrithione, and 5 μM gramicindin were applied and during the exposure the pH was changed from 6.0 to 7.4, a maneuver that promptly affects pHi (Fig. 2d). As can be seen in Fig. 7b,c, the Fmax signal of FluoZin-3 did not change upon the switch from pH 6.0 to 7.4.
The zinc Kd values of FluoZin-3 and Fura-2FF decrease upon a drop in pH
Although the FluoZin-3 Fmax turned out to be insensitive to acidification in the tested range (Fig. 7b,c), the impact of a pH drop on Kd needed to be determined. To this end, Zn-EGTA buffers with a defined [Zn2+] in the 0 – 10 μM range, calculated using the WEBMAXC STANDARD program (Patton 2009), were prepared and the zinc Kd values of FluoZin-3 and Fura-2FF at a pH of 7.2 and 6.1 were characterized in vitro. The zinc Kd values of FluoZin-3 at pH 7.2 and 6.1 were found to be 3.4 nM and 36 nM, respectively (Fig. 8a). The observed drop in FluoZin-3 affinity for Zn2+ upon acidification does not confirm the earlier assertion that the chelator portion of FluoZin-3 molecule is insensitive to protonation (Gee et al. 2002). The zinc Kd values of Fura-2FF at pH 7.2 and 6.1 were found to be 3.6 nM and 31 nM, respectively (Fig. 8b). The data are consistent with an acidification-induced drop of Fura-2 (and other Ca2+ indicators) affinity for Ca2+ (Lattanzio and Bartschat 1991).
Figure 8.
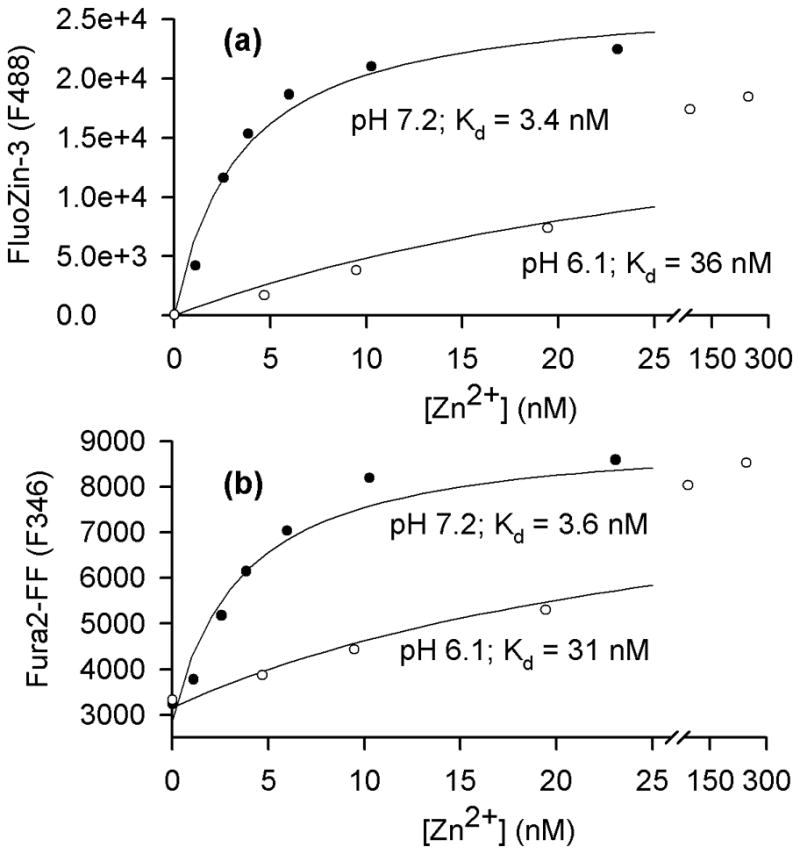
A drop of pH from 7.2 to 6.1 decreases the affinity of FluoZin-3 and Fura-2FF for Zn2+. (a) FluoZin-3 F488 signal was monitored in vitro at different [Zn2+] at a pH of 7.2 and 6.1. The data were fitted as described in the Methods and yielded the indicated apparent Kd values. (b) Analogous data obtained for the Fura-2FF F346 signal.
Approximation of the amount of Zn2+ released from intracellular stores
Using eq. 2 and the Zn2+ Kd measured at pH 6.1, 36 nM (Fig. 8a), one can estimate that the FluoZin-3 signal increase in the experiment shown in Fig. 7b represents about 16 nM [Zn2+]i. However, the released Zn2+ also includes Zn2+ bound to FluoZin-3 and Fura-2FF. To calculate the concentration of the FluoZin-3-Zn complex ([FluoZin-3-Zn]), one needs to estimate the concentration of intracellular FluoZin-3 ([FluoZin-3]). Using the approach described in Methods, it was found that an average [FluoZin-3] in hippocampal neurons was about 1.1 μM. From eq. 3 it was calculated that when [FluoZin-3] is 1.1 μM, [Zn2+]i is 16 nM, and Kd is 36 nM, [FluoZin-3-Zn] is 0.34 μM, meaning that Zn2+ is bound to 31% of intracelluar FluoZin-3. Looking at Fig. 7b, one can notice that the F488 value corresponding to 16 nM [Zn2+]i represents 31% of Fmax, as expected from the eq. 3 calculation. Using an analogous approach for Fura2FF (for details see Methods) it was found that [Fura-2FF] and [Fura-FF-Zn] were about 2.7 μM, and 0.92 μM, respectively. After entering the respective values to eq. 1, one can calculate that [Zn2+ released] = 0.016 μM + 0.34 μM + 0.92 μM = 1.276 μM, meaning that the fluorescent Zn2+ indicators chelated about 99% of released Zn2+.
The accuracy of the above calculation relies in part on calibrations of intracellular Fura-2FF and Fluo-Zin-3 concentrations which, as explained in Methods, are uncertain. Nevertheless, considering that the cells were loaded with very low concentrations of Fura-2FF AM and FluoZin-3 AM (0.1 μM) and for 5 min only, the approximated intracellular Fura-2FF and Fluo-Zin-3 concentrations, 2.7 and 1.1 μM, respectively, are about what one would expect from an extrapolation of the data obtained by others (Dineley et al. 2002; Krężel and Maret 2006) who loaded cells with higher concentrations of the dyes and/or for a longer time (20 – 30 min).
Discussion
Earlier work showed that elevated [Zn2+]i levels in Glu/Gly-treated neurons are promptly cleared from the cytosol in a pH dependent manner – the faster the pHi increase, the faster the [Zn2+]i clearance (Kiedrowski 2011). Since activation of Ca2+ influx via ionotropic glutamate receptors leads to a pHi decrease, it is possible that the Glu/Gly-induced pHi drop promotes intracellular Zn2+ release, which was tested in the present experiments.
The new data causally link a Glu/Gly-induced cytosolic acidification with the mechanism of intracellular Zn2+ release. In fact, a pHi drop alone (without any Ca2+ influx) is a stimulus sufficient to induce an intracellular Zn2+ release in hippocampal neurons (Fig. 7a). Therefore, it appears that the Glu/Gly-induced intracellular Zn2+ release in this model depends on a Ca2+ influx because the latter is necessary to decrease pHi to levels low enough (Fig. 2c) to trigger the mechanism of Zn2+ release. The pHi drop must be large enough, well below 6.7, to trigger an intracellular Zn2+ release, as smaller pHi decreases are ineffective (Fig. 2a). In the absence of a protonophore, short lasting applications of low extracellular pH do not decrease pHi low enough to induce a major intracellular Zn2+ release (compare the data in Fig. 7b versus 7c). The reason that Na+ reduces Glu/Gly-induced intracellular Zn2+ release is that it counteracts a pHi drop (Kiedrowski, 2011). If a pHi drop to 6.0 is imposed in the presence of Na+, Zn2+ is released from intracellular stores, as expected (Fig. 7a,b).
Since intracellular Zn2+ release in this experimental model is triggered by a Ca2+ influx (Fig. 2b), it was surprising to notice that neurons showing the fastest rates of [Ca2+]i increase are the most sluggish in releasing Zn2+ from intracellular stores (Fig. 1a, blue traces). These seemingly paradoxical data can be explained in terms of the pHi decrease being less pronounced in the neurons that increase [Ca2+]i at the fastest rates (Fig. 3b,c). This conclusion is supported by the finding that neurons with compromised energy reserves behave the same way; they increase [Ca2+]i faster (Fig. 4a) but acidify less (Fig. 4c) and release less Zn2+ from intracellular stores (Fig. 4b) than control neurons. It appears that healthier neurons, which are able to cope with large Ca2+ loads for a longer time, acidify more due to the plasmallemal Ca2+ pump and mitochondrial activities and therefore release more Zn2+ from intracellular stores.
The mechanism of acidification-induced intracellular Zn2+ release needs to be further elucidated. Whereas Zn2+ may be displaced from metallothioneins and other zinc-binding ligands by protons (Li et al. 1954; Jiang et al. 2000; Sensi et al. 2003), it is unclear whether this process takes place primarily in the cytosol or in zinc-storing organelles or both. While metallothioneins are considered the primary source of Zn2+ that could be released upon acidification (Shuttleworth and Weiss 2011), the data of Maret and colleagues (Jiang et al. 2000) suggest that metallothioneins release Zn2+ only after pH drops below 5.0. Therefore, the extent to which metallothioneins participate in the here observed intracellular Zn2+ release triggered by a pHi drop to 6.0 (Fig. 7b) remains unclear. One should consider that that a pHi drop could reverse a H+/Zn2+ exchange in the organelles that use this mechanism to accumulate Zn2+ (Colvin 2002; Ohana et al. 2009). Such a reversal would result in a cytosolic [Zn2+] elevation due to the Zn2+ efflux from such organelles.
The pathways of intracellular Zn2+ release may also include mitochondrial Zn2+ fluxes. Dittmer et al. (2009), using cultured hippocampal neurons expressing genetically-encoded Zn2+ sensors, observed that glutamate applications lead to a [Zn2+] drop in the mitochondrial matrix and a [Zn2+] increase in the cytosol. Their data indicate that Zn2+ is being released from the mitochondria to the cytosol and confirm the earlier conclusions of Sensi et al. (2003). Such glutamate-induced mitochondrial Zn2+ fluxes could be strongly affected by a concomitant Ca2+-dependent pH decrease in the cytosol and in the mitochondrial matrix (Bolshakov et al. 2008). However, the extent to which mitochondria represent the intracellular compartments releasing Zn2+ in response to cytosolic acidification is not yet clear. Sensi et al. (2003) reported that brief (10 min) exposures of cortical neurons to an extracellular pH of 6.0 do not induce intracellular Zn2+ release unless a protonophore, carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP), is applied. They concluded that Zn2+ is released from FCCP-depolarized mitochondria and that the resulting [Zn2+]i elevations are enhanced at a lower pHi because many Zn2+-buffering mechanisms, including Zn2+ binding to metallothioneins, are less effective at acidic pH (Frazzini et al. 2007; Sensi et al. 2009). However, the fact that protonophores, such as FCCP, promote [Zn2+]i elevations does not necessarily imply that Zn2+ is released from the mitochondria. FCCP facilitates proton flux through all biological membranes including acidic organelles that store Zn2+. FCCP application per se induces a pHi drop (Wang et al. 1995; Stout et al. 1998). FCCP applied at an acidic extracellular pH, by accelerating proton influx across the plasma membrane, increases the rate of intracellular acidification. FCCP may induce intracellular Zn2+ release by promoting the same acid-sensitive mechanism as gramicidin does (Fig. 7). The role of mitochondria in increasing [Zn2+]i levels following FCCP application may be that the FCCP-depolarized mitochondria do not accumulate Zn2+ (Malaiyandi et al. 2005) that is released from some other intracellular Zn2+ stores in response to a pHi drop.
The mechanisms of Zn2+ homeostasis and their sensitivity to acidification may differ among various neurons. For example, an application of pH 6.0 to hippocampal neurons promptly increased [Zn2+]i (Fig. 7c), whereas this was not observed in cortical neurons (Sensi et al. 2003). In cerebellar granule cells, [Zn2+]i elevations were observed when the exposures to pH 6.0 were prolonged to 30 min (Isaev et al. 2010). Considering that various neuronal populations in the brain are differently innervated by zinc-containing fibers (Palmiter et al. 1996), one may expect that the mechanisms of Zn2+ homeostasis in these neurons quantitatively or qualitatively differ as they evolve to handle different Zn2+ levels.
It is interesting to notice that thalamic neurons which do not receive any Zn2+-containing innervation (Palmiter et al. 1996) do show elevated intracellular Zn2+ levels after the damage caused by a target loss (Land and Aizenman 2005). Apparently, these neurons release Zn2+ from internal stores. Typically, it is speculated that such an intracellular Zn2+ release is triggered by oxidative and/or nitrosative stress (Shuttleworth and Weiss 2011). Support for this idea comes from observations that applications of oxidants (Aizenman et al. 2000) or nitric oxide donors (Berendji et al. 1997; Frederickson et al. 2002) induce intracellular Zn2+ release. The present data do not question the role of reactive oxygen and nitosative species in inducing intracellular zinc release. However, considering the impact of the pHi drop on [Zn2+]i (Fig. 6b,c; 7a,b), the extent to which endogenously produced reactive oxygen/nitrosative species versus pHi drop mediate intracellular Zn2+ release in neurons exposed to ischemic and/or excitotoxic stimuli needs to be determined in future studies.
Ischemic brain tissue acidifies due to ATP hydrolysis (Gault et al. 1994), lactate accumulation (Smith et al. 1986), and insufficient H+ clearance (Hertz 1981). This pHi drop may promote an intracellular Zn2+ release similar to that seen in vitro (Fig. 7b). The present data suggest that about 99% of the internally released Zn2+ was chelated by Fura-2FF and FluoZin-3. However, in neurons not loaded with Zn2+-chelating agents, the released Zn2+ is expected to be buffered or cleared from the cytosol by alternative mechanisms. In particular, Zn2+ sequestration in the mitochondria may play an important role (Malaiyandi et al. 2005), which may have unfortunate consequences for cell viability. Zn2+ was shown to inhibit the bc1 complex of the respiratory chain (Link and von Jagow 1995) and α-ketoglutarate dehydrogenase in the mitochondria (Brown et al. 2000) and to promote mitochondrial permeability transition (Jiang et al. 2001; Bossy-Wetzel et al. 2004). In fact, Isaev et al. (2010) recently linked the acidosis-induced [Zn2+]i elevations to a mechanism of the acidosis-induced death of cerebellar granule cells, which was reduced when Zn2+ was chelated by TPEN. Considering that preischemic hyperglycemia exacerbates pHi drop during ischemia (Siemkowicz and Hansen 1981; Smith et al. 1986) and increases ischemic brain damage (Myers and Yamaguchi 1977; Siemkowicz and Hansen 1978), one may speculate that an enhanced intracellular zinc release plays a role in the ischemic vulnerability of hyperglycemic animals.
Acknowledgments
This work was supported by National Institutes of Health Grants 1R21 NS065305 and 5RO3 NS077095 and American Heart Association Grant-in-Aid 0855825G and was presented on poster 450.03 at the 2011 Society for Neuroscience Meeting. The author is grateful to Dr. C. William Shuttleworth for his critical reading of the manuscript, to Dr. John A. Connor for advice on intracellular [Fura-2FF] calibration, and to Dr. Peter G.W. Gettins for access to PTI QuantaMaster spectrofluorometer. The author declares his involvement in SPOT™ culture kit production at the University of Illinois at Chicago Research Resources Center.
Abbreviations used
- BCECF
2′,7′-bis-(2-carboxyethyl)-5(6)-carboxyfluorescein
- [Ca2+]i and [Zn2+]i
intracellular Ca2+ and Zn2+ concentration
- CaEDTA
calcium EDTA
- 2DG
2-deoxy-D-glucose
- Fmax
maximal F488
- [FluoZin-3-Zn]
concentration of FluoZin-3-Zn complex
- [Fura-2FF-Zn]
concentration of Fura-2FF-Zn complex
- Glu/Gly
cell exposure to 100 μM glutamate plus 10 μM glycine
- pHi
intracellular pH
- pyrithione
1-hydroxypyridine-2-thione zinc salt
- Rmax
maximal F340/F380 ratio
- TPEN
N,N,N′,N′-tetrakis(2-pyridylmethyl)ethylenediamine
- VRO
vanadate + rotenone + oligomycin
References
- Abad MF, Di Benedetto G, Magalhaes PJ, Filippin L, Pozzan T. Mitochondrial pH monitored by a new engineered green fluorescent protein mutant. J Biol Chem. 2004;279:11521–11529. doi: 10.1074/jbc.M306766200. [DOI] [PubMed] [Google Scholar]
- Aizenman E, Stout AK, Hartnett KA, Dineley KE, McLaughlin B, Reynolds IJ. Induction of neuronal apoptosis by thiol oxidation: putative role of intracellular zinc release. J Neurochem. 2000;75:1878–1888. doi: 10.1046/j.1471-4159.2000.0751878.x. [DOI] [PubMed] [Google Scholar]
- Berendji D, Kolb-Bachofen V, Meyer KL, Grapenthin O, Weber H, Wahn V, Kroncke KD. Nitric oxide mediates intracytoplasmic and intranuclear zinc release. FEBS Lett. 1997;405:37–41. doi: 10.1016/s0014-5793(97)00150-6. [DOI] [PubMed] [Google Scholar]
- Bolshakov AP, Mikhailova MM, Szabadkai G, Pinelis VG, Brustovetsky N, Rizzuto R, Khodorov BI. Measurements of mitochondrial pH in cultured cortical neurons clarify contribution of mitochondrial pore to the mechanism of glutamate-induced delayed Ca2+ deregulation. Cell Calcium. 2008;43:602–614. doi: 10.1016/j.ceca.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Talantova MV, Lee WD, Scholzke MN, Harrop A, Mathews E, Gotz T, Han J, Ellisman MH, Perkins GA, Lipton SA. Crosstalk between nitric oxide and zinc pathways to neuronal cell death involving mitochondrial dysfunction and p38-activated K+ channels. Neuron. 2004;41:351–365. doi: 10.1016/s0896-6273(04)00015-7. [DOI] [PubMed] [Google Scholar]
- Brown AM, Kristal BS, Effron MS, Shestopalov AI, Ullucci PA, Sheu KF, Blass JP, Cooper AJ. Zn2+ inhibits alpha-ketoglutarate-stimulated mitochondrial respiration and the isolated alpha-ketoglutarate dehydrogenase complex. J Biol Chem. 2000;275:13441–13447. doi: 10.1074/jbc.275.18.13441. [DOI] [PubMed] [Google Scholar]
- Budd SL, Nicholls DG. A reevaluation of the role of mitochondria in neuronal Ca2+ homeostasis. J Neurochem. 1996a;66:403–411. doi: 10.1046/j.1471-4159.1996.66010403.x. [DOI] [PubMed] [Google Scholar]
- Budd SL, Nicholls DG. Mitochondria, calcium regulation, and acute glutamate excitotoxicity in cultured cerebellar granule cells. J Neurochem. 1996b;67:2282–2291. doi: 10.1046/j.1471-4159.1996.67062282.x. [DOI] [PubMed] [Google Scholar]
- Busa WB. Spectral characterization of the effect of viscosity on Fura-2 fluorescence: excitation wavelength optimization abolishes the viscosity artifact. Cell Calcium. 1992;13:313–319. doi: 10.1016/0143-4160(92)90066-2. [DOI] [PubMed] [Google Scholar]
- Carter RE, Aiba I, Dietz RM, Sheline CT, Shuttleworth CW. Spreading depression and related events are significant sources of neuronal Zn2+ release and accumulation. J Cereb Blood Flow Metab. 2011;31:1073–1084. doi: 10.1038/jcbfm.2010.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colvin RA. pH dependence and compartmentalization of zinc transported across plasma membrane of rat cortical neurons. Am J Physiol Cell Physiol. 2002;282:C317–329. doi: 10.1152/ajpcell.00143.2001. [DOI] [PubMed] [Google Scholar]
- Devinney MJ, 2nd, Reynolds IJ, Dineley KE. Simultaneous detection of intracellular free calcium and zinc using fura-2FF and FluoZin-3. Cell Calcium. 2005;37:225–232. doi: 10.1016/j.ceca.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Dineley KE, Malaiyandi LM, Reynolds IJ. A reevaluation of neuronal zinc measurements: artifacts associated with high intracellular dye concentration. Mol Pharmacol. 2002;62:618–627. doi: 10.1124/mol.62.3.618. [DOI] [PubMed] [Google Scholar]
- Dineley KE, Devinney MJ, 2nd, Zeak JA, Rintoul GL, Reynolds IJ. Glutamate mobilizes [Zn2+] through Ca2+-dependent reactive oxygen species accumulation. J Neurochem. 2008;106:2184–2193. doi: 10.1111/j.1471-4159.2008.05536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmer PJ, Miranda JG, Gorski JA, Palmer AE. Genetically encoded sensors to elucidate spatial distribution of cellular zinc. J Biol Chem. 2009;284:16289–16297. doi: 10.1074/jbc.M900501200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazzini V, Rapposelli IG, Corona C, Rockabrand E, Canzoniero LM, Sensi SL. Mild acidosis enhances AMPA receptor-mediated intracellular zinc mobilization in cortical neurons. Mol Med. 2007;13:356–361. doi: 10.2119/2007-00047.Frazzini. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederickson CJ, Cuajungco MP, LaBuda CJ, Suh SW. Nitric oxide causes apparent release of zinc from presynaptic boutons. Neuroscience. 2002a;115:471–474. doi: 10.1016/s0306-4522(02)00399-8. [DOI] [PubMed] [Google Scholar]
- Gault LM, Lin CW, Lamanna JC, Lust WD. Changes in energy metabolites, cGMP and intracellular pH during cortical spreading depression. Brain Res. 1994;641:176–180. doi: 10.1016/0006-8993(94)91835-x. [DOI] [PubMed] [Google Scholar]
- Gee KR, Zhou ZL, Qian WJ, Kennedy R. Detection and imaging of zinc secretion from pancreatic beta-cells using a new fluorescent zinc indicator. J Am Chem Soc. 2002;124:776–778. doi: 10.1021/ja011774y. [DOI] [PubMed] [Google Scholar]
- Hartley Z, Dubinsky JM. Changes in intracellular pH associated with glutamate excitotoxicity. J Neurosci. 1993;13:4690–4699. doi: 10.1523/JNEUROSCI.13-11-04690.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz L. Features of astrocytic function apparently involved in the response of central nervous tissue to ischemia-hypoxia. J Cereb Blood Flow Metab. 1981;1:143–153. doi: 10.1038/jcbfm.1981.17. [DOI] [PubMed] [Google Scholar]
- Hyrc KL, Bownik JM, Goldberg MP. Ionic selectivity of low-affinity ratiometric calcium indicators: mag-Fura-2, Fura-2FF and BTC. Cell Calcium. 2000;27:75–86. doi: 10.1054/ceca.1999.0092. [DOI] [PubMed] [Google Scholar]
- Irwin RP, Lin S-Z, Long RT, Paul SM. N-methyl-D-aspartate induces a rapid, reversible, and calcium dependent intracellular acidosis in cultured fetal rat hippocampal neurons. J Neurosci. 1994;14:1352–1357. doi: 10.1523/JNEUROSCI.14-03-01352.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaev NK, Stelmashook EV, Lukin SV, Freyer D, Mergenthaler P, Zorov DB. Acidosis-induced zinc-dependent death of cultured cerebellar granule neurons. Cell Mol Neurobiol. 2010;30:877–883. doi: 10.1007/s10571-010-9516-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D, Sullivan PG, Sensi SL, Steward O, Weiss JH. Zn2+ induces permeability transition pore opening and release of pro-apoptotic peptides from neuronal mitochondria. J Biol Chem. 2001;276:47524–47529. doi: 10.1074/jbc.M108834200. [DOI] [PubMed] [Google Scholar]
- Jiang LJ, Vasak M, Vallee BL, Maret W. Zinc transfer potentials of the alpha - and beta-clusters of metallothionein are affected by domain interactions in the whole molecule. Proc Natl Acad Sci U S A. 2000;97:2503–2508. doi: 10.1073/pnas.97.6.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodorov B, Pinelis V, Vergun O, Storozhevykh T, Fajuk D, Vinskaya N, Arsenjeva E, Khaspekov L, Lyzin A, Isayev N, Andreeva N, Victorov I. Dramatic effects of external alkalinity on neuronal calcium recovery following a short-duration glutamate challenge: the role of the plasma membrane Ca2+/H+ pump. FEBS Lett. 1995;371:249–252. doi: 10.1016/0014-5793(95)00894-f. [DOI] [PubMed] [Google Scholar]
- Kiedrowski L. Cytosolic zinc release and clearance in hippocampal neurons exposed to glutamate - the role of pH and sodium. J Neurochem. 2011;117:231–243. doi: 10.1111/j.1471-4159.2011.07194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch RA, Barish ME. Perturbation of intracellular calcium and hydrogen ion regulation in cultured mouse hippocampal neurons by reduction of the sodium ion concentration gradient. J Neurosci. 1994;14:2585–2593. doi: 10.1523/JNEUROSCI.14-05-02585.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh J-Y, Suh SW, Gwag BJ, He YY, Hsu CY, Choi DW. The role of zinc in selective neuronal death after transient global cerebral ischemia. Science. 1996;272:1013–1016. doi: 10.1126/science.272.5264.1013. [DOI] [PubMed] [Google Scholar]
- Krężel A, Maret W. Zinc-buffering capacity of a eukaryotic cell at physiological pZn. J Biol Inorg Chem. 2006;11:1049–1062. doi: 10.1007/s00775-006-0150-5. [DOI] [PubMed] [Google Scholar]
- Land PW, Aizenman E. Zinc accumulation after target loss: an early event in retrograde degeneration of thalamic neurons. Eur J Neurosci. 2005;21:647–657. doi: 10.1111/j.1460-9568.2005.03903.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattanzio FA, Bartschat DK. The effect of pH on rate constants, ion selectivity and thermodynamic properties of fluorescent calcium and magnesium indicators. Biochem Biophys Res Commun. 1991;177:184–191. doi: 10.1016/0006-291x(91)91966-g. [DOI] [PubMed] [Google Scholar]
- Lee JY, Cole TB, Palmiter RD, Koh JY. Accumulation of zinc in degenerating hippocampal neurons of ZnT3-null mice after seizures: evidence against synaptic vesicle origin. J Neurosci. 2000;20:RC79. doi: 10.1523/JNEUROSCI.20-11-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li NC, Gawron O, Bascuas G. Stability of Zinc Complexes with Glutathione and Oxidized Glutathione. J Am Chem Soc. 1954:225–229. [Google Scholar]
- Link TA, von Jagow G. Zinc ions inhibit the QP center of bovine heart mitochondrial bc1 complex by blocking a protonatable group. J Biol Chem. 1995;270:25001–25006. doi: 10.1074/jbc.270.42.25001. [DOI] [PubMed] [Google Scholar]
- Malaiyandi LM, Vergun O, Dineley KE, Reynolds IJ. Direct visualization of mitochondrial zinc accumulation reveals uniporter-dependent and -independent transport mechanisms. J Neurochem. 2005;93:1242–1250. doi: 10.1111/j.1471-4159.2005.03116.x. [DOI] [PubMed] [Google Scholar]
- Mies G, Iijima T, Hossmann KA. Correlation between peri-infarct DC shifts and ischaemic neuronal damage in rat. Neuroreport. 1993;4:709–711. doi: 10.1097/00001756-199306000-00027. [DOI] [PubMed] [Google Scholar]
- Myers RE, Yamaguchi S. Nervous system effects of cardiac arrest in monkeys. Preservation of vision. Arch Neurol. 1977;34:65–74. doi: 10.1001/archneur.1977.00500140019003. [DOI] [PubMed] [Google Scholar]
- Ohana E, Hoch E, Keasar C, Kambe T, Yifrach O, Hershfinkel M, Sekler I. Identification of the Zn2+ binding site and mode of operation of a mammalian Zn2+ transporter. J Biol Chem. 2009;284:17677–17686. doi: 10.1074/jbc.M109.007203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmiter RD, Cole TB, Quaife CJ, Findley SD. ZnT-3, a putative transporter of zinc into synaptic vesicles. Proc Natl Acad Sci U S A. 1996;93:14934–14939. doi: 10.1073/pnas.93.25.14934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton CW. WEBMAXC STANDARD. 2009 http://www.stanford.edu/~cpatton/webmaxc/webmaxcS.htm.
- Poenie M. Alteration of intracellular Fura-2 fluorescence by viscosity: a simple correction. Cell Calcium. 1990;11:85–91. doi: 10.1016/0143-4160(90)90062-y. [DOI] [PubMed] [Google Scholar]
- Sensi SL, Paoletti P, Bush AI, Sekler I. Zinc in the physiology and pathology of the CNS. Nat Rev Neurosci. 2009;10:780–791. doi: 10.1038/nrn2734. [DOI] [PubMed] [Google Scholar]
- Sensi SL, Ton-That D, Sullivan PG, Jonas EA, Gee KR, Kaczmarek LK, Weiss JH. Modulation of mitochondrial function by endogenous Zn2+ pools. Proc Natl Acad Sci U S A. 2003;100:6157–6162. doi: 10.1073/pnas.1031598100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuttleworth CW, Weiss JH. Zinc: new clues to diverse roles in brain ischemia. Trends Pharmacol Sci. 2011;32:480–486. doi: 10.1016/j.tips.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemkowicz E, Hansen AJ. Clinical restitution following cerebral ischemia in hypo-, normo- and hyperglycemic rats. Acta Neurol Scand. 1978;58:1–8. [PubMed] [Google Scholar]
- Siemkowicz E, Hansen AJ. Brain extracellular ion composition and EEG activity following 10 minutes ischemia in normo- and hyperglycemic rats. Stroke. 1981;12:236–240. doi: 10.1161/01.str.12.2.236. [DOI] [PubMed] [Google Scholar]
- Smith ML, von Hanwehr R, Siesjo BK. Changes in extra- and intracellular pH in the brain during and following ischemia in hyperglycemic and in moderately hypoglycemic rats. J Cereb Blood Flow Metab. 1986;6:574–583. doi: 10.1038/jcbfm.1986.104. [DOI] [PubMed] [Google Scholar]
- Stout AK, Raphael HM, Kanterewicz BI, Klann E, Reynolds IJ. Glutamate-induced neuronal death requires mitochondrial calcium uptake. Nature Neuroscience. 1998;1:366–373. doi: 10.1038/1577. [DOI] [PubMed] [Google Scholar]
- Tiffert T, Lew VL. Kinetics of inhibition of the plasma membrane calcium pump by vanadate in intact human red cells. Cell Calcium. 2001;30:337–342. doi: 10.1054/ceca.2001.0241. [DOI] [PubMed] [Google Scholar]
- Trapp S, Lueckermann M, Kaila K, Ballanyi K. Acidosis of hippocampal neurones mediated by a plasmalemmal Ca2+/H+ pump. Neuroreport. 1996;7:2000–2004. doi: 10.1097/00001756-199608120-00029. [DOI] [PubMed] [Google Scholar]
- Vergun O, Han YY, Reynolds IJ. Glucose deprivation produces a prolonged increase in sensitivity to glutamate in cultured rat cortical neurons. Exp Neurol. 2003;183:682–694. doi: 10.1016/s0014-4886(03)00243-7. [DOI] [PubMed] [Google Scholar]
- Wang GJ, Randall RD, Thayer SA. Glutamate-induced intracellular acidification of cultured hippocampal neurons demonstrates altered energy metabolism resulting from Ca2+ loads. J Neurophysiol. 1994;72:2563–2569. doi: 10.1152/jn.1994.72.6.2563. [DOI] [PubMed] [Google Scholar]
- Wang GJ, Richardson SR, Thayer SA. Intracellular acidification is not a prerequisite for glutamate- triggered death of cultured hippocampal neurons. Neurosci Lett. 1995;186:139–144. doi: 10.1016/0304-3940(95)11305-g. [DOI] [PubMed] [Google Scholar]
- Wu ML, Chen JH, Chen WH, Chen YJ, Chu KC. Novel role of the Ca2+-ATPase in NMDA-induced intracellular acidification. Am J Physiol. 1999;277:C717–727. doi: 10.1152/ajpcell.1999.277.4.C717. [DOI] [PubMed] [Google Scholar]