

Abstract
Patients with Krabbe disease, a genetic demyelinating syndrome caused by deficiency of galactosyl-ceramidase and the resulting accumulation of galactosyl-sphingolipids, develop signs of a dying-back axonopathy compounded by a deficiency of large-caliber axons. Here, we show that axonal caliber in Twitcher mice, an animal model for Krabbe disease, is impaired in peripheral axons and is accompanied by a progressive reduction in the abundance and phosphorylation of the three neurofilament (NF) subunits. These changes correlate with an increase in the density of NFs per cross-sectional area in numerous mutant peripheral axons and abnormal increases in the activity of two serine/threonine phosphatases (PP1 and PP2A) in mutant tissue. Similarly, acutely isolated mutant cortical neurons show abnormal phosphorylation of NFs. Psychosine, the neurotoxin accumulated in Krabbe disease, was sufficient to induce abnormal dephosphorylation of NF subunits in a normal motor neuron cell line as well as in acutely isolated normal cortical neurons. This in vitro effect was mediated by PP1 and PP2A, which specifically dephosphorylated NFs. These results demonstrate that the reduced caliber observed in some axons in Krabbe disease involves abnormal dephosphorylation of NFs. We propose that a psychosine-driven pathogenic mechanism through deregulated phosphotransferase activities may be involved in this process.
Keywords: leukodystrophies, Twitcher, Krabbe disease, neurofilament, myelin, dying-back pathology, axonal degeneration, axonal transport, kinases, phosphatases
INTRODUCTION
Krabbe disease, a leukodystrophy caused by the deficiency of galactosylceramidase (GALC) and the accumulation of the lipid-raft-associated neurotoxin psychosine (Igisu and Suzuki, 1984; Aicardi, 1993; Suzuki, 1998; Wenger et al., 2000; White et al., 2009; White et al., 2010), is characterized by central and peripheral demyelination. The disease involves the activation of resident microglia and the continuous infiltration of macrophages, which become multinucleated globoid cells (Kanazawa et al., 2000; Wu et al., 2000). Affected patients are primarily infants, who die within months after diagnosis. Albeit not a cure, cell therapy with hematopoietic precursors (bone marrow- or cord blood-derived cells) has proven to extend patients' survival, especially when performed within weeks after birth. Despite amelioration in some clinical hallmarks, motor and cognitive deficits in treated patients remain largely uncorrected (Kondo et al., 1988; Krivit et al., 1995; Eglitis and Mezey, 1997; Escolar et al., 2005; Galbiati et al., 2009).
One hallmark of Krabbe disease is the rapid deterioration of nerve conduction, which has been historically related with the loss of myelin (Toyoshima et al., 1986; Dolcetta et al., 2005). Interestingly, earlier reports indicated lower numbers of large-caliber axons in the mutant nerves (Hogan et al., 1969; Schlaepfer and Prensky, 1972; Martin et al., 1974; Jacobs et al., 1982), a largely unaddressed defect that may contribute to the compromised nerve conduction observed in Krabbe patients and to the limited neuroprotection observed in most of treated patients. We recently found evidence of a peripheral neuropathy in the Twitcher mouse, the natural murine model of this disease (Castelvetri et al., 2011; Smith et al., 2011). Interestingly, axonal damage started well before that of mutant neurons, which was minimal until the final stage of the disease when nerves were severely demyelinated. These observations raise the possibility that mechanisms controlling the maturation and stability of the axonal compartment (cytoskeletal components, axonal transport) are dysfunctional very early in the mutant mouse.
Expansion of the axonal cytoskeletal diameter is highly influenced by the content and phosphorylation of several neurofilaments (NF) subunits. NFs are intermediate components of the neuron cytoskeleton which are comprised of ~60 kDa (NF-L), ~100 kDa (NF-M) and ~120 kDa (NF-H) subunits (Zhu et al., 1997). NF-M and NF-H form sidearm bulges, which project radially from a rod-like structure primarily composed of NF-L (Hisanaga and Hirokawa, 1989). Lys-Ser-Pro repeats are contained in the carboxyl-end of NF-M and NF-H and their phosphorylation induces charge repulsion forces, separating NFs and thus increasing axonal diameter (de Waegh et al., 1992; Cole et al., 1994). Various kinases and serine/threonine protein phosphatases regulate this process, reviewed in (Perrot et al., 2008).
In this study, we examined for changes in the phosphorylation state of NFs in axons of the sciatic nerve of the Twitcher mouse and in cultures of normal and mutant neurons, and investigated the potential role of psychosine in this process.
MATERIALS AND METHODS
Animals
Twitcher heterozygous mice (C57BL/6J, twi/+) and Thy1.1:YFP H+/+ mice were maintained under standard housing conditions. In vivo experiments were approved by the Animal Care and Use Committee of our institution. The majority of experiments in this study used homozygous Twitcher mice. Where indicated, homozygous Twitcher mice carrying the expression of the yellow fluorescent protein (YFP) under the transcriptional control of the Thy1.1. promoter were used. For this, double-heterozygous TWI+/− thy1.1:YFP+/− mice (generated in our previous study, Castelvetri et al., 2011)were crossed to generate homozygous Twitcher (Twi−//)-Thy1.1 YFP+/−. Genotypes were confirmed by PCR (Sakai et al., 1996; Dolcetta et al., 2006).
Cell cultures
Murine NSC34 motor neuron cells were grown in 5% FBS, DMEM and serum-deprived for 12 hr before treatment. Acutely isolated cortical neurons were prepared from brain cortices from Twitcher and wild type E15.5 embryos and cultured as described before (Bongarzone et al., 1996). Cultures were treated with control vehicle (0.01% ethanol/DMEM), 1 or 5 μM psychosine for up to 3 hr. In some experiments, 10 nM okadaic acid was added to the cultures.
Tissue collection, histology, and immunohistochemistry
Mice were anesthetized and killed by transcardial perfusion with saline on postnatal (P) days P7 to P40. Dissected tissues for immunohistochemistry and histology were post-fixed in 4% paraformaldehyde-PBS for 18 hr, embedded in sucrose, and frozen in OCT, while tissues for psychosine determination and immunoblotting were quickly frozen in liquid nitrogen.
Sciatic nerves were separated and processed for transmission electron microscopy (TEM, see below). Cryosections (20-μm) were mounted onto polylysine-coated slides. For immunofluorescence staining, sections were dried for 15 min at 370C, washed in PBS, blocked and permeabilized in 5% bovine serum albumin, 0.5% Triton X-100/PBS for 1 hr at room temperature and incubated with phosphorylated NF-M/NF-H (SMI31, Covance) or dephosphorylated NF-M/NF-H (SMI32, Covance) mouse monoclonal antibodies diluted in 2% BSA, 0.5% Triton X-100/PBS overnight at 40C, with mild agitation. Slides were incubated with fluorescent secondary antibodies for 1 hr, washed in PBS and mounted with Vectashield (VectorLabs, CA). Confocal microscopy was performed using a confocal laser Meta Leica scanning microscope. For light microscopy of immunostained sections, cryosections were treated with 0.3% (v/v) H2O2 10% (v/v) methanol/PBS, blocked/permeabilized with 0.1% (w/v) casein, 0.1% (v/v) Triton X-100 in PBS for 1 hr, incubated with antibodies against PP1 (Santa Cruz) and PP2A (Cell Signaling) and developed with biotinylated secondary antibodies (VectorLabs).
Western blotting
Tissues were homogenized in lysis buffer (1 mM PMSF, 2 mM sodium orthovanadate, 1 mM NaF, 300 nM okadaic acid, 20 mM Tris-HCl, pH 7.4, 1% Triton X100, 150 mM NaCl, 5 mM MgCl2), briefly sonicated on ice and centrifuged at 5000 rpm for 5 min to remove debris. Protein concentration was quantified (Bradford assay, Biorad) and 10 micrograms of protein were loaded on a Nupage 4–12% Bis-Tris gel (Invitrogen). Gels were run at 80 mV and electrotransferred at 400 mA on PVDF membranes (Biorad). Membranes were blocked in 5% (w/v) milk, 1% (w/v) BSA, 0.05% (v/v) Tween-20 in TBS and probed with primary antibodies against actin (Sigma), phosphorylated NF-M/NF-H (SMI31, Covance), dephosphorylated NF-M/NF-H (SMI32, Covance), NF-L, total NF-M (clone RMO189), PP2A (Cell Signaling) and PP1 (Santa Cruz). Membranes were developed using Enhanced Luminescence kit (Thermo Scientific). Bands were quantified (Image J, NIH), and relative abundance of a particular protein was normalized to actin.
Psychosine determination
Psychosine was extracted from the sciatic nerves and quantified by HPLC-tandem mass spectrometry as described (Galbiati et al. 2009).
Serine/threonine phosphatase activity assay
PP1 and PP2A activities were measured using the Rediplate assay (Molecular Probes) following the manufacturer's instructions. Enzyme activity was expressed as micromol (DiFMUP) per mg protein per minute.
Transmission electron microscopy
Sciatic nerves were quickly removed from 12 and 30 day-old mice, fixed with 2.5% glutaraldehyde, and embedded in Araldite. Ultrathin sections (60-nm) were collected on Formvar-coated 1-hole grids and stained with osmium tetroxide and lead acetate. Samples were observed using a 120kV TEM JEOL JEM-1220 fitted with a tungsten electron source and equipped with a Gatan Es1000W 11MP CCD camera. For quantitative analyses of axonal diameters and neurofilament density, 30 myelinated axons were randomly selected from each sample. A total of 3 nerves per genotype per time point were analyzed. Axonal diameters were measured and images were processed for counting NFs and determination of NF density as described (Cole et al., 1994).
Statistical analysis
Results are expressed as mean ± SE from at least 3 independent samples. Data were analyzed by ANOVA/post hoc paired test or Student's t-test, where appropriate. Correlation analysis was done using the Pearson test.
RESULTS
Decrease in diameter of Twitcher axons
Coronal sections of sciatic nerves collected from WT and Twitcher mice at P12 and P30 were examined by TEM. Axonal diameter was measured and the frequency of axons calculated for mice at each age. Twitcher nerves showed fewer large-caliber axons as compared to controls already at P12 (Fig. 1A, B). Mutant nerves from P30 mice showed severe loss of myelin, edema (Fig. 1D) and a visible reduction in the number of large-caliber axons (Fig. 1D, E). Even those mutant axons with no obvious demyelination showed smaller diameters (Fig. 1D). Frequency analysis confirmed these observations: at P12, mutant nerves contained higher numbers of axons with 2- to 3-micron calibers (Fig. 1C), and at P30, there was a significant (p<0.05) shift to axons of smaller diameters, with the majority of mutant axonal caliber ranging between 1–5 microns (Fig. 1F). In contrast, the majority of WT axons grew in diameter as expected (Fig. 1C, F).
Figure 1. Prevalence of small-diameter fibers in Twitcher sciatic nerves.

A, B, D, E) Electron micrographs of coronal sections of WT and Twitcher sciatic nerves at P12 and P30 show a decrease in axonal diameter in the Twitcher fibers (A) as compared to age-matched WT fibers (B). Severely demyelinated and abundant small diameter axons were clearly observed in older (P30) Twitcher nerves (D). All images are at the same magnification (bar in A=2 μm). Arrows and asterisk in D point to myelinated axons and edema, respectively. C, F) Distribution profiles based on axonal diameter showed a growing increase in small-caliber axons. A minimum of 50 axons was counted per each nerve (n=3 nerves/time point/genotype). Data were analyzed by ANOVA/post hoc paired test. p<0.05.
Lower density of NF in Twitcher axons
To determine whether the observed reduction in diameter of mutant axons involved alterations in the NF cytoskeleton, we examined NF density by TEM. Because NF density is affected by the level of myelination of the axon (Reles and Friede, 1991; de Waegh et al., 1992; Mata et al., 1992; Hsieh et al., 1994), we analyzed only mutant fibers that remained myelinated during the time-frame of our experiments (i.e., P12 and P30). Cross sectional and longitudinal imaging analysis showed that the spacing between NFs increased between P12 and P30 in WT nerve fibers (Fig. 2B, D, F), as expected for normal developing fibers, whereas most Twitcher NFs appeared compacted at both time points (Fig. 2A, C, E), suggesting a failure in the mechanism regulating the spacing of these proteins. Quantitation of these observations by counting the NFs per random region of axoplasm (Cole et al., 1994) revealed the expected decreased density of NFs (number of hexagons containing a given number of NFs/ 3.5 × 10−2 micron2 area of axoplasm) between P12 and P30 in WT nerves (Fig. 3A, 3B) but a consistently high NF density in Twitcher axons at both sampled time points (Fig. 3A, 3B). Immunoblotting analyses of the total amount of NF proteins in sciatic nerves showed significant decreased levels of NF-M protein in the nerves of Twitcher mice (Fig. 3C). Levels of NF-L and NF-H, while showing a decreasing trend, did not reach significance in the mutant nerves. Immunodetection analyses failed to detect the presence of NF proteins in cerebrospinal fluid of Twitcher mice (data not shown). Real-time PCR analysis of each NF mRNA revealed no statistically significant difference in the spinal cord of Twitcher and age-matched WT mice (data not shown), suggesting absence of gene transcription deficits.
Figure 2. Increased packing of NFs in Twitcher axons.

A–D) High-magnification TEM of coronal sections of P12 and P30 sciatic nerves revealed slightly increased NF density at P12 (A) but significantly higher density at P30 in Twitcher axons (C) as compared to WT axons (B and D, respectively). E,F) High-magnification TEM of longitudinal sections of P30 WT (E) and Twitcher (F) sciatic nerves showed several gaps among NF bundles in the WT nerve. Twitcher NFs were tightly packed together, further confirming the increase in NF density in the mutant axons. (Bar =0.5 microns for A,B and =0.250 microns for C–F).
Figure 3. NF abnormalities in Twitcher sciatic nerves.

A,B) NF density in P12 (A) and P30 (B) WT and Twitcher sciatic nerves was quantitated by counting the NFs per hexagonal grid laid on high-magnification EM pictures of sciatic nerve coronal section. Twitcher axons had significantly more hexagons with higher number of NFs at both time points, indicating the increased NF density in Twitcher mice. C) Quantitation of Western blot analysis of NF-L, NF-M and NF-H in P7, P15 and P30 WT and Twitcher sciatic nerves. Levels of all three proteins were decreased at P15 and P30, as assessed using ImageJ software and by normalizing to actin levels. Data are mean±SEM of four independent nerves per genotype per time point. Data were analyzed by ANOVA/post hoc paired test. Asterisk indicates p<0.05.
Reduced phosphorylation of mutant NFs
The higher densities of NFs in the mutant nerve suggested abnormal phosphorylation of NFs. To test this possibility, we first examined coronal sections of WT and Twitcher spinal cords stained for phosphorylated epitopes on NF-M and NF-H using the monoclonal antibody SMI31; the number of SMI31+ fibers was noticeably reduced in Twitcher spinal cords, which was most evident in axons within the ventral white matter (compare Fig. 4A and B with E and F, respectively). Subsequent analysis of longitudinal sections of cords stained using the SMI32 antibody, which binds to dephosphorylated epitopes on NF-M and NF-H, and using postmortem material isolated from the new TWI-YFPax reporter mouse model (Castelvetri et al., 2011), where YFP labels axons and readily reveals axonal dystrophy, showed increased SMI32 immunostaining in Twitcher cords, especially in axons with clear signs of dystrophy (swellings, breaks; Fig. 4C, D and G). In contrast, WT axons were minimally stained with SMI32 (Fig. 4H). Finally, analysis of longitudinal sections of sciatic nerves from TWI-YFPax and age-matched WT-YFPax mice (P30) immunolabeled with the SMI31 antibody indicated a clear reduction in SMI31 binding in the mutant axons (compare Fig. 4I and J with L and M, respectively). This analysis also demonstrated the reduction in the diameter of mutant nerves.
Figure 4. Loss of NF phosphorylation in the Twitcher nervous system.
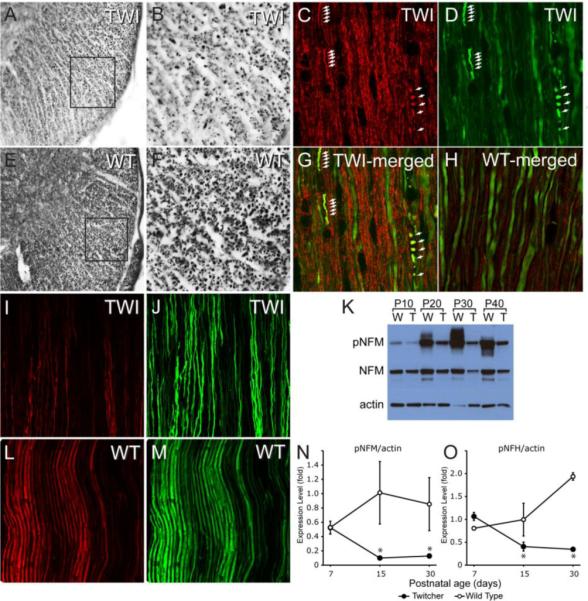
A, B, E, F) Immunohistochemical staining of P30 spinal cord coronal sections from Twitcher (A, enlarged in B) and WT (E, enlarged in F) with monoclonal antibody SMI31 shows a decrease in the density of SMI31+ fibers, especially in the ventral white matter, in Twitcher mice. C, D, G, H) Immunofluorescence staining of spinal cord longitudinal sections from WT-YFPax (H) and Twitcher-YFPax P30 mice (C, D and merged image in G) for SMI32 (red) and YFP (green) shows the stronger reactivity for SMI32 in the Twitcher fibers and the accumulation of dephosphorylated NFs in sites of axonal swellings and breaks (white arrows), suggesting a link between NF dephosphorylation and axonal damage in Twitcher neurons. I, J, L,M) Immunofluorescence staining of sciatic nerve longitudinal sections from WT-YFPax (L and M) and Twitcher-YFPax (I and J) P30 mice for SMI31 (red) and YFP (green) shows stronger SMI31 reactivity in the WT tissue (L) as compared to Twitcher tissue (I), indicating the loss of NF phosphorylation in the Twitcher peripheral nervous system. K) Representative Western blot of phosphorylated NF (pNF-M, obtained with monoclonal antibody SMI31) and total NF-M (obtained with monoclonal antibody RMO189) of P10, P20, P30 and P40 WT and Twitcher sciatic nerves, with actin as a loading control. N,O) Quantitation of Western blot analyses of pNF-M and pNF-M, respectively, in sciatic nerves of P7, P15 and P30 WT and Twitcher mice using ImageJ software and normalizing to actin. Data are mean ± SEM of four replicates. Data were analyzed by ANOVA/post hoc paired test. Asterisk indicates p<0.01.
Further assessment for changes in phosphorylation of NF in Twitcher mice by western blotting of protein extracts from sciatic nerves (obtained between P7 and P40) using antibody SMI31 revealed a clear reduction of phosphorylated NF-M levels at each sampled time point (Fig. 4K). Analyses for the levels of both phosphorylated NF-M (Fig. 4N) and NF-H (Fig. 4O) showed significant (p<0.01) reduction of these cytoskeletal proteins in mutant nerves at P15 and P30 but not at P7. To examine the phosphorylation state of NFs in isolated mutant neurons, cultures of acutely isolated embryonic cortical neurons were prepared from mutant and wild type embryos. We found similar NFs deficiencies in cultured mutant neurons, with decreased phosphorylation of the NF-H (Supplementary fig. 1).
Increased activity of serine/threonine protein phosphatases in Twitcher sciatic nerves
Phosphorylation of NFs is regulated by Erk1/2, CDK5, p35 and SAPK1b kinases and ser/thr protein phosphatases PP1 and PP2A (Perrot et al., 2008). To determine whether PP1 and/or PP2A were involved in abnormal phosphorylation of mutant NFs, we first determined the activation levels for both phosphatases in protein extracts from WT and Twitcher sciatic nerves. Quantitative analysis indicated a significant increase in the activity for both PP1 and PP2A at P30, whereas at P7, only PP2A increased significantly in mutant sciatic nerves (Fig. 5A, B). Further analysis of longitudinal sections of sciatic nerves immunolabeled with antibodies against PP1 (Fig. 5C, E) and PP2A (Fig. 5D, F) also revealed a large increase in PP1 in mutant sciatic nerves (Fig. 5C) and an even greater increase in PP2A (Fig. 5D).
Figure 5. Increased activation of PP1 and PP2A in the Twitcher sciatic nerve.

A, B) Quantitation of PP1 (A) and PP2A (B) enzymatic activity in lysates from P7 and P30 sciatic nerves of WT and Twitcher mice showed that both enzymes in Twitcher nerves were strongly over-activated at P30, while only PP2A was significantly more active in the P7 tissue. Activity is expressed as micromol substrate (DiFMUP)/mg protein/min. Data are mean ± SEM of three replicates. Data were analyzed by ANOVA/post hoc paired test. Asterisk indicates p<0.01. C–F) Representative immunohistochemical staining for PP2A in P30 Twitcher (C,D) and WT (E,F) sciatic nerves shows the higher frequency of fibers intensely stained for PP2A in the Twitcher nerve (black arrows in C and D). Rarely, PP2A+ myelinating Schwann cells were also detected in Twitcher tissue (asterisks in D). Enlarged in D, the presence of PP2A in a mutant axon is readily observed (black arrow in D), surrounded by the myelinating sheath (white arrow in D).
Psychosine is sufficient to increase PP1 and PP2A activity in neuronal cells
To test the possibility that psychosine, a lipid raft-associated sphingolipid (White et al., 2009; 2010), might underlie the observed activation of PP1 and PP2A, we first carried out a correlation analysis of levels of all three molecules in mutant sciatic nerves. Indeed, we found a strong correlation between the elevated levels of psychosine (Fig. 6A) and PP2A activity in the mutant nerve at P7 and P30 (rP7=0.994 and rP30=1, respectively), whereas the correlation with PP1 levels was significant only at P30 (rP30=0.932). To test whether psychosine is sufficient to modify phosphatase activities, PP1 and PP2A activity was measured in the normal immortalized NSC34 motor neuronal cells exposed to various concentrations of psychosine. Doses were chosen based on previous work on Twitcher granular neurons (Castelvetri et al. 2011). Concentrations of 1 and 5 μM psychosine were chosen to emulate early and late stages of the disease, respectively. Activities of both PP1 and PP2A were significantly increased (Fig. 6B, Fig. 6C) in a psychosine concentration-dependent manner.
Figure 6. Psychosine induces activity of PP1 and PP2A.

A) Quantitation of psychosine by mass spectrometry in P30 WT and Twitcher sciatic nerves indicated significantly higher levels of psychosine already at P7 in the Twitcher tissue, with accumulation progressively increasing over the course of the disease. Data are mean ± SEM from 4 nerves per time point per genotype. Data were analyzed by ANOVA/post hoc paired test. Asterisk indicates p<0.01. B,C) Quantitation of PP1 (B) and PP2A (C) enzymatic activities in NSC34 cells treated with 0, 1 or 5 μM psychosine for 3 hr, showed that psychosine induces a dose-dependent increase in phosphatase activity. Activity is expressed as μmol substrate (DiFMUP)/mg protein/min. Data are mean ± SEM of three replicates. Data were analyzed by t-test of Student. Asterisk indicates p<0.05.
To determine whether psychosine leads to dephosphorylation (i.e, the enzymatic removal of phosphate groups by dephosphorylating activities of specific phosphatases), two experiments were performed. First, cultures of acutely isolated cortical neurons prepared from control wild type embryos were exposed to psychosine. Immunoblotting analysis using SMI31 antibodies showed a significant reduction of phosphorylated NF-H in psychosine-treated neurons (Supplementary fig. 2). In the second experiment, we assessed the activities of PP1 and PP2A in NSC34 cells incubated with 5 μM psychosine and with 10 nM okadaic acid, a broad phosphatase inhibitor that is non-toxic at this concentration (Atkinson et al., 2009). Induction of PP1 activity was efficiently blocked by 10 nM okadaic acid in these cultures, while blockage of PP2A activity was less efficient (Fig. 7A). Western analyses for changes in levels of phosphorylated NFs using protein extracts from treated and control cells labeled with SMI31 monoclonal antibodies showed that psychosine was sufficient to induce significant dephosphorylation of NF-M, while the presence of okadaic acid almost completely inhibited this dephosphorylation (Fig. 7B and C).
Figure 7. Okadaic acid protects NF from dephosphorylation triggered by psychosine.
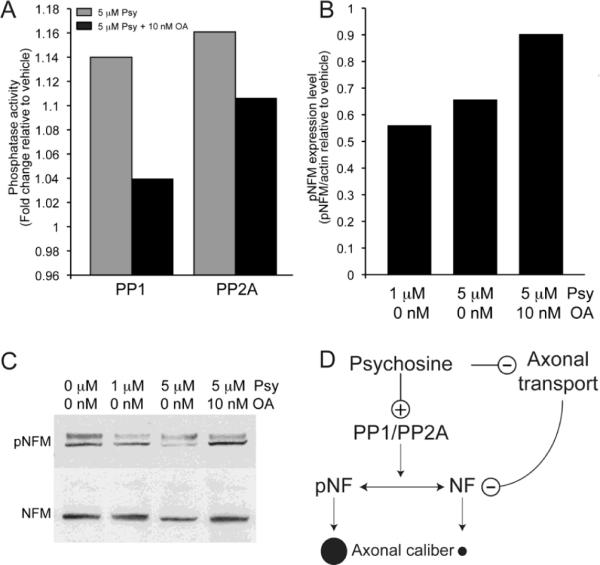
A) NSC34 cells incubated with 5 μM psychosine and 10 nM okadaic acid (OA) for 3 hr showed significant inhibition of both PP1 and PP2A activity. Data are expressed as fold-change of phosphatase enzymatic activity relative to the vehicle (n=3). B, C) Analysis of NSC34 cells treated with vehicle, 1 or 5 μM psychosine for 3 hr and processed for immunoblotting detection of pNF-M and total NF-M indicated that psychosine triggers NF-M dephosphorylation (quantitation in B using ImageJ and normalizing the amount of pNF-M with respect to actin; representative Western blot in C), and that OA prevents this dephosphorylation. Data are expressed as fold-change relative to the vehicle (n=3). D) In a proposed model for the effect of psychosine on NFs and axonal diameter, psychosine triggers the activities of PP1 and PP2A, which, in turn dephosphorylate NFs. Loss of NF phosphorylation hampers the ability of Twitcher axons to grow radially, explaining the prevalence of small-diameter fibers in Twitcher peripheral nerves. This pathogenic mechanism is compounded by abnormal transport of NFs in the axon.
DISCUSSION
Our analyses of the molecular basis for the reduction of axonal caliber in the Twitcher mouse model of Krabbe disease demonstrate: 1) deficient phosphorylation of the NF cytoskeleton in axons of the Twitcher mouse during postnatal development and in acutely isolated mutant neurons; 2) increased dephosphorylating activities of PP1 and PP2A in peripheral mutant nerves; and 3) the ability of psychosine alone to trigger dephosphorylation of NFs by inducing activation of these two phosphatases in vitro. This is the first report to establish a molecular link between the presence of the neurotoxin psychosine and a deficiency in axon cytoskeletal maturation.
Abnormalities in the Twitcher axonal cytoskeleton
The functional impact of large-caliber axon loss in Krabbe disease has remained controversial and unaddressed (Hogan et al., 1969; Schlaepfer and Prensky, 1972; Martin et al., 1974; Jacobs et al., 1982). TEM morphometric analysis of mutant sciatic nerves confirmed a deficient growth of the axonal radius. Axonal growth is a vital process for the maturation of the nervous system and its failure may contribute to various neurological phenotypes (Takei et al., 2000; Eng et al., 2001; Wang et al., 2006; Lin et al., 2011). For example, tremor, ataxia, progressive impairment of locomotion skills and muscle atrophy appear in Twitcher mice at about P20. The cause of these symptoms has been historically linked to the ongoing demyelination (Jacobs et al., 1982; Kobayashi et al., 1988; Taniike and Suzuki, 1994). However, a study done by Olmstead (1987) reported a subtle neurological phenotype in young mutants. Further, our recent work revealed axonal dystrophy in Twitcher mice as young as 1 week old, suggesting a much earlier and more complex mechanism of disease (Castelvetri et al., 2011; Smith et al., 2011). The prevalence of small-diameter axons throughout the postnatal life of this mutant points to an impairment in the growth and/or the maintenance of large myelinated axons. In the developing nervous system, axonal diameter of myelinated axons increases up to 15-fold after the neuron successfully establishes its synaptic connections. Although myelin is fundamental for saltatory conduction, the increase in axonal diameter is a critical step to accommodate faster action potential conduction. Defects in radial growth substantially decrease the propagation speed of action potentials. Thus, the prevalence of small-diameter fibers in Twitcher nerves may contribute significantly to the decreased conduction velocities in this mutant (Toyoshima et al., 1986; Dolcetta et al., 2005) and to one or more of the observed neurological symptoms such as muscle atrophy (Shen et al., 2001).
Axonal radial growth is closely related to myelination (Aguayo et al., 1977; Windebank et al., 1985; Kirkpatrick and Brady, 1994), so that demyelination may also contribute to the decrease in axonal size in Twitcher nerves. However, thinner axons were already more frequent at P12 in the absence of substantial demyelination. Because our analyses were performed on myelinated axons, our data suggest myelin-independent deficits in axonal radial growth in this mutant, although we cannot exclude the possibility that GALC deficiency per se introduces subtle defects during early stages of myelination that affect axonal growth.
Abnormal dephosphorylation of NFs in Twitcher axons
NFs are critical for the establishment and maintenance of axonal caliber in mammalian neurons (de Waegh and Brady, 1991; Reles and Friede, 1991; de Waegh et al., 1992; Hsieh et al., 1994) and appear to play an important role in several unrelated neurodegenerative disorders, such as amyotrophic lateral sclerosis (Hirano et al., 1984), Parkinson disease (Pappolla, 1986) and Charcot-Marie-Tooth disease (Brownlees et al., 2002). Cytoskeletal alterations were evident in Twitcher nerves, with increased NF densities paralleling axonal caliber decreases. This observation raises the question about the molecular cause(s) of deficient axonal radial growth. While alterations in gene expression, stoichiometry, and post-translational modifications of NFs can alter axonal stability and caliber (Elder et al., 1998; Perrot et al., 2008), we found no significant changes in mRNA and proteins levels for any NF form in spinal cord of Twitcher mice (data not shown) but did find decreasing amounts of NF-M within mutant sciatic nerves at P15 and P30. A similar decrease in NFs in a mouse model lacking one copy of each NF protein led to a 50% decrease in axonal diameter (Nguyen et al., 2000). Thus, the reduced axonal caliber in the Twitcher nerve may be caused, at least in part, by decreased amounts of NFs contained within the axons.
The reason for the lower abundance of NF-M in the Twitcher sciatic nerves, without change in the corresponding gene transcript, remains unclear. NFs are accumulated in the axon by means of molecular motor-assisted transport (Shea and Flanagan, 2001; Wagner et al., 2004; Theiss et al., 2005). Defects in motility mechanisms may hamper their axonal localization and hence contribute to abnormal calibers (Yabe et al., 1999; Roy et al., 2000; Shah et al., 2000; Wagner et al., 2004; Theiss et al., 2005). The coexistence of reduced NFs and axonal dystrophy in peripheral nerves of the Twitcher mutant (Castelvetri et al., 2011) is consistent with defective axonal transport mechanisms in these mice. Furthermore, our previous study implicated psychosine as a potential pathogenic modulator of axonal transport, a process regulated by an array of coordinated activities of various kinases and phosphatases (Morfini et al., 2002; Morfini et al., 2006; Ikeda et al., 2011). Importantly, deregulated axonal transport appears to play a pathogenic role in various non-related neurological diseases, creating the conditions for axonal dystrophy, dying-back axonopathy, and eventual neuronal demise (Szebenyi et al., 2003; Morfini et al., 2009). A better understanding of axonal transport mechanisms in Krabbe leukodystrophy will help in the design of improved and more efficient therapeutic options.
Deregulation of phosphatases and dephosphorylation of NFs in the Twitcher nerve
Axonal caliber is highly dependent on the level of phosphorylation of NF sidearms (Nixon et al., 1994; Pant and Veeranna, 1995; Gotow, 2000). Addition of phosphates in sidearms of NF-M and NF-H creates clouds of negative charges, which repel filaments and increase axon caliber. Expression of non-phosphorylatable NF-M results in severe loss of large-diameter fibers (Garcia et al., 2003; Rao et al., 2003) due to a decreased repulsion between adjacent filaments (Kumar and Hoh, 2004). Our results demonstrating that Twitcher NFs are significantly dephosphorylated in the sciatic nerve support the idea that abnormal dephosphorylation of NFs decreases axonal caliber. Dephosphorylated NFs were localized in axonal swellings and varicosities, which are sites of axonal stress and damage (Coleman, 2005). Thus, dephosphorylation of mutant NFs may occur differentially along the axon, with focal points of axonal instability in mutant fibers.
The regulated activities of kinases and phosphatases determine the level, the location and the temporal profile of NF phosphorylation (Perrot et al., 2008). Consequently, an imbalance in phosphotransferase activities may result in more or less phosphorylation of NFs, leading to changes in their assembly. Our analysis of PP1 and PP2A, which account for ~15% and ~65% of the turnover of phosphorylation of NFs (Strack et al., 1997), indicated elevated activities of both phosphatases in axons of the mutant sciatic nerves, particularly during the final stages of the disease. These findings suggest that the decreased frequency of large-caliber axons in Krabbe disease is due mainly to altered phosphorylation of the axonal cytoskeleton. However, our laboratory has ongoing studies to determine whether Erk1/2, CDK5, p35 and SAPK1b (kinases that affect NF phosphorylation) are also altered in this disease. The extent to which the phosphatase phenotype affects other neuronal functions in Krabbe disease is unclear. Phosphatases have a wide range of functions, which are not limited to the physiology of NFs (Virshup and Shenolikar, 2009), and abnormal phosphatase activities may deregulate other signaling pathways in neurons, oligodendrocytes, Schwann cells and microglial cells, introducing deficits that affect their survival and function permanently and irreversibly. For example, PP1 has been characterized as a regulator of fast axonal transport (Morfini et al., 2004). The relevance of these metabolic pathways for neuronal health has prompted studies to determine the status of these kinases as well as additional effects of PP1 and PP2A in Krabbe disease.
Psychosine is sufficient to cause NF dephosphorylation in vitro
Based on our findings that: 1) psychosine accumulation was significantly correlated with increased PP1 and PP2A activities in the sciatic nerve of Twitcher mice; 2) exposure of NSC34 cells and cortical neurons to psychosine induced NF dephosphorylation and 3) increased PP1 and PP2A activities in NSC34 cells was induced by psychosine, while co-incubation with okadaic acid blocked psychosine-effect, we propose that psychosine is a pathogenic sphingolipid capable to trigger NF dephosphorylation by recruitment of PP1 and PP2A.
The mechanism by which psychosine activates both phosphatases is still unclear. Phosphatase activity depends heavily on the recruitment and coordination of various regulatory subunits (Lechward et al., 2001; Cohen, 2002; Virshup and Shenolikar, 2009). Interestingly, ceramide was recently found to bind inhibitor 2, a natural inhibitory subunit regulating endogenous activation of phosphatases (Mukhopadhyay et al., 2009), but it is unknown whether psychosine binds to inhibitor 2 in the Twitcher axon and thereby releases the catalytic subunits of phosphatases. Psychosine may also mediate steric rearrangements of the phosphatase complex by modifying membrane microdomains (White et al., 2009). PP2A is known to bind to lipid rafts (Berrou and Bryckaert, 2009), where cholesterol levels modulate its activity. Psychosine might also modulate the spatial orientation of NF sidearms, making them more accessible to phosphatases. In this context, certain phospholipids have been found to bind to NF-L (Kim et al. 2011). These and other possible mechanisms await further studies.
We propose a pathogenic model to explain the mechanism of the decrease in axonal caliber in Twitcher nerves (Fig. 7D) whereby psychosine accumulation facilitates higher activities of PP1 and PP2A, which, in turn, dephosphorylate NF-M and NF-H, leading to reduced radial growth of Twitcher axons. In addition, our model proposes that psychosine affects the transport of NFs into the axon, decreasing the availability of these cytoskeletal proteins for proper axonal growth. Because these neuronal deficits may be untreatable by traditional hematopoietic-based therapies used for Krabbe disease, studies of neuronal and axonal neuroprotection to modulate phosphatase activities may be relevant in the design of more efficient therapies for this leukodystrophy.
Supplementary Material
Highlights
-
▶
Axonal diameter is decreased in the nerves of the mouse model of Krabbe disease.
-
▶
Neurofilaments are less phosphorylated in the Twitcher mouse.
-
▶
Protein phosphatase 1 and 2A are responsible for the NF dephosphorylation in the Twitcher mouse.
-
▶
Psychosine causes the dephosphorylation of NFs by inducing the activity of protein phosphatases in vitro.
Acknowledgments
Acknowledgements: This study was partially funded by grants from NIH (RNS065808A), the Morton Cure paralysis foundation and the Board of Trustees at the University of Illinois to ERB.
Abbreviations
- GALC
galactosyl-ceramidase
- NF
neurofilament
- NFL
neurofilament light polypeptide
- NFM
neurofilament medium polypeptide
- NFH
neurofilament heavy polypeptide
- PP1
protein phosphatase 1
- PP2A
protein phosphatase 2A
- TEM
transmission electron microscopy
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Aguayo AJ, Kasarjian J, Skamene E, Kongshavn P, Bray GM. Myelination of mouse axons by Schwann cells transplanted from normal and abnormal human nerves. Nature. 1977;268:753–755. doi: 10.1038/268753a0. [DOI] [PubMed] [Google Scholar]
- Aicardi J. The inherited leukodystrophies: a clinical overview. J Inherit Metab Dis. 1993;16:733–743. doi: 10.1007/BF00711905. [DOI] [PubMed] [Google Scholar]
- Atkinson T, Whitfield J, Chakravarthy B. The phosphatase inhibitor, okadaic acid, strongly protects primary rat cortical neurons from lethal oxygen-glucose deprivation. Biochem Biophys Res Commun. 2009;378:394–398. doi: 10.1016/j.bbrc.2008.11.036. [DOI] [PubMed] [Google Scholar]
- Berrou E, Bryckaert M. Recruitment of protein phosphatase 2A to dorsal ruffles by platelet-derived growth factor in smooth muscle cells: dephosphorylation of Hsp27. Exp Cell Res. 2009;315:836–848. doi: 10.1016/j.yexcr.2008.12.002. [DOI] [PubMed] [Google Scholar]
- Bongarzone ER, Foster LM, Byravan S, Verity AN, Landry CF, Schonmann V, Amur-Umarjee S, Campagnoni AT. Conditionally Immortalized Neural Cell Lines: Potential Models for the Study of Neural Cell Function. Methods. 1996;10:489–500. doi: 10.1006/meth.1996.0126. [DOI] [PubMed] [Google Scholar]
- Brownlees J, Ackerley S, Grierson AJ, Jacobsen NJ, Shea K, Anderton BH, Leigh PN, Shaw CE, Miller CC. Charcot-Marie-Tooth disease neurofilament mutations disrupt neurofilament assembly and axonal transport. Hum Mol Genet. 2002;11:2837–2844. doi: 10.1093/hmg/11.23.2837. [DOI] [PubMed] [Google Scholar]
- Castelvetri LC, Givogri MI, Zhu H, Smith B, Lopez-Rosas A, Qiu X, van Breemen R, Bongarzone ER. Axonopathy is a compounding factor in the pathogenesis of Krabbe disease. Acta Neuropathol. 2011;122:35–48. doi: 10.1007/s00401-011-0814-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen PT. Protein phosphatase 1-targeted in many directions. J Cell Sci. 2002;115:241–256. doi: 10.1242/jcs.115.2.241. [DOI] [PubMed] [Google Scholar]
- Cole JS, Messing A, Trojanowski JQ, Lee VM. Modulation of axon diameter and neurofilaments by hypomyelinating Schwann cells in transgenic mice. J Neurosci. 1994;14:6956–6966. doi: 10.1523/JNEUROSCI.14-11-06956.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman M. Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci. 2005;6:889–898. doi: 10.1038/nrn1788. [DOI] [PubMed] [Google Scholar]
- de Waegh SM, Brady ST. Local control of axonal properties by Schwann cells: neurofilaments and axonal transport in homologous and heterologous nerve grafts. J Neurosci Res. 1991;30:201–212. doi: 10.1002/jnr.490300121. [DOI] [PubMed] [Google Scholar]
- de Waegh SM, Lee VM, Brady ST. Local modulation of neurofilament phosphorylation, axonal caliber, and slow axonal transport by myelinating Schwann cells. Cell. 1992;68:451–463. doi: 10.1016/0092-8674(92)90183-d. [DOI] [PubMed] [Google Scholar]
- Dolcetta D, Amadio S, Guerrini U, Givogri MI, Perani L, Galbiati F, Sironi L, Del Carro U, Roncarolo MG, Bongarzone E. Myelin deterioration in Twitcher mice: motor evoked potentials and magnetic resonance imaging as in vivo monitoring tools. J Neurosci Res. 2005;81:597–604. doi: 10.1002/jnr.20574. [DOI] [PubMed] [Google Scholar]
- Dolcetta D, Perani L, Givogri MI, Galbiati F, Amadio S, Del Carro U, Finocchiaro G, Fanzani A, Marchesini S, Naldini L, Roncarolo MG, Bongarzone E. Design and optimization of lentiviral vectors for transfer of GALC expression in Twitcher brain. J Gene Med. 2006;8:962–971. doi: 10.1002/jgm.924. [DOI] [PubMed] [Google Scholar]
- Eglitis MA, Mezey E. Hematopoietic cells differentiate into both microglia and macroglia in the brains of adult mice. Proc Natl Acad Sci U S A. 1997;94:4080–4085. doi: 10.1073/pnas.94.8.4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elder GA, Friedrich VL, Jr., Kang C, Bosco P, Gourov A, Tu PH, Zhang B, Lee VM, Lazzarini RA. Requirement of heavy neurofilament subunit in the development of axons with large calibers. J Cell Biol. 1998;143:195–205. doi: 10.1083/jcb.143.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng SR, Gratwick K, Rhee JM, Fedtsova N, Gan L, Turner EE. Defects in sensory axon growth precede neuronal death in Brn3a-deficient mice. J Neurosci. 2001;21:541–549. doi: 10.1523/JNEUROSCI.21-02-00541.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escolar ML, Poe MD, Provenzale JM, Richards KC, Allison J, Wood S, Wenger DA, Pietryga D, Wall D, Champagne M, Morse R, Krivit W, Kurtzberg J. Transplantation of umbilical-cord blood in babies with infantile Krabbe's disease. N Engl J Med. 2005;352:2069–2081. doi: 10.1056/NEJMoa042604. [DOI] [PubMed] [Google Scholar]
- Galbiati F, Givogri MI, Cantuti L, Lopez Rosas A, Cao H, van Breemen R, Bongarzone ER. Combined hematopoietic and lentiviral gene-transfer therapies in newborn Twitcher mice reveal contemporaneous neurodegeneration and demyelination in Krabbe disease. J Neurosci Res. 2009;87:1748–59. doi: 10.1002/jnr.22006. [DOI] [PubMed] [Google Scholar]
- Garcia ML, Lobsiger CS, Shah SB, Deerinck TJ, Crum J, Young D, Ward CM, Crawford TO, Gotow T, Uchiyama Y, Ellisman MH, Calcutt NA, Cleveland DW. NF-M is an essential target for the myelin-directed “outside-in” signaling cascade that mediates radial axonal growth. J Cell Biol. 2003;163:1011–1020. doi: 10.1083/jcb.200308159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotow T. Neurofilaments in health and disease. Med Electron Microsc. 2000;33:173–199. doi: 10.1007/s007950000019. [DOI] [PubMed] [Google Scholar]
- Hirano A, Donnenfeld H, Sasaki S, Nakano I. Fine structural observations of neurofilamentous changes in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 1984;43:461–470. doi: 10.1097/00005072-198409000-00001. [DOI] [PubMed] [Google Scholar]
- Hisanaga S, Hirokawa N. The effects of dephosphorylation on the structure of the projections of neurofilament. J Neurosci. 1989;9:959–966. doi: 10.1523/JNEUROSCI.09-03-00959.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan GR, Gutmann L, Chou SM. The peripheral neuropathy of Krabbe's (globoid) leukodystrophy. Neurology. 1969;19:1094–1100. doi: 10.1212/wnl.19.11.1094. [DOI] [PubMed] [Google Scholar]
- Hsieh ST, Crawford TO, Griffin JW. Neurofilament distribution and organization in the myelinated axons of the peripheral nervous system. Brain Res. 1994;642:316–326. doi: 10.1016/0006-8993(94)90937-7. [DOI] [PubMed] [Google Scholar]
- Igisu H, Suzuki K. Progressive accumulation of toxic metabolite in a genetic leukodystrophy. Science. 1984;224:753–755. doi: 10.1126/science.6719111. [DOI] [PubMed] [Google Scholar]
- Ikeda K, Zhapparova O, Brodsky I, Semenova I, Tirnauer JS, Zaliapin I, Rodionov V. CK1 activates minus-end-directed transport of membrane organelles along microtubules. Mol Biol Cell. 2011;22:1321–1329. doi: 10.1091/mbc.E10-09-0741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs JM, Scaravilli F, De Aranda FT. The pathogenesis of globoid cell leucodystrophy in peripheral nerve of the mouse mutant twitcher. J Neurol Sci. 1982;55:285–304. doi: 10.1016/0022-510x(82)90127-7. [DOI] [PubMed] [Google Scholar]
- Kanazawa T, Nakamura S, Momoi M, Yamaji T, Takematsu H, Yano H, Sabe H, Yamamoto A, Kawasaki T, Kozutsumi Y. Inhibition of cytokinesis by a lipid metabolite, psychosine. J Cell Biol. 2000;149:943–950. doi: 10.1083/jcb.149.4.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SK, Kim H, Yang YR, Suh PG, Chang JS. Phosphatidylinositol phosphates directly bind to neurofilament light chain (NF-L) for the regulation of NF-L self assembly. Exp Mol Med. 43:153–160. doi: 10.3858/emm.2011.43.3.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick LL, Brady ST. Modulation of the axonal microtubule cytoskeleton by myelinating Schwann cells. J Neurosci. 1994;14:7440–7450. doi: 10.1523/JNEUROSCI.14-12-07440.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S, Katayama M, Satoh J, Suzuki K, Suzuki K. The twitcher mouse. An alteration of the unmyelinated fibers in the PNS. Am J Pathol. 1988;131:308–319. [PMC free article] [PubMed] [Google Scholar]
- Kondo A, Hoogerbrugge PM, Suzuki K, Poorthuis BJ, Van Bekkum DW, Suzuki K. Pathology of the peripheral nerve in the twitcher mouse following bone marrow transplantation. Brain Res. 1988;460:178–183. doi: 10.1016/0006-8993(88)91220-6. [DOI] [PubMed] [Google Scholar]
- Krivit W, Lockman LA, Watkins PA, Hirsch J, Shapiro EG. The future for treatment by bone marrow transplantation for adrenoleukodystrophy, metachromatic leukodystrophy, globoid cell leukodystrophy and Hurler syndrome. J Inherit Metab Dis. 1995;18:398–412. doi: 10.1007/BF00710052. [DOI] [PubMed] [Google Scholar]
- Kumar S, Hoh JH. Modulation of repulsive forces between neurofilaments by sidearm phosphorylation. Biochem Biophys Res Commun. 2004;324:489–496. doi: 10.1016/j.bbrc.2004.09.076. [DOI] [PubMed] [Google Scholar]
- Lechward K, Awotunde OS, Swiatek W, Muszynska G. Protein phosphatase 2A: variety of forms and diversity of functions. Acta Biochim Pol. 2001;48:921–933. [PubMed] [Google Scholar]
- Lin L, Lee VM, Wang Y, Lin JS, Sock E, Wegner M, Lei L. Sox11 regulates survival and axonal growth of embryonic sensory neurons. Dev Dyn. 2011;240:52–64. doi: 10.1002/dvdy.22489. [DOI] [PubMed] [Google Scholar]
- Martin JJ, Ceuterick C, Martin L, Leroy JG, Nuyts JP, Joris C. [Globoid cell leucodystrophy (Krabbe's disease). Peripheral nerve lesion (author's transl)] Acta Neurol Belg. 1974;74:356–375. [PubMed] [Google Scholar]
- Mata M, Kupina N, Fink DJ. Phosphorylation-dependent neurofilament epitopes are reduced at the node of Ranvier. J Neurocytol. 1992;21:199–210. doi: 10.1007/BF01194978. [DOI] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Elluru R, Ratner N, Brady ST. Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. Embo J. 2002;21:281–293. doi: 10.1093/emboj/21.3.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G, Pigino G, Szebenyi G, You Y, Pollema S, Brady ST. JNK mediates pathogenic effects of polyglutamine-expanded androgen receptor on fast axonal transport. Nat Neurosci. 2006;9:907–916. doi: 10.1038/nn1717. [DOI] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Brown H, Pant HC, Pigino G, DeBoer S, Beffert U, Brady ST. A novel CDK5-dependent pathway for regulating GSK3 activity and kinesin-driven motility in neurons. Embo J. 2004;23:2235–2245. doi: 10.1038/sj.emboj.7600237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini GA, Burns M, Binder LI, Kanaan NM, LaPointe N, Bosco DA, Brown RH, Jr., Brown H, Tiwari A, Hayward L, Edgar J, Nave KA, Garberrn J, Atagi Y, Song Y, Pigino G, Brady ST. Axonal transport defects in neurodegenerative diseases. J Neurosci. 2009;29:12776–12786. doi: 10.1523/JNEUROSCI.3463-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay A, Saddoughi SA, Song P, Sultan I, Ponnusamy S, Senkal CE, Snook CF, Arnold HK, Sears RC, Hannun YA, Ogretmen B. Direct interaction between the inhibitor 2 and ceramide via sphingolipid-protein binding is involved in the regulation of protein phosphatase 2A activity and signaling. FASEB J. 2009;23:751–763. doi: 10.1096/fj.08-120550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen MD, Lariviere RC, Julien JP. Reduction of axonal caliber does not alleviate motor neuron disease caused by mutant superoxide dismutase 1. Proc Natl Acad Sci U S A. 2000;97:12306–12311. doi: 10.1073/pnas.97.22.12306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA, Paskevich PA, Sihag RK, Thayer CY. Phosphorylation on carboxyl terminus domains of neurofilament proteins in retinal ganglion cell neurons in vivo: influences on regional neurofilament accumulation, interneurofilament spacing, and axon caliber. J Cell Biol. 1994;126:1031–1046. doi: 10.1083/jcb.126.4.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmstead CE. Neurological and neurobehavioral development of the mutant `twitcher' mouse. Behav Brain Res. 1987;25:143–53. doi: 10.1016/0166-4328(87)90007-6. [DOI] [PubMed] [Google Scholar]
- Pant HC, Veeranna Neurofilament phosphorylation. Biochem Cell Biol. 1995;73:575–592. doi: 10.1139/o95-063. [DOI] [PubMed] [Google Scholar]
- Pappolla MA. Lewy bodies of Parkinson's disease. Immune electron microscopic demonstration of neurofilament antigens in constituent filaments. Arch Pathol Lab Med. 1986;110:1160–1163. [PubMed] [Google Scholar]
- Perrot R, Berges R, Bocquet A, Eyer J. Review of the multiple aspects of neurofilament functions, and their possible contribution to neurodegeneration. Mol Neurobiol. 2008;38:27–65. doi: 10.1007/s12035-008-8033-0. [DOI] [PubMed] [Google Scholar]
- Rao MV, Campbell J, Yuan A, Kumar A, Gotow T, Uchiyama Y, Nixon RA. The neurofilament middle molecular mass subunit carboxyl-terminal tail domains is essential for the radial growth and cytoskeletal architecture of axons but not for regulating neurofilament transport rate. J Cell Biol. 2003;163:1021–1031. doi: 10.1083/jcb.200308076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reles A, Friede RL. Axonal cytoskeleton at the nodes of Ranvier. J Neurocytol. 1991;20:450–458. doi: 10.1007/BF01252273. [DOI] [PubMed] [Google Scholar]
- Roy S, Coffee P, Smith G, Liem RK, Brady ST, Black MM. Neurofilaments are transported rapidly but intermittently in axons: implications for slow axonal transport. J Neurosci. 2000;20:6849–6861. doi: 10.1523/JNEUROSCI.20-18-06849.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai N, Inui K, Tatsumi N, Fukushima H, Nishigaki T, Taniike M, Nishimoto J, Tsukamoto H, Yanagihara I, Ozono K, Okada S. Molecular cloning and expression of cDNA for murine galactocerebrosidase and mutation analysis of the twitcher mouse, a model of Krabbe's disease. J Neurochem. 1996;66:1118–1124. doi: 10.1046/j.1471-4159.1996.66031118.x. [DOI] [PubMed] [Google Scholar]
- Schlaepfer WW, Prensky AL. Quantitative and qualitative study of sural nerve biopsies in Krabbe's disease. Acta Neuropathol. 1972;20:55–66. doi: 10.1007/BF00687902. [DOI] [PubMed] [Google Scholar]
- Shah JV, Flanagan LA, Janmey PA, Leterrier JF. Bidirectional translocation of neurofilaments along microtubules mediated in part by dynein/dynactin. Mol Biol Cell. 2000;11:3495–3508. doi: 10.1091/mbc.11.10.3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shea TB, Flanagan LA. Kinesin, dynein and neurofilament transport. Trends Neurosci. 2001;24:644–648. doi: 10.1016/s0166-2236(00)01919-6. [DOI] [PubMed] [Google Scholar]
- Shen JS, Watabe K, Ohashi T, Eto Y. Intraventricular administration of recombinant adenovirus to neonatal twitcher mouse leads to clinicopathological improvements. Gene Ther. 2001;8:1081–1087. doi: 10.1038/sj.gt.3301495. [DOI] [PubMed] [Google Scholar]
- Smith B, Galbiati F, Cantuti-Castelvetri L, Givogri MI, Lopez-Rosas A, Bongarzone ER. Peripheral neuropathy in the Twitcher mouse involves the activation of axonal caspase 3. ASN Neuro. 2011;3:213–222. doi: 10.1042/AN20110019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack S, Westphal RS, Colbran RJ, Ebner FF, Wadzinski BE. Protein serine/threonine phosphatase 1 and 2A associate with and dephosphorylate neurofilaments. Brain Res Mol Brain Res. 1997;49:15–28. doi: 10.1016/s0169-328x(97)00117-4. [DOI] [PubMed] [Google Scholar]
- Suzuki K. Twenty five years of the “psychosine hypothesis”: a personal perspective of its history and present status. Neurochem Res. 1998;23:251–259. doi: 10.1023/a:1022436928925. [DOI] [PubMed] [Google Scholar]
- Szebenyi G, Morfini GA, Babcock A, Gould M, Selkoe K, Stenoien DL, Young M, Faber PW, MacDonald ME, McPhaul MJ, Brady ST. Neuropathogenic forms of huntingtin and androgen receptor inhibit fast axonal transport. Neuron. 2003;40:41–52. doi: 10.1016/s0896-6273(03)00569-5. [DOI] [PubMed] [Google Scholar]
- Takei Y, Teng J, Harada A, Hirokawa N. Defects in axonal elongation and neuronal migration in mice with disrupted tau and map1b genes. J Cell Biol. 2000;150:989–1000. doi: 10.1083/jcb.150.5.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniike M, Suzuki K. Spacio-temporal progression of demyelination in twitcher mouse: with clinico-pathological correlation. Acta Neuropathol. 1994;88:228–236. doi: 10.1007/BF00293398. [DOI] [PubMed] [Google Scholar]
- Theiss C, Napirei M, Meller K. Impairment of anterograde and retrograde neurofilament transport after anti-kinesin and anti-dynein antibody microinjection in chicken dorsal root ganglia. Eur J Cell Biol. 2005;84:29–43. doi: 10.1016/j.ejcb.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Toyoshima E, Yeager AM, Brennan S, Santos GW, Moser HW, Mayer RF. Nerve conduction studies in the Twitcher mouse (murine globoid cell leukodystrophy) J Neurol Sci. 1986;74:307–318. doi: 10.1016/0022-510x(86)90116-4. [DOI] [PubMed] [Google Scholar]
- Virshup DM, Shenolikar S. From promiscuity to precision: protein phosphatases get a makeover. Mol Cell. 2009;33:537–545. doi: 10.1016/j.molcel.2009.02.015. [DOI] [PubMed] [Google Scholar]
- Wagner OI, Ascano J, Tokito M, Leterrier JF, Janmey PA, Holzbaur EL. The interaction of neurofilaments with the microtubule motor cytoplasmic dynein. Mol Biol Cell. 2004;15:5092–5100. doi: 10.1091/mbc.E04-05-0401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang J, Mori S, Nathans J. Axonal growth and guidance defects in Frizzled3 knock-out mice: a comparison of diffusion tensor magnetic resonance imaging, neurofilament staining, and genetically directed cell labeling. J Neurosci. 2006;26:355–364. doi: 10.1523/JNEUROSCI.3221-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger DA, Rafi MA, Luzi P, Datto J, Costantino-Ceccarini E. Krabbe disease: genetic aspects and progress toward therapy. Mol Genet Metab. 2000;70:1–9. doi: 10.1006/mgme.2000.2990. [DOI] [PubMed] [Google Scholar]
- White AB, Givogri MI, Lopez-Rosas A, Cao H, van Breemen R, Thinakaran G, Bongarzone ER. Psychosine accumulates in membrane microdomains in the brain of krabbe patients, disrupting the raft architecture. J Neurosci. 2009;29:6068–6077. doi: 10.1523/JNEUROSCI.5597-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White AB, Givogri MI, Lopez Rosas A, Qiu X, van Breemen R, Bongarzone ER. Persistence of psychosine in brain lipid rafts is a limiting factor in the therapeutic recovery of a mouse model for Krabbe disease. J Neurosci Res. 2010;89:352–64. doi: 10.1002/jnr.22564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windebank AJ, Wood P, Bunge RP, Dyck PJ. Myelination determines the caliber of dorsal root ganglion neurons in culture. J Neurosci. 1985;5:1563–1569. doi: 10.1523/JNEUROSCI.05-06-01563.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YP, Matsuda J, Kubota A, Suzuki K, Suzuki K. Infiltration of hematogenous lineage cells into the demyelinating central nervous system of twitcher mice. J Neuropathol Exp Neurol. 2000;59:628–639. doi: 10.1093/jnen/59.7.628. [DOI] [PubMed] [Google Scholar]
- Yabe JT, Pimenta A, Shea TB. Kinesin-mediated transport of neurofilament protein oligomers in growing axons. J Cell Sci. 1999;112(Pt 21):3799–3814. doi: 10.1242/jcs.112.21.3799. [DOI] [PubMed] [Google Scholar]
- Zhu Q, Couillard-Després S, Julien JP. Delayed maturation of myelinated axons in mice lacking neurofilaments. Exp. Neurol. 1997;148:299–316. doi: 10.1006/exnr.1997.6654. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.