

Abstract
Numerous studies have examined gene expression profiles in post-mortem human brain samples from individuals with schizophrenia compared to healthy controls, to gain insight into the molecular mechanisms of the disease. While some findings have been replicated across studies, there is a general lack of consensus of which genes or pathways are affected. It has been unclear if these differences are due to the underlying cohorts, or methodological considerations. Here we present the most comprehensive analysis to date of expression patterns in the prefrontal cortex of schizophrenic compared to unaffected controls. Using data from seven independent studies, we assembled a data set of 153 affected and 153 control individuals. Remarkably, we identified expression differences in the brains of schizophrenics that are validated by up to seven laboratories using independent cohorts. Our combined analysis revealed a signature of 39 probes that are up-regulated in schizophrenia and 86 down-regulated. Some of these genes were previously identified in studies that were not included in our analysis, while others are novel to our analysis. In particular, we observe gene expression changes associated with various aspects of neuronal communication, and alterations of processes affected as a consequence of changes in synaptic functioning. A gene network analysis predicted previously unidentified functional relationships among the signature genes. Our results provide evidence for a common underlying expression signature in this heterogeneous disorder.
Keywords: schizophrenia, gene expression, microarray, postmortem brain, prefrontal cortex
Introduction
Schizophrenia is a severe psychotic disorder that affects approximately one percent of the population worldwide (1). Many groups have attempted to identify changes in gene expression in the brains of schizophrenics, often focusing on the prefrontal cortex (2–4). Such studies have suggested several altered molecular processes including (but not limited to) synaptic machinery and mitochondrial-related transcripts (5–8), immune function (9) and a reduction in oligodendrocyte and myelination-related genes (10–12). The variety and scope of these processes, found in different subject cohorts, raises the question as to whether there are underlying commonalities in molecular signatures among schizophrenics. Such commonalities are presupposed by most genetic studies, which look for alleles overrepresented in large numbers of schizophrenic individuals (13–15). It is important to establish if there are any common features of the disease at the molecular level.
The diversity of results in transcriptome studies can be attributed to many sources. Besides differences in the sampled cohorts and disease heterogeneity, discrepancies between transcriptome studies can be due to methodological differences in sample preparation, choice of platform, and data analysis. There are issues that are especially pertinent to the analysis of post-mortem human brain tissue. One is the confounding effect of factors such as age, gender and medication. Such factors are often associated with relatively large gene expression changes (16), while psychiatric illnesses such as schizophrenia are associated with small effect sizes. If these factors are not correctly controlled for, they can mask or masquerade as expression patterns associated with the disease. Standard practice involves minimizing the effects of such factors either in the experimental design by sample matching or treating these factors as covariates in regression models. It is also increasingly appreciated that technical artifacts such as ‘batch effects’ can result in substantial variability (17–20). In addition, post-mortem brain tissue is a limited resource, leading to small sample sizes with low statistical power. For this reason, most studies have not applied multiple test correction, and perform validation only on the same RNA samples that were used for profiling. All of these issues are likely to contribute to the differences in findings across studies. We propose that a good way to address these problems is to re-analyze and meta-analyze the studies in question, a task we undertake in this paper.
The use of meta-analyses to combine high-throughput genomics studies has become increasingly used in neuropsychiatry (14, 17, 21–23). Combining datasets across studies increases power and facilitates the identification of gene expression changes that are consistent and reliable, reducing false positives. In a meta-analysis, multiple studies are statistically pooled to provide an overall estimate of significance of an effect, highlighting important yet subtle variations. While meta-analysis has been used in the study of gene expression data (24–26), to our knowledge only a few studies have done so with post-mortem human brain data (17, 22, 27, 28). A cross-study analysis of psychosis was conducted across seven datasets using samples from the Stanley Medical Research Institute (SMRI) post-mortem brain collections (22). Additionally, the SMRI report results from a cross-study analysis across schizophrenia datasets in their online genomics database (http://stanleygenomics.org), computing ‘consensus’ fold changes while adjusting for confounding variables. However, the studies used in these analyses use samples from the same two brain collections and are therefore not entirely independent. More recently, a comparative analysis was conducted across two independent schizophrenia cohorts; probes were identified as differentially expressed within each study and the intersecting probes between the two studies were reported (29). Thus while there have been attempts to meta-analyze schizophrenia expression profiling data, there has not yet been an integration using the primary data of more than two independent microarray studies.
In the current study we present a cross-study analysis of seven microarray datasets comprising a total of 153 schizophrenia samples and 153 normal controls. We applied a linear modeling approach to control for factors such as age, brain pH and batch effects, and applied multiple testing corrections to control the false discovery rate. We show that we are able to detect small yet consistent and statistically significant changes. Careful control of extraneous factors using probe-specific statistical modeling, results in gene expression changes associated with the disease effect. Our results confirm some previously reported expression changes in schizophrenia in addition to identifying potential new targets suggesting alterations in synaptic function.
Materials and Methods
Data pre-processing and quality control
Genome-wide expression data sets were selected on the basis of microarray platform, use of prefrontal cortex (BA 9, 10 or 46), the availability of information on covariates such as age, and finally the availability of the raw data. Each dataset is comprised of a cohort of neuropathologically normal subjects and a cohort of schizophrenia subjects, as diagnosed and reported in their respective studies (Table 1). Sources for data include the Stanley Medical Research Institute (SMRI), the Harvard Brain Bank, and the Gene Expression Omnibus (GEO). GEO studies were identified by extensive manual and keyword searches. While the SMRI has additional data sets, these represent repeated runs of the samples from the same subjects, so we selected one dataset to represent each of the two SMRI brain collections. Two additional studies were obtained from the authors (30, 31). Datasets consisted of single-channel intensity data generated from two Affymetrix platforms, but only probe sets on the HG-U133A chip from each dataset were used for analysis. Probe sets were re-annotated at the sequence level by alignment to the hg19 genome assembly, using methods essentially as described in (20), and also cross-referenced with problematic probe lists provided by http://masker.nci.nih.gov/ev/. The raw data (“CEL”) files from all the datasets were pooled together and expression levels were summarized, log transformed and normalized by using the R Bioconductor ‘affy’ package (R Development Core Team, 2005) using default settings for the RMA algorithm. Data was also processed using four other pre-processing methods for evaluating the robustness our meta-signatures (see Supplementary Text). We decided to retain standard RMA as the method on which to centre our analysis, because RMA has been shown independently to be a high performer on gold standard data sets (32–34). Sample outliers were then identified and removed from each dataset based on inter-sample correlation analysis (see Supplement), resulting in the removal of 13 samples (2 of these are the same outliers identified in a previous analysis of SMRI data; http://stanleygenomics.org). Batch information was obtained using the ‘scan date’ stored in the CEL files; chips run on different days were considered different batches. The final data matrix consisted of expression values for 22,215 probes sets and 306 samples. Sample characteristics for the subjects were collected and are summarized in Table 2.
Table 1.
Schizophrenia Datasets
| Dataset | Reference | Microarray Platform | Brain region(s) | No. of Subjects CTL:SZ |
|---|---|---|---|---|
| Stanley Bahn | SMRI database | HG-U133A | Frontal BA46 | 31 : 34 |
| Stanley AltarC | SMRI database | HG-U133A | Frontal BA46/10 | 11 : 9 |
| Mclean | HBTRC | HG-U133A | Prefrontal cortex (BA9) |
26 : 19 |
| Mirnics | Garbett K. et al, 2008 (30) | HG-U133A/B | Prefrontal cortex (BA46) |
6 : 9 |
| Haroutunian | Katsel P. et al, 2005 (31) | HG-U133A/B | Frontal (BA10/46) |
29 : 31 |
| GSE17612 | Maycox P. et al, 2009 (29) | HG-U133 Plus 2.0 | Anterior prefrontal cortex (BA10) |
21: 26 |
| GSE21138 | Narayan S. et al, 2008 (52) | HG-U133 Plus 2.0 | Frontal (BA46) | 29 : 25 |
SMRI, Stanley Medical Research Institute; HBTRC, Harvard Brain Tissue Resource Centre (Mclean66 collection)
Table 2.
Summary of demographic variables across combined cohort
| Control | Schizophrenia | P-value | |
|---|---|---|---|
| Number of Subjects | 153 | 153 | |
| Age | 56.25 ± 20 | 55.27 ± 19 | p = 0.67 |
| Sex | 101M : 52F | 113M : 40F | p = 0.1 |
| Brain pH | 6.5 ± 0.28 | 6.39 ± 0.29 | p = 0.001 |
| PMI | 21.95 ± 15.3 | 22.65 ± 15.2 | p = 0.69 |
F, female; M, male; PMI, post-mortem interval. There were 319 samples collected across seven datasets of which 306 passed quality control analysis. The summary demographics (mean ± standard deviation) and t- test p-values for group differences are shown for those subjects used in the analysis. For sex we report the p-value generated from a chi-squared test for equality of proportions.
Statistical Modeling
Gene expression values for each probe set were modeled using a standard fixed effects linear model (FEM) framework. We treated Disease, Age, Brain pH, Batch date and Study as fixed effects for which unknown constants are to be estimated from the data. We also employed a model selection procedure, in which each probe set was modeled using the full model including all five factors, as well as various sub-models (details in Supplemental Methods; our approach is similar to that used in (35)). For each probe set, the t-statistic for the disease effect was then extracted from the selected model and p-values were computed using one-sided tests, preformed independently for the two alternative null hypotheses (i.e. gene expression does not increase with schizophrenia and gene expression does not decrease with schizophrenia). The resulting p-values for the up- and down-regulated signatures were further adjusted for multiple testing using the q-value method (36) to control the false discovery rate (FDR).
Literature-derived signatures
Our signatures were compared to probe lists obtained from the original publication for each of the datasets used in our analysis. As the two SMRI datasets were unpublished, gene lists were compiled from the SMRI online genomics database. For the Mclean dataset we used the list of ‘significant probes’ as reported in (29). For the Haroutunian data we chose to use probes selected at the ‘low stringency criteria’ described in (31). Details on each of these gene sets can be found in Table 4 (probes were excluded if they were not on the HG-U133A chip). Additional signatures for comparison were obtained for published schizophrenia expression profiling studies, and a list of the top 45 candidate schizophrenia genes reported in the SZGene database (13). Agreement of the meta-signature ranking with each validation gene set was assessed using receiver operating characteristic (ROC) curve analysis described in greater detail in Supplementary Methods.
Table 4.
Comparison of meta-signatures with findings from original study
| Significance Criteria | Down-regulated | Up-regulated | |||||
|---|---|---|---|---|---|---|---|
| Dataset | Probes | AUC | Overlap | Probes | AUC | Overlap | |
| Stanley AltarC | p < 0.05 | 848 | 0.70 | 14 | 34 | 0.78 | 0 |
| Stanley Bahn | p < 0.05 | 69 | 0.85 | 6 | 91 | 0.89 | 5 |
| Mclean (29) | p < 0.05, intensity > 30 | 570 | 0.75 | 13 | 300 | 0.76 | 7 |
| Mirnics (30) | p < 0.05, |ALR| > 0.58 | 7 | 0.55 | 0 | 4 | 0.94 | 1 |
| Haroutunian (BA10) (31) |
p < 0.05, |FC| > 1.4, present calls > 60% |
5 | 0.5 | 0 | 14 | 0.66 | 1 |
| Haroutunian (BA46) (31) |
p < 0.05, |FC| > 1.4, present calls > 60% |
50 | 0.21 | 0 | 11 | 0.59 | 0 |
| GSE17612 (29) | p < 0.05, intensity > 30 | 548 | 0.74 | 22 | 466 | 0.71 | 7 |
|
GSE21138 (52) (Short DOI) |
p < 0.05; |FC| > 1.25 | 482 | 0.50 | 1 | 173 | 0.57 | 1 |
|
GSE21138 (52) (Int DOI) |
p < 0.05; |FC| >1.25 | 132 | 0.60 | 0 | 78 | 0.69 | 0 |
|
GSE21138 (52) (Long DOI) |
p < 0.05; |FC| > 1.25 | 37 | 0.63 | 1 | 89 | 0.63 | 1 |
DOI, duration of illness; AUC, area under the curve; FC, fold change
The findings from each dataset are summarized including only probes used in our analysis. The ‘Probes’ column indicates number of probes found from the study based on study specific significance criteria. AUC values were computed from an ROC analysis of each gene set against the corresponding ranked meta-signatures. Overlap values report the number of probes in each gene set that overlaps with probes from the meta-signatures at q < 0.1.
Functional and network analysis
We analyzed each signature for enrichment of Gene Ontology (GO) terms (37), using the gene score re-sampling (GSR) method in ErmineJ (38, 39). We also evaluated the path-length and node degree (number of associations) properties of the meta-signature genes in a large human protein-protein interaction network (PPIN) obtained by aggregating data from multiple sources(40–45). The network contains 100,623 unique interactions among 11,697 genes. Path lengths in the network were measured using Dijkstra’s algorithm (46). Statistical significance was assessed by reference to an empirical null distribution obtained by randomly sampled 10000 gene sets of similar size and node degree.
Results
Schizophrenia and control groups had no significant differences in age and PMI, and the number of males and females between the groups were fairly well matched (Table 2). Brain pH was significantly different (t-test; p = 0.001). P-value distributions for each demographic variable were also assessed to help determine the selection of factors used as fixed effects for our model. We found it was necessary to correct for “batch effects” (technical artifacts caused by running chips on different days or even years (20)), as they contributed the vast majority of variance in gene expression (Supplementary Figure 1). Each factor was considered in a model selection procedure (see Methods and Supplementary Methods), and a final set of linear models were used to identify probe sets that were differential expressed between schizophrenic and control samples. After multiple test correction we identified a meta-signature of 39 up-regulated and 86 down-regulated probes at an FDR of 0.1 (Supplementary Table 2, Table 3). If we assess the number of unique genes that appear in each signature we obtain a list of 25 up-regulated and 70 down-regulated genes. These numbers highlight several cases of a gene which appears in our signature more than once, suggesting higher confidence in the finding of expression changes for those genes. Figure 1 shows the expression levels top down-regulated probe we identified. As expected, expression changes were small (~ 15% expression change), and more evident in some datasets. As required by our modeling procedure, the direction of expression changes is mostly consistent.
Figure 1. Example of consistent expression changes for a gene across data sets.

Expression data within each dataset after covariate correction is presented for the top down-regulated gene NECAB3. Plots are labelled with the associated dataset. Samples were separated into disease and control cohorts and expression was plotted as a boxplot. Individual sample values were overlaid on with red squares representing control individuals and blue triangles representing schizophrenics.
To test the robustness of these findings, we used a jackknife procedure, sequentially removing one of the seven studies and performing the meta-analysis on the remaining six, for each study in turn. We expected that results highly influenced by a single data set would not be stable across jackknife runs. Each leave-out iteration resulted in a new meta-signature, which was then ranked by q-value and compared against the final meta-signature (Supplementary Table 3). The range of rank correlations among jackknife iterations (0.87 – 0.99) illustrates the robustness of our meta-signatures, demonstrating that our results are not highly biased by any single dataset. The lowest correlations were observed upon removal of the Bahn and GSE21138 datasets (0.88 and 0.87, respectively) suggesting that these datasets may be contributing a slightly stronger signal, particularly to the up-regulated signature. The lack of significant genes at a q < 0.1 in the signature for those jackknife runs corroborates this finding. Finally, the top 100 probes were taken from each jackknife signature and an intersection set was retained to form a ‘core signature’ of 16 down-regulated and 14 up-regulated probes (Table 3). We consider these probes to be the most reliable findings from our study as they are relatively insensitive to the choice of data sets used. In Figure 2, we have assembled the ‘core signatures’ and plotted expression levels within each dataset with samples separated into control and schizophrenia groups. For some studies we observe a more obvious gradient between the two groups illustrating expression change, and for others the difference is more subtle.
Table 3.
Core signatures retained after jackknife validation
|
A: Up-regulated in schizophrenia | ||||
|---|---|---|---|---|
| Probe | Gene Symbol | Gene Description | Fold Change | Q-value |
| 210057_at | SMG1g | SMG1 homolog, phosphatidylinositol 3-kinase- related kinase |
1.04 | 0.009 |
| 202619_s_at | PLOD2▲ | procollagen-lysine, 2-oxoglutarate 5-dioxygenase 2 |
1.12 | 0.05 |
| 203548_s_at | LPL▲ | lipoprotein lipase | 1.11 | 0.009 |
| *202975_s_at | RHOBTB3 b | Rho-related BTB domain containing | 1.16 | 0.063 |
| *216048_s_at | 3 | 1.11 | 0.02 | |
| *213015_at | BBX b | bobby sox homolog (Drosophila) | 1.05 | 0.082 |
| 213016_at | 1.11 | 0.086 | ||
| 219426_at | EIF2C3 b | |||
| 213187_x_at | FTL | ferritin, light polypeptide | 1.11 | 0.022 |
| 207543_s_at | P4HA1g | prolyl 4-hydroxylase, alpha polypeptide I |
1.12 | 0.074 |
| 218345_at | TMEM176A | transmembrane protein 176A | 1.10 | 0.071 |
| 221503_s_at | KPNA3 | karyopherin alpha 3 (importin alpha 4) |
1.02 | 0.072 |
| 209069_s_at | LOC440093b, m H3F3B |
multiple gene mappings | 1.11 | 0.020 |
| 209747_at | BCYRN1b, g TGFB3 |
Multiple gene mappings | 1.11 | 0.072 |
|
B: Down-regulated in schizophrenia | ||||
|---|---|---|---|---|
| Probe | Gene Symbol | Gene Description | Fold Change | Q-value |
| 210720_s_at | NECAB3 | N-terminal EF-hand calcium binding protein |
0.92 | 0.006 |
| 212646_at | RFTN1 | raftlin, lipid raft linker 1 | 0.91 | 0.006 |
| 220807_at | HBQ1 | hemoglobin, theta 1 | 0.91 | 0.007 |
|
*206355_at 206356_s_at |
GNAL | guanine nucleotide binding protein G(olf) subunit alpha |
0.91 0.90 |
0.007 0.048 |
| 220741_s_at | PPA2a, g | pyrophosphatase (inorganic) 2 | 0.92 | 0.009 |
| 204679_at | KCNK1▲ | potassium channel, subfamily K, member 1 | 0.88 | 0.087 |
| 206215_at | OPCML | opiate binding cell adhesion molecule | 0.94 | 0.074 |
| *219032_x_at | OPN3m | opsin 3 | 0.89 | 0.027 |
| *212987_at | FBXO9▲ a, b, g | F-box protein 9 | 0.88 | 0.033 |
| 206290_s_at | RGS7 | regulator of G-protein signaling 7 | 0.89 | 0.043 |
| 203719_at | ERCC1 | DNA excision repair protein | 0.94 | 0.042 |
| 202688_at | TNFSF10b | tumor necrosis factor (ligand) superfamily, member 10 | 0.83 | 0.075 |
| 218653_at | SLC25A15 | solute carrier family 25 (mitochondrial carrier; ornithine transporter) member 15 |
0.94 | 0.042 |
| 205510_s_at | GABPB1 b, g FLJ10038 |
multiple gene mappings | 0.93 | 0.027 |
| *213924_at | Unknowna | Unknown | 0.89 | 0.006 |
Figure 2. Expression changes in the ‘core signatures’.

For each probe in the core signatures (meaning they are retained as significant even after the removal of any single study), the corresponding data from each study was extracted and converted to a heat map. Expression values were normalized across all samples within each dataset, and as in Figure 1 the data are corrected for the covariates such as batch and age. Rows represent probes and are labeled with its unique gene mapping if one exists. Columns represent samples. Grey bars represent the control brain samples, and the black bar represents the schizophrenia samples. Light values in the heat map indicate higher expression values.
To assess the sensitivity of our results to the choice of pre-processing algorithm we re-analyzed our data with four different methods (see Supplemental Text). We obtained good agreement between the results of each method and our final meta-signatures despite dramatic changes to the preprocessing procedure (Supplementary Table 4). Additionally, we took the intersection of significant probes from each of the different methods to assemble a list of probes that are completely insensitive to the choice of preprocessing method. This list comprises a total of 5 up-regulated and 8 down-regulated probes, highlighting novel genes and genes that have been previously implicated in independent studies (Table 3; Supplementary Table 2).
The set of differentially expressed genes identified from our analysis implicates a variety of genes and functional groups, many of which have been previously reported in the literature. For example, down-regulation of mu-crystallin (CRYM), potassium channel subfamily K member 1 (KCNK1), F-box protein 9 (FBXO9) and up-regulation of lipoprotein lipase (LPL) and lysyl hydroxylase 2 (PLOD2) are concordant with findings from previous studies (7, 9, 12, 47). We manually evaluated the significant genes in our list (q < 0.1) individually according to literature reports and Uniprot definitions for each, to characterize genes into high-level functional categories. In the down-regulated signature we found genes to cluster into functional groups pertaining to various molecular mechanisms of neuronal communication. On the pre-synaptic side we find genes involved in cell adhesion (for example, OPCML), and neurotransmitter secretion (for example, APBA2, PCSK2). We also find genes involved in signalling pathways that elicit metabotropic effects (for example, GNAL, OPN3, CRHR, RGS7, GNB5). Concordant with previous studies, we also identified various genes involved in oxidative phosphorylation (for example, CYP26B1, COQ4, SLC25A15, ATP5C1, SLC25A12) and ubiquitination (for example, FBXO9, COPS7B, USP19, TACC2, DCAF8). From our up-regulated signature we find a number of transcription-related genes (for example, BAZ1A, CBFA2T2, BBX, ANP32A) and genes involved in translation (for example, EIF3E, EIF2C3, PAIP2B). Other genes include cell organization/maintenance factors (for example, PKP4, PLOD2) and various stress response genes (for example, SMG1). Additionally for both signatures we find a small group of genes with unknown function.
We performed a functional analysis to systematically detect enrichment of biological processes, using Gene Ontology (GO) annotations. After multiple test correction, we were unable to identify any significant terms using the over-representation method (ORA), but significant terms found using the threshold-free GSR algorithm (39) corroborate findings from the above manual evaluation. For genes with decreasing expression levels in schizophrenia, the top GO categories included those involved in energy metabolism, and ubiquitination, neurotransmitter transport and various metabolic processes. The schizophrenia up-regulated genes showed enrichment in various immune-related GO categories in addition to terms related to cellular localization (full results from this analysis can be found in Supplementary Table 5).
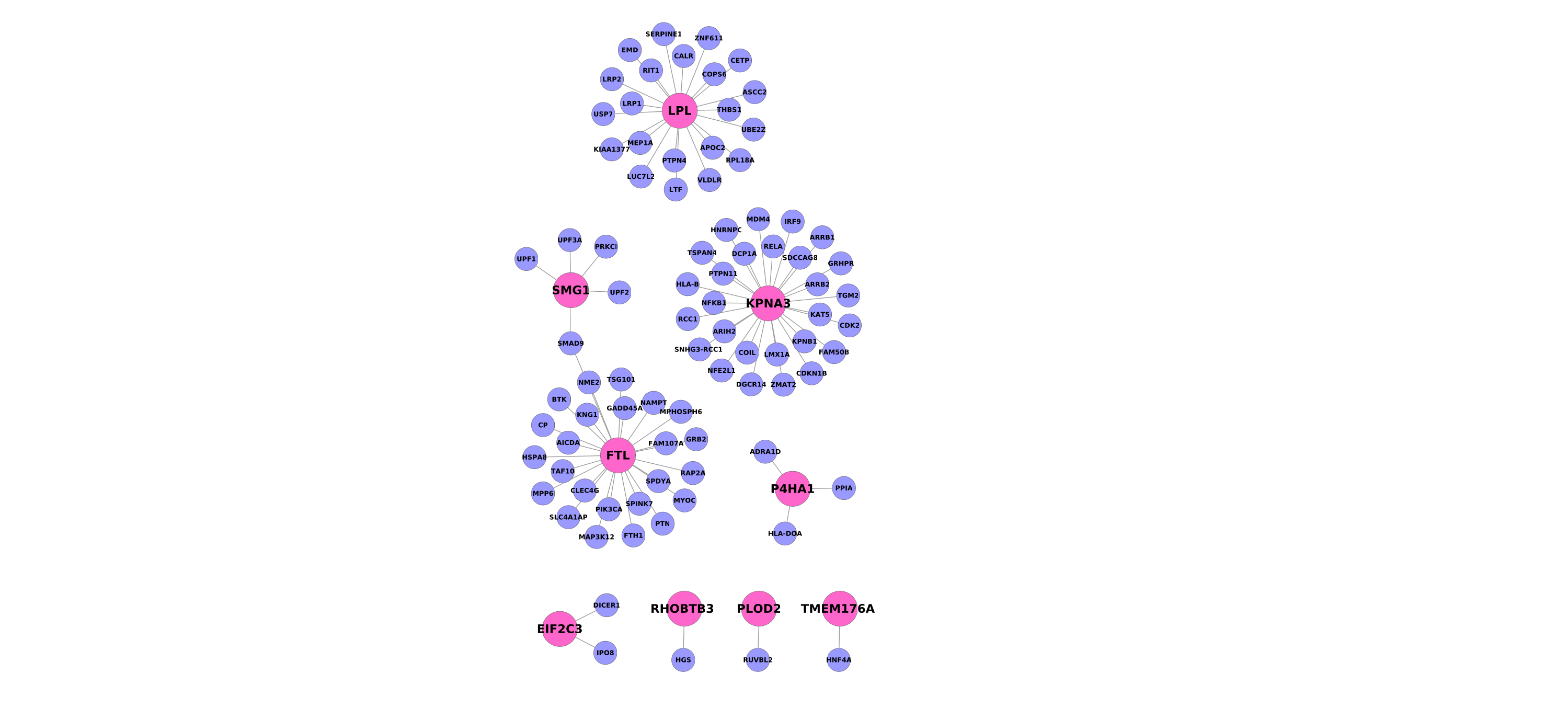
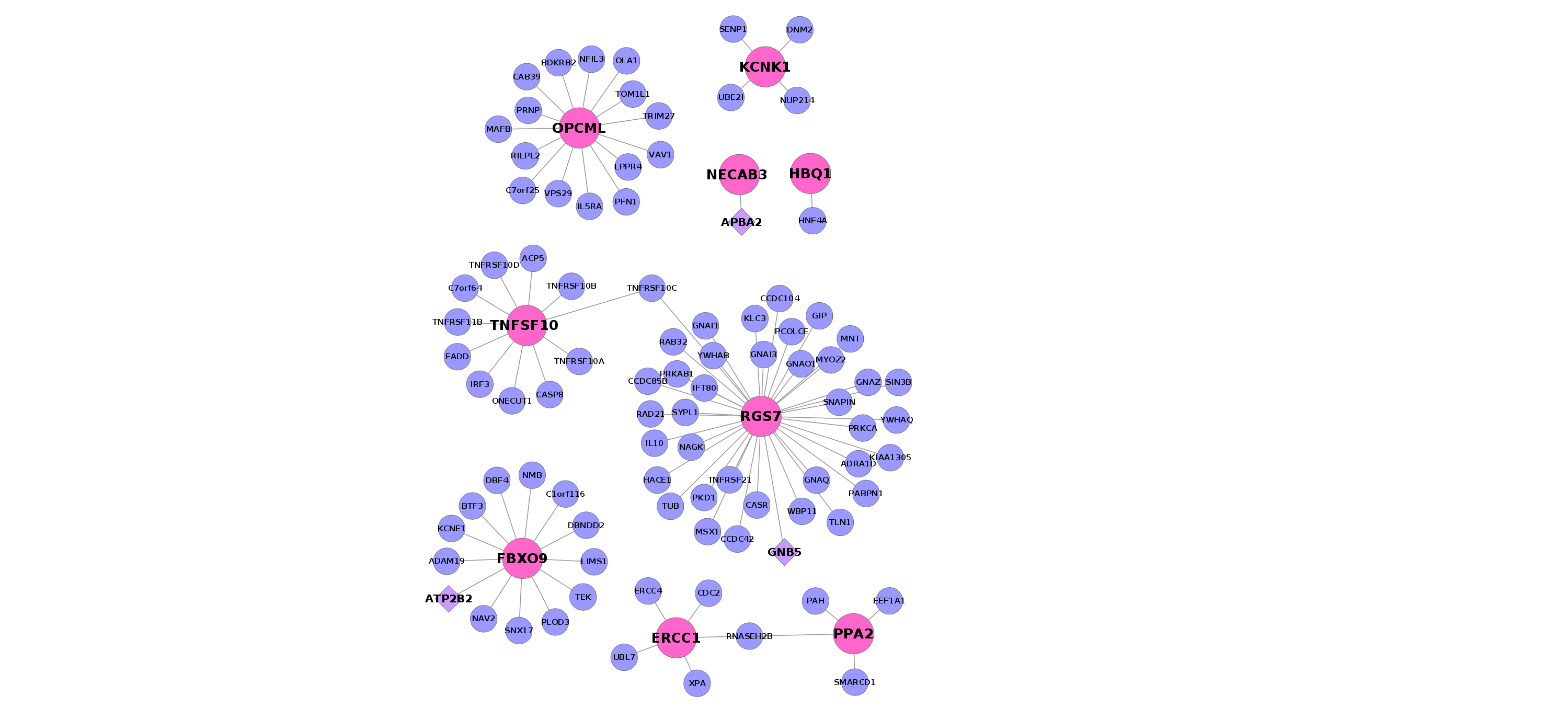
Because the genes we identified were functionally diverse, we hypothesized there might be additional insight gained at the level of gene networks. In particular we asked whether the signature genes had any unusual properties in their protein interaction patterns, compared to carefully selected groups of background genes (see Methods). We specifically looked at within-group connectivity, node degree (the number of connections) and path lengths between genes. Our most striking finding is that the genes within our set were significantly closer to one another in the network than expected by chance (p<0.02). This relationship suggests a higher likelihood of functional relationships among the signature genes (41, 48).
In contrast, the signature genes did not possess a particularly high node degree within the network (23rd percentile in the whole network), that is, they tend not to be ‘hubs’. We illustrate these properties for the up and down-regulated “core” signature genes in Supplementary Figure 2.
We also evaluated each meta-signature against modules of co-expressed genes in the human cortex as reported in(49). Details on this analysis can be found in the Supplemental Methods. Our up- and down-regulated signatures significantly overlap with the “turquoise” and “brown” modules (p < 0.01 and p < 0.05 respectively; Supplementary Table 6). These are modules of interest as they display a notable extent of preservation across datasets in (49), suggesting that differential expression of our signature genes may be disrupting core networks in the human brain. This also reinforces the importance of gene network structure analysis in determining the basis of this disorder.
To characterize our schizophrenia signatures with respect to cellular organization in the cortex we cross-referenced our ranked meta-signatures with published lists of CNS cell type markers (50). An ROC analysis of the meta-signatures for astrocytes, oligodendrocytes and neurons revealed no preferential association with our ranked meta-signatures. However, evaluating only the significant probes (q<0.1) in our signatures, we find an enrichment of probes mapping to neuronal markers in the down-regulated signature (Supplementary Table 2). While our linear modeling approach controlled for the effects of age and brain pH, we checked our signatures against gene lists for pH and age from our previous study of normal post-mortem human brain (16). The overlap was significant only for our down-regulated signature, which contains 33 genes previously identified to be down-regulated by age. Because our profiles are age-corrected and our cohorts age-matched, this suggests overlap in expression changes in age and schizophrenia rather than a confounding effect. We also sought to address factors that were not accounted for in our linear modeling, such as medication effects and alcohol and drug abuse. Using gene lists provided from the SMRI Online Genomics Database (http://www.stanleygenomics.org), we extracted significant gene lists (p < 0.001; FC>1.2) pertaining to the effects of lifetime alcohol use (23 genes), lifetime drug use (26 genes), and lifetime antipsychotics (69 genes) in subjects with schizophrenia. A comparison of each of these lists to our meta-signatures identified only two overlapping genes. We found KCNK1, which is present in our down-regulated signature, also increases with lifetime alcohol use. From the up-regulated signature the gene LPL, appears to increase with lifetime antipsychotic use and decrease with increased drug use.
Each meta-signature was evaluated against the top 45 candidate schizophrenia genes reported in the SZGene database (http://www.szgene.org/). Agreement of the meta-signature ranking with the SZGene set was assessed using receiver operating characteristic (ROC) curve analysis. The SZGene list appears to be randomly distributed across our ranking. We also computed a simple overlap between the 45 candidate genes and our results, identifying OPCML as the only common gene.
We were interested in comparing our re-analysis of these seven data sets to the “hit lists” provided by the data set providers. We first tested whether our meta-signature gene rankings were enriched for genes reported by the original study, using ROC analysis (Table 4; see Supplementary Methods). We observed high AUC scores for most gene sets; however the Haroutunian and GSE21138 studies exhibited exceptionally low scores, possibly in part because the original studies have an added dimension of variability as gene sets were generated for stratified cohorts as opposed to a case versus control comparison. While high AUCs suggest some similarity in the results, a more sensitive analysis examines just the very top of the rankings. We therefore computed the overlap of each reference gene set with the meta-signature of genes collected at q<0.1. This reveals a handful of probes in each study that also show up in our significant gene lists (Table 4). We also re-analyzed each individual dataset using our linear modeling approach. This allowed a more fair evaluation of the contribution of each to the final meta-signatures, since the original studies used a variety of methods for gene selection. After correcting for multiple testing, only two of the data sets (Altar and Haroutunian) yielded significant genes at q < 0.1. We therefore considered the top 100 probes from each dataset, and computed overlaps with our meta-signatures (Supplementary Table 7). The overlap is highest with the Bahn and GSE21138 datasets, which is in accord with the finding that these datasets contribute a stronger signal to the meta-signature than the others. Despite being the only two data sets which have significant differential expression after multiple test correction, the Altar and Haroutunian results showed very little overlap with the final meta-signature. We note that considering the 7 data sets independent of our meta-signature, there was no overlap among their top 100 probes. Similarly, there was little correlation of the overall rankings of probes among the data sets (Supplementary Table 8). Overall these results suggest that our re-analysis is concordant with the analysis conducted by the original study authors, subject to important differences likely attributable to our analytic approach (for example, correction for batch effects), and only revealing commonalities through meta-analysis which contribute weakly to the findings of the individual studies.
Discussion
In this study we present expression changes associated with schizophrenia consistent across up to seven independent cohorts of subjects. To our knowledge, the degree of validation and confirmation inherent in our analysis is unprecedented. Unlike previous studies, which use PCR assays to check results on the same RNA samples used for microarrays, or which compare at most two cohorts, we identified changes in expression that are shared across independent subject cohorts, analyzed by laboratories distributed around the world. Our study provides a new window into the molecular changes that might underlie schizophrenia.
The larger number of down-regulated probes (86 vs. 39) is in agreement with previous reports (2, 8, 9). Many of the genes we have identified have been previously reported to be expressed in the brain, with some genes showing neuronal specificity. Some of the genes we report as differentially expressed have been previously implicated in schizophrenia, either through expression profiling studies of schizophrenia (KCNK1, CRYM, FBXO9), or genetic association studies (OPCML (15)). We also identify three genes in our signature (up-regulated genes WNK1 and ABCA1 and down-regulated gene SNN) that overlap with results from a comparative analysis of two of the studies we used (29). Additionally, we found functional gene groups discussed in previous expression studies of schizophrenia. Many of the same metabolic processes were observed in a study of 71 different metabolic genes groups in schizophrenia (7). Also in agreement, various energy pathway genes were found previously in DLPFC studies of schizophrenia (47, 51). Over-expression of immune responses from our GSR analysis is also concordant with recent findings of over-expression in genes related to immune function in schizophrenia (9, 52, 53).Thus, our results are supportive of at least some previous findings and reveal a previously unrecognized similarity across studies.
Our meta-signatures contain a number of interesting new candidate genes, particularly our down-regulated meta-signature which potentially reflects alterations in neuronal communication. NOVA1 is a regulator of RNA splicing recently found to inhibit splicing of exon6 from the dopamine receptor D2 gene resulting in D2L, the long isoform of the receptor (54). With NOVA1 decreasing in expression in schizophrenia, inhibition may be repressed leading to higher than normal levels of the spliced D2S isoform which is involved in neuron firing and dopamine release. The DLGAP1 gene encodes a protein interacting with PSD-95 and a complex of other proteins in the postsynaptic density. Decreased expression of this scaffold protein may have consequences for anchoring and organizing receptors and signalling molecules on the postsynaptic side. Moreover, we have identified several genes associated with calcium signalling (CACNB3), binding (SLC25A12, NECAB3) and homeostasis (CCL3, ATP2B2), processes of likely relevance to schizophrenia (55). We have also identified genes that associate with the G-protein coupled receptor (GPCR) signalling pathway. One example is GNAL, a gene encoding for the alpha subunit of the G-protein Golf, expressed in many regions of the brain. Given the critical roles of G-proteins it is plausible that GNAL (and other GPCR related genes) may have a role in the pathophysiology of schizophrenia (56). GNAL expression has not been previously shown to be affected by schizophrenia, but it is located in a chromosomal region (18p.2) that has been linked to schizophrenia and bipolar disorder. More specifically, a di-nucleotide repeat in intron 5 of the GNAL gene has been linked to schizophrenia in some families (57). These expression changes concerning synaptic function may reduce neuronal energy demand in the brains of affected patients thus providing explanation for the down-regulation of various oxidative phosphorylation and energy metabolism genes that we observe.
We also sought to examine whether our signature genes could be inferred to share some previously unknown function, making use of gene network analysis. One way to do this is by the principle of “guilt by association”, which states that genes with shared function are more likely to interact (58). However, the meta-signature genes have a fairly low number of interaction partners, making “guilt” difficult to ascertain. Another property to examine is path length in the network, where genes that have short paths between them might be more functionally related. In general, low node degrees would imply higher path lengths among the genes, but was not the case for our gene set. That is, the signature genes are linked by unusually short paths in the network. Additionally, we found each of our meta-signatures revealed a significant overlap with previously identified gene co-expression modules in the human cortex (49). This suggests a relationship among the genes that is not reflected in current annotations and a network analysis of these schizophrenia genes will need to be investigated in greater detail in future studies.
We found that some of the down-regulated schizophrenia genes overlap with genes that decrease in expression with age. Many of the biological processes affected by age also tend to appear as affected processes in schizophrenia, both in this study and existing profiling studies (7, 9, 12, 47, 51). These findings suggest that many genes affected by age are also affected by schizophrenia, but also raise the possibility of confounding effects. As these effects could be confounded, one could filter the list of schizophrenia candidate genes from our results by simply removing known age- and pH-affected genes from the final signature (leaving 31 up- and 51 down-regulated probes) to investigate these effects more thoroughly.
Our results should be interpreted in the context of several caveats. First, our approach is specifically designed to find concordant results across studies, and does not detract from the potential novel findings that might be found in any single data set. We do suggest that genes found to be commonly differentially expressed by multiple studies are of particularly high value in identifying underlying etiological influences in schizophrenia. As is the case for all postmortem brain studies, we also cannot be sure that the expression changes we have identified are direct effects of the illness or are secondary to an underlying pathology. An additional caveat is that because we were unable to obtain medication or illicit drug use information for all subjects, we were not able to incorporate this information into our analysis. To help address this we compared our signatures against gene lists derived from a recent review on convergent antipsychotic mechanisms(59). We observed no overlap with our signatures. In addition to antipsychotics, the use and abuse of other recreational drugs and smoking are also compounds that can confound the study of disease-related gene expression. Due to a lack of sufficient information on these factors we were unable to strictly control for them in our analysis. However, using gene lists provided from the SMRI Online Genomics Database (http://www.stanleygenomics.org) we were able to make comparisons to address some of these factors and identified two overlapping genes. While the small number of overlapping genes is suggestive that we have identified genes in our signature that are not affected by such extraneous factors; we acknowledge that we cannot entirely exclude the possibility that the gene expression changes we have identified are still in some way influenced.
In conclusion, we have contributed the most comprehensive meta-analysis of schizophrenia expression profiling studies to date. Our most striking finding is that despite the heterogeneity of the disorder, we were able to detect a common signature of schizophrenia. Additionally, we have elaborated on the biological relevance of our gene list, illustrating a need for further genetic study to fully enhance our understanding of the direct implication of these changes in expression with the illness. The signatures we identified are consistent with current hypotheses of molecular dysfunction in schizophrenia, including alterations in synaptic transmission and energy metabolism. However, the diversity of genes we found suggests that systems biology approaches, exemplified by the analysis of gene network structure, will be of value in determining the basis of this disorder. The approaches used in our study should be applicable to other neuropsychiatric disorders if sufficient data are available.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgements
We would like to thank the groups and institutions who made their data available, including Dr. Karoly Mirnics (Vanderbilt), Dr. Vahram Haroutunian (Mt Sinai), the SMRI and the Harvard Brain bank. This study would not have been possible without their generosity. Supported by a grant from the National Institutes of Health to PP (GM076990). MM was partly supported by the MIND Foundation of BC for Schizophrenia Research. JG is partly supported by CIHR and the Michael Smith Foundation for Health Research. PP is also supported by a career award from the Michael Smith Foundation for Health Research, a CIHR New Investigator award, and the Canadian Foundation for Innovation.
Footnotes
Conflicts of Interest
No conflict of interest.
Supplementary information is available at Molecular Psychiatry's website
Contributor Information
Meeta Mistry, Email: mmistry@bioinformatics.ubc.ca.
Jesse Gillis, Email: jesse.gillis@gmail.com.
Paul Pavlidis, Email: paul@chibi.ubc.ca.
References
- 1.Jablensky A. Epidemiology of schizophrenia: the global burden of disease and disability. Eur Arch Psychiatry Clin Neurosci. 2000;250(6):274–285. doi: 10.1007/s004060070002. Epub 2001/01/12. [DOI] [PubMed] [Google Scholar]
- 2.Iwamoto K, Kato T. Gene expression profiling in schizophrenia and related mental disorders. Neuroscientist. 2006;12(4):349–361. doi: 10.1177/1073858406287536. Epub 2006/07/15. [DOI] [PubMed] [Google Scholar]
- 3.Mirnics K, Levitt P, Lewis DA. Critical appraisal of DNA microarrays in psychiatric genomics. Biol Psychiatry. 2006;60(2):163–176. doi: 10.1016/j.biopsych.2006.02.003. Epub 2006/04/18. [DOI] [PubMed] [Google Scholar]
- 4.Pongrac J, Middleton FA, Lewis DA, Levitt P, Mirnics K. Gene expression profiling with DNA microarrays: advancing our understanding of psychiatric disorders. Neurochem Res. 2002;27(10):1049–1063. doi: 10.1023/a:1020904821237. Epub 2002/12/05. [DOI] [PubMed] [Google Scholar]
- 5.Altar CA, Jurata LW, Charles V, Lemire A, Liu P, Bukhman Y, et al. Deficient hippocampal neuron expression of proteasome, ubiquitin, and mitochondrial genes in multiple schizophrenia cohorts. Biol Psychiatry. 2005;58(2):85–96. doi: 10.1016/j.biopsych.2005.03.031. Epub 2005/07/26. [DOI] [PubMed] [Google Scholar]
- 6.Iwamoto K, Bundo M, Kato T. Altered expression of mitochondria-related genes in postmortem brains of patients with bipolar disorder or schizophrenia, as revealed by large-scale DNA microarray analysis. Hum Mol Genet. 2005;14(2):241–253. doi: 10.1093/hmg/ddi022. Epub 2004/11/26. [DOI] [PubMed] [Google Scholar]
- 7.Middleton FA, Mirnics K, Pierri JN, Lewis DA, Levitt P. Gene expression profiling reveals alterations of specific metabolic pathways in schizophrenia. J Neurosci. 2002;22(7):2718–2729. doi: 10.1523/JNEUROSCI.22-07-02718.2002. Epub 2002/03/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mirnics K, Middleton FA, Marquez A, Lewis DA, Levitt P. Molecular characterization of schizophrenia viewed by microarray analysis of gene expression in prefrontal cortex. Neuron. 2000;28(1):53–67. doi: 10.1016/s0896-6273(00)00085-4. Epub 2000/11/22. [DOI] [PubMed] [Google Scholar]
- 9.Arion D, Unger T, Lewis DA, Levitt P, Mirnics K. Molecular evidence for increased expression of genes related to immune and chaperone function in the prefrontal cortex in schizophrenia. Biol Psychiatry. 2007;62(7):711–721. doi: 10.1016/j.biopsych.2006.12.021. Epub 2007/06/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aston C, Jiang L, Sokolov BP. Microarray analysis of postmortem temporal cortex from patients with schizophrenia. J Neurosci Res. 2004;77(6):858–866. doi: 10.1002/jnr.20208. Epub 2004/08/31. [DOI] [PubMed] [Google Scholar]
- 11.Dracheva S, Davis KL, Chin B, Woo DA, Schmeidler J, Haroutunian V. Myelin-associated mRNA and protein expression deficits in the anterior cingulate cortex and hippocampus in elderly schizophrenia patients. Neurobiol Dis. 2006;21(3):531–540. doi: 10.1016/j.nbd.2005.08.012. Epub 2005/10/11. [DOI] [PubMed] [Google Scholar]
- 12.Hakak Y, Walker JR, Li C, Wong WH, Davis KL, Buxbaum JD, et al. Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc Natl Acad Sci U S A. 2001;98(8):4746–4751. doi: 10.1073/pnas.081071198. Epub 2001/04/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Allen NC, Bagade S, McQueen MB, Ioannidis JP, Kavvoura FK, Khoury MJ, et al. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat Genet. 2008;40(7):827–834. doi: 10.1038/ng.171. Epub 2008/06/28. [DOI] [PubMed] [Google Scholar]
- 14.Mathieson I, Munafo MR, Flint J. Meta-analysis indicates that common variants at the DISC1 locus are not associated with schizophrenia. Mol Psychiatry. 2011 doi: 10.1038/mp.2011.41. Epub 2011/04/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O'Donovan MC, Craddock N, Norton N, Williams H, Peirce T, Moskvina V, et al. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat Genet. 2008;40(9):1053–1055. doi: 10.1038/ng.201. Epub 2008/08/05. [DOI] [PubMed] [Google Scholar]
- 16.Mistry M, Pavlidis P. A cross-laboratory comparison of expression profiling data from normal human postmortem brain. Neuroscience. 2010;167(2):384–395. doi: 10.1016/j.neuroscience.2010.01.016. Epub 2010/02/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Torkamani A, Dean B, Schork NJ, Thomas EA. Coexpression network analysis of neural tissue reveals perturbations in developmental processes in schizophrenia. Genome Res. 2010;20(4):403–412. doi: 10.1101/gr.101956.109. Epub 2010/03/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choi KH, Higgs BW, Wendland JR, Song J, McMahon FJ, Webster MJ. Gene expression and genetic variation data implicate PCLO in bipolar disorder. Biol Psychiatry. 2011;69(4):353–359. doi: 10.1016/j.biopsych.2010.09.042. Epub 2010/12/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu C, Cheng L, Badner JA, Zhang D, Craig DW, Redman M, et al. Whole-genome association mapping of gene expression in the human prefrontal cortex. Mol Psychiatry. 2010;15(8):779–784. doi: 10.1038/mp.2009.128. Epub 2010/03/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barnes M, Freudenberg J, Thompson S, Aronow B, Pavlidis P. Experimental comparison and cross-validation of the Affymetrix and Illumina gene expression analysis platforms. Nucleic acids research. 2005;33(18):5914–5923. doi: 10.1093/nar/gki890. Epub 2005/10/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baum AE, Hamshere M, Green E, Cichon S, Rietschel M, Noethen MM, et al. Meta-analysis of two genome-wide association studies of bipolar disorder reveals important points of agreement. Mol Psychiatry. 2008;13(5):466–467. doi: 10.1038/mp.2008.16. Epub 2008/04/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choi KH, Elashoff M, Higgs BW, Song J, Kim S, Sabunciyan S, et al. Putative psychosis genes in the prefrontal cortex: combined analysis of gene expression microarrays. BMC Psychiatry. 2008;8:87. doi: 10.1186/1471-244X-8-87. Epub 2008/11/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y, Blackwood DH, Caesar S, de Geus EJ, Farmer A, Ferreira MA, et al. Meta-analysis of genome-wide association data of bipolar disorder and major depressive disorder. Mol Psychiatry. 2011;16(1):2–4. doi: 10.1038/mp.2009.107. Epub 2010/03/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dawany NB, Tozeren A. Asymmetric microarray data produces gene lists highly predictive of research literature on multiple cancer types. BMC Bioinformatics. 2010;11:483. doi: 10.1186/1471-2105-11-483. Epub 2010/09/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leek JT, Storey JD. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007;3(9):1724–1735. doi: 10.1371/journal.pgen.0030161. Epub 2007/10/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, et al. Large-scale meta-analysis of cancer microarray data identifies common transcriptional profiles of neoplastic transformation and progression. Proc Natl Acad Sci U S A. 2004;101(25):9309–9314. doi: 10.1073/pnas.0401994101. Epub 2004/06/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Magalhaes JP, Curado J, Church GM. Meta-analysis of age-related gene expression profiles identifies common signatures of aging. Bioinformatics. 2009;25(7):875–881. doi: 10.1093/bioinformatics/btp073. Epub 2009/02/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elashoff M, Higgs BW, Yolken RH, Knable MB, Weis S, Webster MJ, et al. Meta-analysis of 12 genomic studies in bipolar disorder. J Mol Neurosci. 2007;31(3):221–243. doi: 10.1385/jmn:31:03:221. Epub 2007/08/30. [DOI] [PubMed] [Google Scholar]
- 29.Maycox PR, Kelly F, Taylor A, Bates S, Reid J, Logendra R, et al. Analysis of gene expression in two large schizophrenia cohorts identifies multiple changes associated with nerve terminal function. Mol Psychiatry. 2009;14(12):1083–1094. doi: 10.1038/mp.2009.18. Epub 2009/03/04. [DOI] [PubMed] [Google Scholar]
- 30.Garbett K, Gal-Chis R, Gaszner G, Lewis DA, Mirnics K. Transcriptome alterations in the prefrontal cortex of subjects with schizophrenia who committed suicide. Neuropsychopharmacol Hung. 2008;10(1):9–14. Epub 2008/09/06. [PubMed] [Google Scholar]
- 31.Katsel P, Davis KL, Gorman JM, Haroutunian V. Variations in differential gene expression patterns across multiple brain regions in schizophrenia. Schizophr Res. 2005;77(2–3):241–252. doi: 10.1016/j.schres.2005.03.020. Epub 2005/06/01. [DOI] [PubMed] [Google Scholar]
- 32.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19(2):185–193. doi: 10.1093/bioinformatics/19.2.185. Epub 2003/01/23. [DOI] [PubMed] [Google Scholar]
- 33.Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic acids research. 2003;31(4):e15. doi: 10.1093/nar/gng015. Epub 2003/02/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4(2):249–264. doi: 10.1093/biostatistics/4.2.249. Epub 2003/08/20. [DOI] [PubMed] [Google Scholar]
- 35.Kent WJ. BLAT--the BLAST-like alignment tool. Genome Res. 2002;12(4):656–664. doi: 10.1101/gr.229202. Epub 2002/04/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 2003;100(16):9440–945. doi: 10.1073/pnas.1530509100. Epub 2003/07/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–29. doi: 10.1038/75556. Epub 2000/05/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gillis J, Mistry M, Pavlidis P. Gene function analysis in complex data sets using ErmineJ. Nat Protoc. 2010;5(6):1148–1159. doi: 10.1038/nprot.2010.78. Epub 2010/06/12. [DOI] [PubMed] [Google Scholar]
- 39.Lee HK, Braynen W, Keshav K, Pavlidis P. ErmineJ: tool for functional analysis of gene expression data sets. BMC Bioinformatics. 2005;6:269. doi: 10.1186/1471-2105-6-269. Epub 2005/11/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chatr-aryamontri A, Ceol A, Palazzi LM, Nardelli G, Schneider MV, Castagnoli L, et al. MINT: the Molecular INTeraction database. Nucleic acids research. 2007;35(Database issue):D572–D574. doi: 10.1093/nar/gkl950. Epub 2006/12/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chua HN, Sung WK, Wong L. Exploiting indirect neighbours and topological weight to predict protein function from protein-protein interactions. Bioinformatics. 2006;22(13):1623–1630. doi: 10.1093/bioinformatics/btl145. Epub 2006/04/25. [DOI] [PubMed] [Google Scholar]
- 42.Gilbert D. Biomolecular interaction network database. Brief Bioinform. 2005;6(2):194–198. doi: 10.1093/bib/6.2.194. Epub 2005/06/25. [DOI] [PubMed] [Google Scholar]
- 43.Lynn DJ, Winsor GL, Chan C, Richard N, Laird MR, Barsky A, et al. InnateDB: facilitating systems-level analyses of the mammalian innate immune response. Mol Syst Biol. 2008;4:218. doi: 10.1038/msb.2008.55. Epub 2008/09/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Prasad TS, Kandasamy K, Pandey A. Human Protein Reference Database and Human Proteinpedia as discovery tools for systems biology. Methods Mol Biol. 2009;577:67–79. doi: 10.1007/978-1-60761-232-2_6. Epub 2009/09/01. [DOI] [PubMed] [Google Scholar]
- 45.Razick S, Magklaras G, Donaldson IM. iRefIndex: a consolidated protein interaction database with provenance. BMC. Bioinformatics. 2008;9:405. doi: 10.1186/1471-2105-9-405. Epub 2008/10/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dijkstra EW. A note on two problems in connexion with graphs. Numerische Mathematik. 1959;1:269–271. [Google Scholar]
- 47.Glatt SJ, Everall IP, Kremen WS, Corbeil J, Sasik R, Khanlou N, et al. Comparative gene expression analysis of blood and brain provides concurrent validation of SELENBP1 up-regulation in schizophrenia. Proc Natl Acad Sci U S A. 2005;102(43):15533–15538. doi: 10.1073/pnas.0507666102. Epub 2005/10/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou X, Kao MC, Wong WH. Transitive functional annotation by shortest-path analysis of gene expression data. Proc Natl Acad Sci USA. 2002;99(20):12783–12788. doi: 10.1073/pnas.192159399. Epub 2002/08/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oldham MC, Konopka G, Iwamoto K, Langfelder P, Kato T, Horvath S, et al. Functional organization of the transcriptome in human brain. Nature neuroscience. 2008;11(11):1271–1282. doi: 10.1038/nn.2207. Epub 2008/10/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28(1):264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. Epub 2008/01/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Prabakaran S, Swatton JE, Ryan MM, Huffaker SJ, Huang JT, Griffin JL, et al. Mitochondrial dysfunction in schizophrenia: evidence for compromised brain metabolism and oxidative stress. Mol Psychiatry. 2004;9(7):684–697. 43. doi: 10.1038/sj.mp.4001511. Epub 2004/04/21. [DOI] [PubMed] [Google Scholar]
- 52.Narayan S, Tang B, Head SR, Gilmartin TJ, Sutcliffe JG, Dean B, et al. Molecular profiles of schizophrenia in the CNS at different stages of illness. Brain Res. 2008;1239:235–248. doi: 10.1016/j.brainres.2008.08.023. Epub 2008/09/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saetre P, Emilsson L, Axelsson E, Kreuger J, Lindholm E, Jazin E. Inflammation-related genes up-regulated in schizophrenia brains. BMC Psychiatry. 2007;7:46. doi: 10.1186/1471-244X-7-46. Epub 2007/09/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Park E, Iaccarino C, Lee J, Kwon I, Baik SM, Kim M, et al. Regulatory roles of hnRNP M and Nova-1 in the alternative splicing of the dopamine D2 receptor pre-mRNA. J Biol Chem. 2011 doi: 10.1074/jbc.M110.206540. Epub 2011/05/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eyles DW, McGrath JJ, Reynolds GP. Neuronal calcium-binding proteins and schizophrenia. Schizophr Res. 2002;57(1):27–34. doi: 10.1016/s0920-9964(01)00299-7. Epub 2002/08/08. [DOI] [PubMed] [Google Scholar]
- 56.Manji HK. G proteins: implications for psychiatry. Am J Psychiatry. 1992;149(6):746–760. doi: 10.1176/ajp.149.6.746. Epub 1992/06/01. [DOI] [PubMed] [Google Scholar]
- 57.Schwab SG, Hallmayer J, Lerer B, Albus M, Borrmann M, Honig S, et al. Support for a chromosome 18p locus conferring susceptibility to functional psychoses in families with schizophrenia, by association and linkage analysis. Am J Hum Genet. 1998;63(4):1139–1152. doi: 10.1086/302046. Epub 1998/10/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Oliver S. Guilt-by-association goes global. Nature. 2000;403(6770):601–603. doi: 10.1038/35001165. Epub 2000/02/25. [DOI] [PubMed] [Google Scholar]
- 59.Thomas EA. Molecular profiling of antipsychotic drug function: convergent mechanisms in the pathology and treatment of psychiatric disorders. Mol Neurobiol. 2006;34(2):109–128. doi: 10.1385/MN:34:2:109. Epub 2007/01/16. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.