

Abstract
The protective antigen is a key component of the anthrax toxin, as it allows entry of the enzymatic components edema factor and lethal factor into the host cell, through the formation of a membrane spanning pore. This event is absolutely critical for the pathogenesis of anthrax, and although we have yet to understand the mechanism of pore formation, recent developments have provided key insights into how this process may occur. Based on the available data, a model is proposed for the kinetic steps for protective antigen conversion from prepore to pore. In this model, the driving force for pore formation is the formation of the phi (ϕ)-clamp, a region that forms a leak-free seal around the translocating polypeptide. Formation of the ϕ-clamp elicits movements within the prepore that provide steric freedom for the subsequent conformational changes required to form the membrane spanning pore.
Keywords: anthrax, toxin, protective antigen, pore, pH, membrane protein, review
Introduction
Anthrax disease is caused by the gram positive bacterium Bacillus anthracis, and this year marks the 10th anniversary of the anthrax attacks that occurred in September and October of 2001. There were 11 confirmed cases of inhalational anthrax due to exposure to aerosolized anthrax spores, resulting in the deaths of five people. Although administration of antibiotics during the 2001 attacks was effective in saving lives,1 major efforts since then have been directed toward improving early diagnosis and treatment of the disease.2 While anthrax has the potential to be used as a biological weapon, it is also a threat to agriculture worldwide. Recently, animal outbreaks have occurred in Africa, Australia, Bangladesh, Brazil, Canada, China, Sweden, and the United States. Under more harsh environmental conditions (lack of nutrients, dry conditions), the bacterium forms a dormant spore which is very hardy and can last in the soil for an indefinite amount of time. To cause infection, however, the spore must germinate to an actively growing vegetative state, but requires the right physicochemical conditions (pH > 6, temperature > 15°C).3, 4 The conversion from a dormant spore to a vegetative state was seen in the recent re-emergence of anthrax in Australia, where flooding unearthed anthrax spores that had been dormant for decades, eventually infecting and causing the deaths of over 53 cattle and 1 horse.5
There are three primary routes of infection that B. anthracis uses—the skin (cutaneous), the gut (gastrointestinal) or the lung (inhalational), the former two the primary mechanisms of entry into animals from either contact with spores on the skin, or through ingesting contaminated soil.6 In inhalational anthrax, the inhaled spores are engulfed by macrophages and carried to the mediastinal lymph nodes, and eventually burst out of the cell and spread into the bloodstream.7–9 The incubation period for germination can be quite long, and is the reason for the 60-day long time course of antibiotic treatment for an anthrax infection. This allows a sufficient amount of time for spore germination and exposure of the bacterium to antibiotic.10, 11
Shortly after germination, the bacterium will secrete an antiphagocytic poly-d-glutamic acid capsule,12 and a set of three proteins: edema factor (EF), lethal factor (LF), and protective antigen (PA).13, 14 These three proteins are alone nontoxic, but can assemble together in the bloodstream or on host cell surfaces to form the anthrax toxin, which belongs to the broader class of toxins called AB toxins.15 Both EF and LF of the anthrax toxin correspond to the “A” components, and PA corresponds to the “B” component. PA attaches to host cells, recruiting EF and LF to the cell surface, and forms a pore inside the cell that allows entry of EF and LF into the cytosol. PA also provides protective immunity against anthrax infection in animals,16 and is the major component of the current licensed anthrax vaccine, anthrax vaccine absorbed (AVA), given to active military personnel in the United States.
In early studies of the anthrax toxin, the individual components EF, LF, and PA were purified from the supernatant of cultures of the Sterne strain of B. anthracis, a strain that lacks the pXO2 plasmid which encodes the poly-d-glutamic acid capsule required for virulence.17 In studies on primates, injection of PA, EF or LF alone, or the complex of PA + EF (called edema toxin, ETx) did not cause lethality (although negative effects were observed with the injection of PA alone18). However, injection of PA + LF (called lethal toxin, LTx) resulted in fairly rapid physiological changes, including anaphylaxis, respiratory difficulties, and changes in cortical brain activity, leading to the death of the animal within 30 hours.18, 19 There are currently no antitoxin treatments available against the anthrax toxin, and therefore it is absolutely critical that antibiotic treatment be given as early as possible, before the secretion of these toxin components.
EF is an 89 kDa calcium-calmodulin-dependent adenylate cyclase, which raises intracellular cAMP levels20 and has been shown to alter cellular chemotaxis.21 LF is a zinc-metalloproteinase that cleaves mitogen activated protein kinase kinases (MAPKKs),22 and has been shown to inhibit the release of cytokines.23, 24 However, recent studies using lung epithelial cells also indicate that LF disrupts the cytoskeletal and microtubule networks needed to maintain the integrity of the epithelial barrier.25 PA is an 83 kDa, calcium binding, four-domain monomeric protein that interacts with host cells and undergoes several changes that lead ultimately to the formation of a membrane spanning pore, and is the main focus of this review.
PA-Receptor Binding
From early studies it was suggested that PA was the component that interacted with host cells,19 and later the receptor that bound to PA was identified as anthrax toxin receptor or “ATR,” previously identified as tumor endothelial marker 8 (TEM8).26, 27 A second cell surface receptor was subsequently identified, called capillary morphogenesis protein 2 (CMG2) or anthrax toxin receptor 2 (ATR2).28 More recently, heterodimeric complexes of integrin β1 have also been shown to bind PA and promote internalization.29 Natural ligands for CMG2 have been shown to include type IV collagen and laminin which comprise the basal lamina,30 and for TEM8 the α3 subunit of collagen type VI.31 Exactly how PA effectively competes with these natural ligands for binding to these receptors is not well understood.
The receptors TEM8, CMG2, and integrin β1 all contain an integrin I (inserted) domain or von-Willebrand factor A (vWA) domain, that contains acidic residues that coordinate a magnesium ion on the surface of the receptor, forming a metal-ion-dependent adhesion site (MIDAS).32 The three-dimensional structures of the full length PA,33 and heptameric prepore,34 bound to CMG2 [Figs. 1 and 2(A), respectively], confirmed earlier biochemical studies indicating that the receptor bound primarily to domain 4 of PA (residues 595-735).26, 35 Domain 4 contains an essential aspartic acid (D683), which completes the coordination shell to the MIDAS site magnesium (Fig. 1). Mutation of this aspartic acid to asparagine (D683N) abrogates binding to TEM8, but surprisingly not to CMG2.36 Domain 4 constitutes the majority of the binding interface, and buries ∼1300 Å2 of surface area, sufficient for binding to both TEM8 and CMG2.26, 33, 37 However, the crystal structures of PA or prepore bound to CMG2 also revealed that a small loop from domain 2 (the domain 2β3-2β4 loop, Fig. 1) is inserted into a groove on the surface of CMG2, and contributes another ∼600 Å2 of surface area to the binding interface. This additional surface area explains in part why the D683N mutation in PA does not prevent binding to CMG2, as well as the higher affinity of PA for CMG2 versus TEM8 (KD ∼ 200 pM vs. 200 nM, respectively).36, 38
Figure 1.
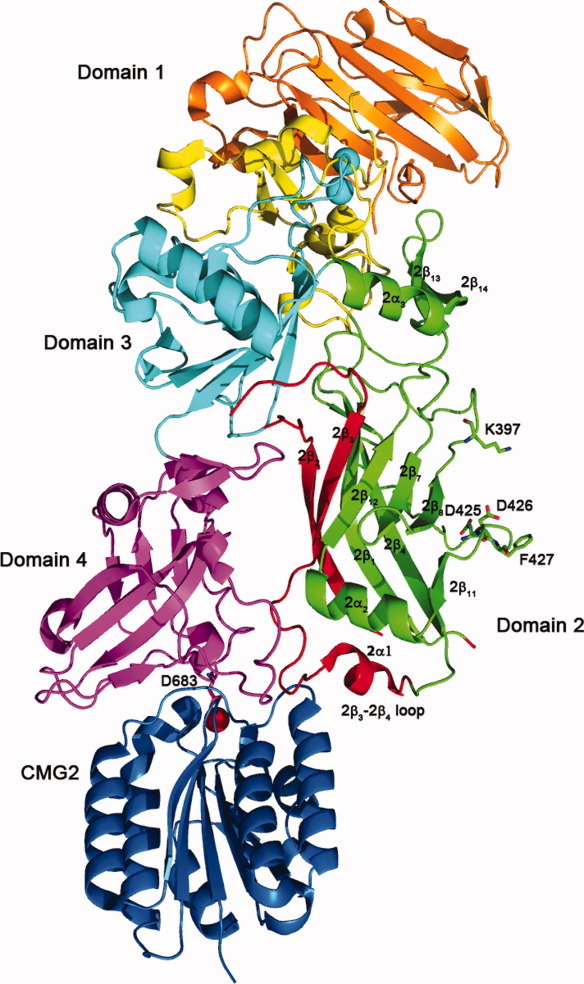
Structure of the PA bound to CMG2 (PDB: 1T6B).33 The PA20 region of domain 1 is colored orange, with domain 1′ shown in yellow, and calcium ions shown as blue spheres; domain 2 is green, and the region which comprises the β-barrel portion of the pore (residues 275-352) is shown in red; domain 3 is shown in cyan, and domain 4 is shown in magenta. D683 is shown from domain 4 coordinating a magnesium ion shown in red, which lies in the MIDAS site of CMG2 (shown in blue). The residues that form the phi (ϕ) clamp – K397, D425, D426, and F427 are also shown.
Figure 2.
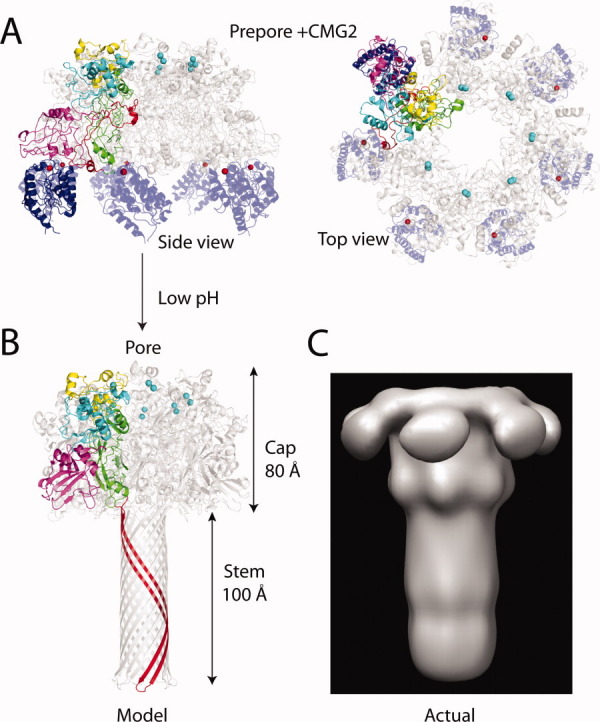
(A) Prepore and pore structures. Heptameric prepore bound to CMG2 (PDB:1TZN),34 with color coding identical to Figure 1 (but without PA20). Only one subunit of the heptamer is colored, the remaining subunits are shown in gray. CMG2 is in blue. Shown are both a side view and a top view of the prepore. (B) The final pore structure as modeled by Nguyen,65 with the long β-barrel stem from a single subunit stretching from residues 275-352 is shown in red. (C) EM structure of the pore.66 Note the absence of domain 4, and the differences in the β-barrel stem from the model of Nguyen. Figure was generated using Chimera.67
PA—Formation of the Prepore
After PA has bound to the receptor on the cell surface, PA undergoes proteolytic processing by a furin-like protease, which cleaves off the first 167 amino acids of PA within domain 1, termed PA20.39 This leaves behind domain 1′ (residues 168-258) and the remaining 3 domains, constituting a 63 kDa fragment (termed PA63). PA63 can assemble on the surface of cells into a heptameric40, 41 or octameric42 donut shaped structure called the prepore.
The crystallographic structure of the heptameric prepore [Fig. 2(A)] revealed the monomer–monomer contact interfaces which involve interactions that stretch between domain 1′ to domain 4.34, 41 Surprisingly, aside from the loss of PA20, there is little change in the backbone conformation between monomeric PA and the heptameric prepore. There is also little change in the structure of the prepore upon binding CMG2.34 Consistent with the lack of change in backbone conformation in the free and bound states, studies on the kinetics of association of PA63 into heptamers showed that the rate constants for association do not change upon binding to CMG2, indicating that receptor binding does not significantly influence the structure of the prepore.38
Through mutagenesis studies, domain 3 has been shown to be particularly important for the formation of the prepore, where a single point mutation at position 512, D512A, largely blocked heptameric prepore formation.43 Although the resolution of the crystal structure of the heptameric prepore is low (∼3.6 Å),34 the structure indicates that D512 lies at the beginning of a short α-helix, where the side-chain carboxyl of D512 is positioned to form an intramolecular hydrogen bond with the backbone amide NH of L514. The L514 NH also forms a hydrogen bond with the amide carbonyl of T240 in an adjacent α-helix from a neighboring PA63 monomer, and this intermolecular helix-to-helix interaction may be an important determinant in stabilizing the prepore structure.
Although the form most well studied is the heptameric prepore ((PA63)7), in an elegant series of experiments PA was shown to be capable of forming octameric prepore ((PA63)8) complexes. Kintzer et al. used a combination of electron microscopy, mass spectrometry, crystallography, electrophysiology and other biochemical methods to show the presence of octamers both in solution and on cell surfaces.42 At 20°C the octamer species is only present at a very small percentage, with the predominate form being the heptamer. However, shifting the temperature to 37°C causes the heptamers to aggregate, while the octamers remain soluble.
Once the prepore has formed, binding sites are created for EF and LF, which bind within the region of domain 1′.44–46 In studies by Ezzell, and later by Panchal, cleavage of PA to PA63 has been shown to occur in the bloodstream, where presumably the prepore forms and binds to either the EF or LF components.47, 48 In fact, their studies indicate that very little to no monomeric PA (83 kDa) exists in the bloodstream after infection, and only the PA63 fragment is present. In contrast, studies by Moayeri and coworkers showed that in rats injected with PA, very little cleavage of PA occurs after 6 hours within the bloodstream, but in mice injected with PA, cleavage of PA to PA63 was fast (within 5 min).49 Whether the PA63 observed represents the heptameric or octameric prepore is not known. However, since the heptameric structure forms SDS-resistant oligomers at pH values below 7.5 and is unstable at 37°C,34, 50, 51 conditions which favor the octameric prepore, the latter is more likely to be the form of PA63 encountered in the studies by Ezzell, Panchal and Moayeri.52 Therefore, it may be that there are two potential mechanisms for cell entry of lethal and edema toxin. In one mechanism, a cell surface receptor binds to PA (which is stable at physiological pH and temperature), and is then cleaved by a furin-like protease to PA63, which allows the heptameric or octameric prepore structure to form and subsequent binding of EF and LF; in a second mechanism, the prepore (either heptameric or octameric) is formed in the bloodstream, and the prepore, either alone or bound to EF or LF, binds to the receptor.53 It has been suggested that the heptameric form of PA may bind cells more local to the site of infection, while the octameric form, which in more stable, may act at sites more distant from the point of infection.52
Endocytosis
Once the prepore has formed and bound to the cell surface, the toxin is internalized by receptor-mediated endocytosis. Internalization occurs through regions of the plasma membrane called lipid rafts, which are rich in cholesterol and sphingomyelin.54–56 Endocytosis has also been shown to be mediated by clathrin, which is required for the uptake of other cellular proteins, most notably the low-density lipoprotein (LDL) by the LDL receptor.57 However, a key difference is that while the LDL receptor is naturally present in lipid rafts, the toxin receptor only associates with lipid rafts after PA has bound to the receptor, and after proteolytic processing and formation of the prepore.54, 55, 58 Recent studies have also shown that the intracellular portion of the receptor TEM8 may interact directly with actin, where it changes the conformation of the vWA domain on the extracellular side from a high affinity state to a low affinity state.59 Conversely, interaction of PA with TEM8 dissociates the receptor from actin, presumably to allow oligomerization into the prepore and clustering into clathrin-coated pits.56 Recent studies by Zornetta and coworkers also suggest a role for caveolae, also present in lipid rafts, to be important in cellular uptake of the toxin.60 Once the toxin is endocytosed, it is trafficked to late endosomes, which become acidic.54, 61
Formation of the Membrane Spanning Pore—Identification of Domain 2 as the Major Contributor to the Structure of the Pore
Within the acidified endosome (pH ∼5-6), the prepore undergoes a major conformational change to form a membrane spanning pore, but exactly how this process occurs is the subject of much study. The structure that is ultimately formed is an extended, 14-stranded β-barrel pore which is similar to that of the α-hemolysin pore from Staphylococcal aureus.62 Studies by Benson, and later extended by Nassi, using cysteine scanning mutagenesis and labeling with the membrane impermeable probe methanethiosulfonate ethyltrimethylammonium (MTS-ET), showed that the residues which comprise the “stem” of the pore extend from residue 275 to 352 (the red colored ribbons in Fig. 1).63, 64 This includes the domain 2β2-2β3 strands, the 2β2-2β3 loop that includes the transmembrane region (residues 303-324), and the 2β3-2β4 loop. Based on these biochemical experiments and the structure of the α-hemolysin pore, Nguyen generated a model of the pore which was 180 Å in length (cap and β-barrel stem included), with the 14-stranded β-barrel stem extending 100 Å away from the 80 Å cap region [Fig. 2(B)].65 Using negative stain electron microscopy (EM), Katayama and coworkers have provided the first direct images of the pore, bound to the chaperone GroEL or in lipid nanodiscs.66, 68 The pore dimensions are similar to that predicted from the biochemical data of Nassi and Benson, which again shows the pore forming a ∼100 Å long β-barrel stem [Fig. 2(C)]. However, the pore structure as observed by the EM analysis shows several differences from the model, including a constriction in the cap region, that is, near to where F427 is predicted to be located. This residue is conserved among PA homologs, and forms a key structure in the lumen of the pore called the phi (ϕ)-clamp. This residue is critical for translocation of EF and LF into the cell, and forms a leak-free seal around a translocating polypeptide chain.69, 70 In addition, domain 4 was not resolved in the EM analysis, and is likely to be highly dynamic in the pore structure. In any case, domain 2 and possibly other regions within PA must undergo a major conformational change to form the extended β-barrel structure, and this is most evident from the EM studies.68
To form the pore, the residues that comprise the domain 2β2-2β3 strands as well as the 2β2-2β3 and 2β3-2β4 loops linking these strands must peel away from the core of domain 2 (Fig. 1). As pointed out by Santelli and coworkers, there are many steric factors that impede this process. The domain 2β3-2β4 loop, which is bound in a groove on the surface of CMG2, must dissociate from the receptor to allow movement of the 2β2-2β3 strands away from the core of domain 2. Also inhibiting this process is domain 4, which is buttressed against domain 2. The crystal structure indicates that in order for pore formation to occur, domain 4 must at least partially move away from domain 2.33, 50
How does this process work? What is the initiating step? Because the prepore to pore conversion process can occur within the pH range of ∼7-7.5, the earliest hypotheses suggested that pore formation occurred as a consequence of the protonation of histidine residues.71 Histidine has a side-chain pKa of ∼6-6.5, which is dependent on local environment. There are 10 histidine residues in PA, five of which are located in domain 2, and four of these five reside in the β strands that comprise the β-barrel of the pore. Thus, protonation of one or more of these histidines was postulated to initiate the conformational change to a pore. However, in an effort to determine the role of histidine protonation in the process of pore formation, experiments have been carried out in which PA was labeled biosynthetically with the histidine analog 2-fluorohistidine (2-FHis), which has a side-chain pKa of ∼172 and should resist protonation at pH values required for pore formation (∼5). In these studies, no difference was observed in the pH values required to form a pore between the WT and 2-FHis-labeled proteins.73 In addition to these experiments, Mourez and coworkers used cysteine scanning mutagenesis to determine which residues in PA were critical for function, making 568 individual mutations to cysteine. The majority of the mutations that resulted in defects in cytotoxicity were localized to domain 2. While many of these mutations failed to convert the prepore to an SDS-resistant pore, none of the histidine mutations showed defects in cytotoxicity (except for H304C, which was not expressed).74 Furthermore, before the mutagenesis study by Mourez, Miller and coworkers had shown that deletion of residues 302-325 from PA did not affect the pH required to form an SDS-resistant pore, suggesting that H304 and H310 are not part of the pH sensing mechanism.50
The study by Mourez was carried out to identify key residues that specifically prevent the prepore to pore conversion process and translocation and that are dominant-negative, in which co-oligomerization of potentially a single defective mutant with WT PA63 monomers (ratio of 1 mutant: 6 WT) prevents one or both of these processes from occurring.74 Prior to that study, Sellman and coworkers had identified three key conserved residues (K397, D425, and F427), that upon mutation led specifically to either a block in pore formation (K397A and K397D; D425A, D425N, D425E, and D425K mutations) or a block in translocation (F427A).75, 76 Also, if these mutant proteins are allowed to co-oligomerize with the WT PA63, the resulting prepores are rendered nonfunctional, indicating that the mutations have a dominant negative effect on the prepore structure.73 K397 is located in a loop linking domain 2 β-strands 7 and 8 (2β7-2β8 loop), while D425 and F427 are located in a loop linking domain 2 β-strands 10 and 11 (2β10-2β11 loop). Importantly, these strands are not part of the domain 2β2-2β3 strands that form the β-barrel portion of the pore, and in fact are on the opposite side of domain 2 facing the lumen of the prepore (see Figs. 1 and 3). Conservative mutations of D425 to either asparagine or glutamate (isosteric and isoelectric changes) block low pH-induced pore formation, and as a consequence translocation of LFN, the N-terminal PA binding domain of LF, through the pore. Importantly, studies by Sellman and coworkers showed that mutations at D425 and K397 did not change the ability of PA to bind to cells or form prepores, or alter the cleavage pattern by trypsin to form PA63, strongly suggesting that these mutations did not affect proper folding (although local effects on structure could not be resolved).
Figure 3.
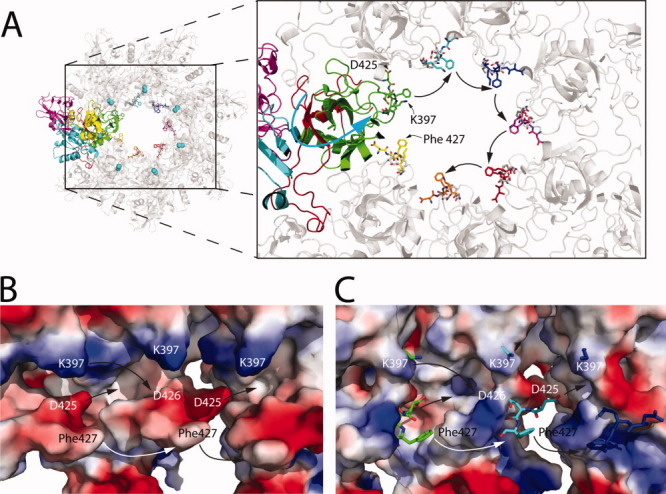
(A) Formation of the ϕ-clamp. Structure of the heptameric prepore (PDB:1TZO),34 with color coding according to Figure 2(A), except that CMG2 is absent. Notice that there are additional residues represented as colored sticks that are within the lumen of the prepore. These residues are from the structure of the monomer of PA bound to CMG2 (PDB: 1T6B), and was generated by creating a structural alignment between PA bound to CMG2 and heptameric prepore. The alignment was carried out using the SSM superpose algorithm in Coot (v 0.1.6).77 Additionally, we show K397 and D425 from 1TZO, represented as gray sticks. Note that the domain 2β2-2β3 strands are on the opposite side of where K397, D425, D426, and F427 are located. The counter-clockwise movement of the subunit proposed here is indicated by the blue arrow. Residues that correspond to 1T6B and 1TZO are indicated. (B) Vacuum electrostatic potential surface of a heptamer comprised of solely 1T6B molecules, generated by the Coot alignment. This is purely a hypothetical model showing the potentials created by K397, D425, D426, and F427 together. In C, the vacuum electrostatic potential generated from the crystal structure of the heptameric prepore 1TZO is shown. For contrast, residues K397, D425, D426, and F427 from the electrostatic potential map in (B) are shown as sticks in C. In both (B) and (C), movement of the subunits together is indicated by the arrows.
These studies have been extended recently by Janowiak and coworkers, who developed a clever strategy to isolate single-subunit mutant containing heptameric prepore molecules (1:6, mutant:WT).78 In that study, a biotinylated form of the D425A or F427A mutant was co-oligomerized with an excess (∼20-fold) of WT PA, and purified using avidin affinity chromatography. Under these conditions, the ratio of 1 mutant to 6 WT monomers per prepore was confirmed, and the D425A single mutation was shown to block low pH (pH 5.5) induced pore formation, highlighting again the importance of this residue in the prepore to pore conversion process. The F427A mutant had a marginal effect on pore formation, but largely blocked translocation of LFN.
F427 has been shown to be particularly important in protein translocation as mentioned above, forming the ϕ-clamp.70 However, F427 seems to also play an important role in the formation of the pore, if the right mutation at this site is made. For instance, mutation of F427 to Gly, Asp or Arg strongly inhibited low-pH induced pore formation.79 While the specific mechanism for how F427 may facilitate pore formation has yet to be resolved, these authors suggest that pi-stacking interactions between adjacent F427 rings may be a driving force for the formation of the pore. Alternatively, Melnyk and Collier showed that K397 and D426 are important for translocation of LFN across the membrane, and postulated that these residues form a conserved intermolecular salt bridge in the pore state that correctly orients F427 into a functional ϕ-clamp conformation.80 Since K397 was identified in previous experiments by Sellman and coworkers to be critical for pore formation, perhaps the formation of this salt bridge is an additional driving force for the formation of the pore. In Figure 3(B,C), we have created a vacuum electrostatic map of the region including K397, D425, D426, and F427, showing the potential positioning of F427 in the lumen of the prepore. Since D426 and F427 are missing electron density in the structures of the heptamer (PDB:1TZO (without CMG2) and 1TZN34), we have added these residues as sticks by overlaying the structure of the monomeric form of PA bound to CMG2 (PDB:1T6B, which has electron density for D426 and F427) and 1TZO. Clearly, there are cavities and room for movement of the side-chains closer together in the pore state.
pH Changes in the Structure of the Monomer as Clues to Regions Sensitive to pH
We know that the consequence of lowering the pH on the prepore structure is pore formation, but what is the consequence of lowering the pH on the monomeric PA structure? Again, aside from PA20, the structures of PA and the prepore overlay quite well, so it may be that there are regions that are sensitive to pH in the monomer structure that provide clues as to which regions in the prepore are also sensitive to pH. Interestingly, regions within the monomeric form of PA have been identified as sensitive to pH using X-ray crystallography, and include the domain 2β3-2β4 loop that binds to the receptor.41 The X-ray structure of PA at pH 7.5 and 6 showed that the 2β3-2β4 loop (Fig. 1) becomes disordered at pH 6. The crystal structure of a homolog of PA, C2-II toxin, solved at pH 4.3, showed little change in the overall structure compared to PA (at pH 6), except that the electron density for domain 4 and for the domain 2β3-2β4 loop was missing.81 These studies indicate that while the overall structure of the monomer is relatively stable to variations in pH, the structure of the domain 2β3-2β4 loop is sensitive to pH, and may aid in facilitating the prepore to pore conversion.
Effect of the Receptor on the Prepore to Pore Conversion
Aside from being a part of the pore, the domain 2β3-2β4 loop has been shown to play a key role in determining the pH requisite for pore formation. Previous studies by Miller and coworkers have shown that if pore formation is monitored on the surface of CHO-K1 cells rather than in solution, the pH requisite for pore formation is ∼1 pH unit less (pH ∼6).50 Lacy and coworkers showed that binding of the vWA domain of the receptor CMG2 to the heptameric prepore lowers the pH requisite for pore formation, from a pH of 7.5 in the absence of the receptor to a pH of ∼5-6 when bound to the receptor.34 Importantly, mutation of residues in CMG2 which destabilize intermolecular interactions with the domain 2β3-2β4 loop result in higher pH values required for pore formation, indicating that this loop plays an important role in dictating the pH requisite for pore formation.36, 82 Using histidine hydrogen–deuterium exchange (HDX), binding of the receptor reduces the rate of hydrogen exchange for most of the histidine residues in monomeric PA, in particular domain 2, including one (H299), that is, distant (∼40Å) from the binding interface.83 This suggests that receptor binding reduces the dynamics of PA, and may strengthen hydrogen bonding interactions throughout the protein, aiding to stabilize the protein against variations in pH.
As mentioned above, the prepore needs to at least partially dissociate from the receptor to sterically allow pore formation to occur. Studies by Rainey and coworkers showed using immunoprecipitation that both CMG2 and TEM8 dissociate from the prepore at pH values concomitant with pore formation (pH ∼5 for CMG2, and pH ∼6 for TEM8).84 Receptor dissociation at low pH was also supported by NMR studies, but interestingly the receptor did not release from the monomeric form of PA at low pH, only the heptameric prepore form. It was argued that since the binding interfaces between monomer and heptameric prepore change very little upon receptor binding, it is unlikely that the protonation of residues within the receptor is the cause of low-pH induced receptor dissociation.85 Furthermore, studies on the isolated domain 4 indicate that the receptor remains bound at pH values that are required for pore formation.37 Taking into account all of these observations, it was argued that receptor release may only occur as a consequence of pore formation.85
Additional evidence that receptor release occurs as a consequence of pore formation has come from recent studies using the 2-FHis-labeled PA, but conducting experiments in the presence of the vWA domain of CMG2. As mentioned, the 2-FHis-labeled prepore is able to form pores at pH values identical to the WT protein. However, when the 2-FHis-labeled prepore is bound to the vWA domain of CMG2, pore formation is largely blocked (a small percentage of pores could form, at pH values similar to that of the WT prepore bound to CMG2).83 Using NMR, it was shown that the receptor did not release at low pH, suggesting that the inability to form a pore was related to an inability to release the receptor. It was argued that there is a structural tightening of the prepore (reduced dynamics) when the receptor binds,86 and that this tightening strengthens hydrogen bonds to the imidazole nitrogens of the histidine residues. Because the 2-FHis residues have a low pKa (∼1), protonation is more difficult, and thus hydrogen bonds to the 2-FHis imidazole nitrogens are likely to be more stable.83 It was suggested, based on that study, that histidine protonation is a trigger for pore formation, but only when bound to the receptor. However, given the studies by Sellman and others, it is unlikely to be the initiating step (see below). Thus, it is hypothesized that the receptor prevents conformational changes that destabilize the prepore to pH, including the conformational changes observed in the domain 2β3-2β4 loop, which, as mentioned is a region that is sensitive to pH.
A Proposed Model for Pore Formation
Based on these cumulative observations, we propose the following kinetic steps for anthrax prepore to pore conversion, which we show in Figure 4, beginning with a subunit of the heptameric prepore (for descriptive reasons) bound to CMG2. STEP 1, at low pH, residues which include K397, D425, D426, and F427, form a narrow iris which becomes the ϕ-clamp. This is based largely on the observations by Sellman, Mourez, Sun and Janowiak and coworkers, that mutation of D425, K397 or, under certain instances F427, blocked pore formation in the absence of the receptor, and in the absence of folding defects, indicating that independent of receptor binding, changes in structure surrounding these residues must occur to initiate pore formation. This structure is stabilized by the formation of favorable intermolecular salt bridges,76, 80 or pi–pi interactions between adjacent Phe427 rings.79 The movement toward the formation of the ϕ-clamp would cause rotation of the individual subunits in a counter-clockwise rotation (Fig. 3), and provides a driving force such that the domain 2-domain 4 interface is shifted away from one another, allowing the domain 2β3-2β4 loop to partially dissociate from the receptor. Both processes (formation of the ϕ-clamp and dissociation of the domain 2 2β3-2β4 loop) would likely occur concomitantly.
Figure 4.
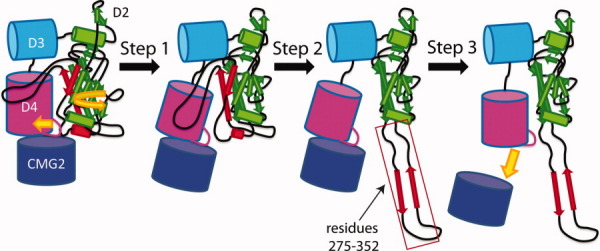
Model of pore formation. In STEP 1, low pH triggers the formation of the ϕ-clamp, which forces a counter-clockwise movement of the subunit and partial receptor dissociation. In STEP 2, the domain 2β2-2β3 strands (comprising residues 275 to 352) peel away from the core of domain 2. In STEP 3, the receptor dissociates.
Early formation of the ϕ-clamp would play an important functional role, since the ϕ-clamp is expected to form a leak-free seal around the N-terminal regions of EF and LF. Formation of the ϕ-clamp as an initial step, followed by insertion of the N-terminal domain of either EF or LF,87 but before the formation of the transmembrane β-barrel, would prevent the flow of ions (H+) from the acidic endosome into the more basic cytosol, preventing dissipation of the gradient required for translocation.69 In addition, it is known that elimination of domain 4 from domain 2 changes the pH requisite for pore formation (in the absence of receptor) to pH > 8, indicating that domain 4 stabilizes the prepore to variations in pH.68 Partial dissociation from domain 4 and from the receptor allows more steric freedom of movement such that the domain 2β2-2β3 strands can dissociate from the core of domain 2.
STEP 2, the domain 2β2-2β3 strands peel away from the core of domain 2 like a banana peeling from the fruit. Based on our studies using the 2-FHis-labeled prepore, this process is likely to be facilitated by histidine protonation, but only when bound to the receptor. Preliminary mutagenesis experiments indicate that at least four of the five histidine residues in domain 2 must be mutated together to prevent pore formation, and as with the 2-FHis-labeled prepore, these combined mutations only prevent pore formation when bound to the receptor (unpublished observations).
STEP 3, the receptor dissociates. In our NMR studies, CMG2 can remain bound to the isolated domain 4 at pH values concomitant with pore formation,37 and thus it may be that pore formation induces a structural change in domain 4 that causes receptor release. Alternatively, it may be that once the pore has formed, domain 4 is sterically prevented from associating with the receptor, perhaps by partially filling the void left by the movement of the domain 2β3-2β4 strands. Once the β strands have peeled away from domain 2, they come together spontaneously to form the β-barrel stem and transmembrane pore [Fig. 2(C)], the barrel inserts into the membrane, and the pore is complete.
Although experimental evidence for the stepwise formation of the pore is lacking, the fact that mutation of residues that form the ϕ-clamp prevents pore formation even when the receptor is not bound would indicate that the formation of the ϕ-clamp is an initiating step in the process. Alternatively, it may be that the domain 2β3-2β4 loop, which is also sensitive to pH, dissociates from the surface of CMG2 and this allows freedom of movement such that the ϕ-clamp may form. In either case, if the ϕ-clamp does form before the unfurling of the β-strands from domain 2, it would support a more general mechanism in which the ion-selectivity filter region of a pore forms initially before acquisition of the ion-conducting pore state. Such is the case for the voltage-gated potassium channel Kv1.3, for instance, where the reentrant pore conformation is attained before oligomerization into the tetramer and formation of a folded functional channel.88
Once the pore has formed, EF and LF must unfold their three-dimensional structures for passage through the pore into the cell.69, 89 The pore can form independent of binding to EF or LF, and is the likely cause of cytotoxicity in the absence of the enzymatic components.90, 91 Although as yet there is no direct evidence that EF and LF translocate through the pore into the cytosol, introduction of disulfide bonds into LF blocks translocation.92 In addition, recent studies by Zornetta and coworkers utilizing GFP fusions between EF or LF, have shown that the movement of the fusion protein into the cytosol is dependent on the ability of the fusion protein to efficiently unfold.60 The recent structure of the prepore form of the octamer, bound to LFN, has provided key insight into how the prepore interacts with the enzymatic components46 (for a recent excellent review, see Thoren and Krantz93). Advancements along this line continue to be made in deciphering not only the mechanisms of translocation of EF and LF, but for providing much needed knowledge on the general mechanisms of protein unfolding and translocation through membranes.
Acknowledgments
The author would like to thank the members of the Bann laboratory for their helpful comments on this review: Kiran K. Andra, Fatemah Chadegani, Alex S. Williams, and Letisha J. Ferris. He also thank Prof. Mark T. Fisher (University of Kansas Medical School) for graciously providing the EM image of the pore. The author would like to especially thank his wife Jodie for her support and reading of this review.
References
- 1.Jernigan JA, Stephens DS, Ashford DA, Omenaca C, Topiel MS, Galbraith M, Tapper M, Fisk TL, Zaki S, Popovic T, Meyer RF, Quinn CP, Harper SA, Fridkin SK, Sejvar JJ, Shepard CW, McConnell M, Guarner J, Shieh WJ, Malecki JM, Gerberding JL, Hughes JM, Perkins BA. Bioterrorism-related inhalational anthrax: the first 10 cases reported in the United States. Emerg Infect Dis. 2001;7:933–944. doi: 10.3201/eid0706.010604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chitlaru T, Altboum Z, Reuveny S, Shafferman A. Progress and novel strategies in vaccine development and treatment of anthrax. Immunol Rev. 2011;239:221–236. doi: 10.1111/j.1600-065X.2010.00969.x. [DOI] [PubMed] [Google Scholar]
- 3.Cote CK, Bozue J, Twenhafel N, Welkos SL. Effects of altering the germination potential of Bacillus anthracis spores by exogenous means in a mouse model. J Med Microbiol. 2009;58:816–825. doi: 10.1099/jmm.0.008656-0. [DOI] [PubMed] [Google Scholar]
- 4.Van Ness GB. Ecology of anthrax. Science. 1971;172:1303–1307. doi: 10.1126/science.172.3990.1303. [DOI] [PubMed] [Google Scholar]
- 5.Durrheim DN, Freeman P, Roth I, Hornitzky M. Epidemiologic questions from anthrax outbreak, Hunter Valley, Australia. Emerg Infect Dis. 2009;15:840–842. doi: 10.3201/eid1505.081744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bell JH, Fee E, Brown TM. Anthrax and the wool trade. 1902. Am J Public Health. 2002;92:754–757. doi: 10.2105/ajph.92.5.754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stojkovic B, Torres EM, Prouty AM, Patel HK, Zhuang L, Koehler TM, Ballard JD, Blanke SR. High-throughput, single-cell analysis of macrophage interactions with fluorescently labeled Bacillus anthracis spores. Appl Environ Microbiol. 2008;74:5201–5210. doi: 10.1128/AEM.02890-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cleret A, Quesnel-Hellmann A, Vallon-Eberhard A, Verrier B, Jung S, Vidal D, Mathieu J, Tournier JN. Lung dendritic cells rapidly mediate anthrax spore entry through the pulmonary route. J Immunol. 2007;178:7994–8001. doi: 10.4049/jimmunol.178.12.7994. [DOI] [PubMed] [Google Scholar]
- 9.Guidi-Rontani C, Weber-Levy M, Labruyere E, Mock M. Germination of Bacillus anthracis spores within alveolar macrophages. Mol Microbiol. 1999;31:9–17. doi: 10.1046/j.1365-2958.1999.01137.x. [DOI] [PubMed] [Google Scholar]
- 10.Friedlander AM, Welkos SL, Pitt ML, Ezzell JW, Worsham PL, Rose KJ, Ivins BE, Lowe JR, Howe GB, Mikesell P, Lawrence WB. Postexposure prophylaxis against experimental inhalation anthrax. J Infect Dis. 1993;167:1239–1243. doi: 10.1093/infdis/167.5.1239. [DOI] [PubMed] [Google Scholar]
- 11.Jefferds MD, Laserson K, Fry AM, Roy S, Hayslett J, Grummer-Strawn L, Kettel-Khan L, Schuchat A. Adherence to antimicrobial inhalational anthrax prophylaxis among postal workers, Washington, D.C., 2001. Emerg Infect Dis. 2002;8:1138–1144. doi: 10.3201/eid0810.020331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Green BD, Battisti L, Koehler TM, Thorne CB, Ivins BE. Demonstration of a capsule plasmid in Bacillus anthracis. Infect Immun. 1985;49:291–297. doi: 10.1128/iai.49.2.291-297.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beall FA, Taylor MJ, Thorne CB. Rapid lethal effect in rats of a third component found upon fractionating the toxin of Bacillus anthracis. J Bacteriol. 1962;83:1274–1280. doi: 10.1128/jb.83.6.1274-1280.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stanley JL, Smith H. The three factors of anthrax toxin: their immunogenicity and lack of demonstrable enzymic activity. J Gen Microbiol. 1963;31:329–337. doi: 10.1099/00221287-31-2-329. [DOI] [PubMed] [Google Scholar]
- 15.Barth H, Aktories K, Popoff MR, Stiles BG. Binary bacterial toxins: biochemistry, biology, and applications of common Clostridium and Bacillus proteins. Microbiol Mol Biol Rev. 2004;68:373–402. doi: 10.1128/MMBR.68.3.373-402.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strange RE, Thorne CB. Further purification studies on the protective antigen of Bacillus anthracis produced in vitro. J Bacteriol. 1958;76:192–202. doi: 10.1128/jb.76.2.192-202.1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ivins BE, Ezzell JW, Jr, Jemski J, Hedlund KW, Ristroph JD, Leppla SH. Immunization studies with attenuated strains of Bacillus anthracis. Infect Immun. 1986;52:454–458. doi: 10.1128/iai.52.2.454-458.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vick JA, Lincoln RE, Klein F, Mahlandt BG, Walker JS, Fish DC. Neurological and physiological responses of the primate to anthrax toxin. J Infect Dis. 1968;118:85–96. doi: 10.1093/infdis/118.1.85. [DOI] [PubMed] [Google Scholar]
- 19.Fish DC, Mahlandt BG, Dobbs JP, Lincoln RE. Purification and properties of in vitro-produced anthrax toxin components. J Bacteriol. 1968;95:907–918. doi: 10.1128/jb.95.3.907-918.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leppla SH. Bacillus anthracis calmodulin-dependent adenylate cyclase: chemical and enzymatic properties and interactions with eucaryotic cells. Adv Cyclic Nucleotide Protein Phosphorylation Res. 1984;17:189–198. [PubMed] [Google Scholar]
- 21.Hong J, Doebele RC, Lingen MW, Quilliam LA, Tang WJ, Rosner MR. Anthrax edema toxin inhibits endothelial cell chemotaxis via Epac and Rap1. J Biol Chem. 2007;282:19781–19787. doi: 10.1074/jbc.M700128200. [DOI] [PubMed] [Google Scholar]
- 22.Duesbery NS, Webb CP, Leppla SH, Gordon VM, Klimpel KR, Copeland TD, Ahn NG, Oskarsson MK, Fukasawa K, Paull KD, Vande Woude GF. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science. 1998;280:734–737. doi: 10.1126/science.280.5364.734. [DOI] [PubMed] [Google Scholar]
- 23.Pellizzari R, Guidi-Rontani C, Vitale G, Mock M, Montecucco C. Lethal factor of Bacillus anthracis cleaves the N-terminus of MAPKKs: analysis of the intracellular consequences in macrophages. Int J Med Microbiol. 2000;290:421–427. doi: 10.1016/S1438-4221(00)80056-9. [DOI] [PubMed] [Google Scholar]
- 24.Pellizzari R, Guidi-Rontani C, Vitale G, Mock M, Montecucco C. Anthrax lethal factor cleaves MKK3 in macrophages and inhibits the LPS/IFNgamma-induced release of NO and TNFalpha. FEBS Lett. 1999;462:199–204. doi: 10.1016/s0014-5793(99)01502-1. [DOI] [PubMed] [Google Scholar]
- 25.Lehmann M, Noack D, Wood M, Perego M, Knaus UG. Lung epithelial injury by B. anthracis lethal toxin is caused by MKK-dependent loss of cytoskeletal integrity. PLoS One. 2009;4:e4755. doi: 10.1371/journal.pone.0004755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bradley KA, Mogridge J, Mourez M, Collier RJ, Young JA. Identification of the cellular receptor for anthrax toxin. Nature. 2001;414:225–229. doi: 10.1038/n35101999. [DOI] [PubMed] [Google Scholar]
- 27.St Croix B, Rago C, Velculescu V, Traverso G, Romans KE, Montgomery E, Lal A, Riggins GJ, Lengauer C, Vogelstein B, Kinzler KW. Genes expressed in human tumor endothelium. Science. 2000;289:1197–1202. doi: 10.1126/science.289.5482.1197. [DOI] [PubMed] [Google Scholar]
- 28.Scobie HM, Rainey GJ, Bradley KA, Young JA. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc Natl Acad Sci USA. 2003;100:5170–5174. doi: 10.1073/pnas.0431098100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martchenko M, Jeong SY, Cohen SN. Heterodimeric integrin complexes containing beta1-integrin promote internalization and lethality of anthrax toxin. Proc Natl Acad Sci USA. 2010;107:15583–15588. doi: 10.1073/pnas.1010145107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bell SE, Mavila A, Salazar R, Bayless KJ, Kanagala S, Maxwell SA, Davis GE. Differential gene expression during capillary morphogenesis in 3D collagen matrices: regulated expression of genes involved in basement membrane matrix assembly, cell cycle progression, cellular differentiation and G-protein signaling. J Cell Sci. 2001;114:2755–2773. doi: 10.1242/jcs.114.15.2755. [DOI] [PubMed] [Google Scholar]
- 31.Nanda A, Carson-Walter EB, Seaman S, Barber TD, Stampfl J, Singh S, Vogelstein B, Kinzler KW, St Croix B. TEM8 interacts with the cleaved C5 domain of collagen alpha 3(VI) Cancer Res. 2004;64:817–820. doi: 10.1158/0008-5472.can-03-2408. [DOI] [PubMed] [Google Scholar]
- 32.Lee JO, Rieu P, Arnaout MA, Liddington R. Crystal structure of the A domain from the alpha subunit of integrin CR3 (CD11b/CD18) Cell. 1995;80:631–638. doi: 10.1016/0092-8674(95)90517-0. [DOI] [PubMed] [Google Scholar]
- 33.Santelli E, Bankston LA, Leppla SH, Liddington RC. Crystal structure of a complex between anthrax toxin and its host cell receptor. Nature. 2004;430:905–908. doi: 10.1038/nature02763. [DOI] [PubMed] [Google Scholar]
- 34.Lacy DB, Wigelsworth DJ, Melnyk RA, Harrison SC, Collier RJ. Structure of heptameric protective antigen bound to an anthrax toxin receptor: a role for receptor in pH-dependent pore formation. Proc Natl Acad Sci USA. 2004;101:13147–13151. doi: 10.1073/pnas.0405405101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Varughese M, Teixeira AV, Liu S, Leppla SH. Identification of a receptor-binding region within domain 4 of the protective antigen component of anthrax toxin. Infect Immun. 1999;67:1860–1865. doi: 10.1128/iai.67.4.1860-1865.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scobie HM, Wigelsworth DJ, Marlett JM, Thomas D, Rainey GJ, Lacy DB, Manchester M, Collier RJ, Young JA. Anthrax toxin receptor 2-dependent lethal toxin killing in vivo. PLoS Pathog. 2006;2:e111. doi: 10.1371/journal.ppat.0020111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Williams AS, Lovell S, Anbanandam A, El-Chami R, Bann JG. Domain 4 of the anthrax protective antigen maintains structure and binding to the host receptor CMG2 at low pH. Protein Sci. 2009;18:2277–2286. doi: 10.1002/pro.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wigelsworth DJ, Krantz BA, Christensen KA, Lacy DB, Juris SJ, Collier RJ. Binding stoichiometry and kinetics of the interaction of a human anthrax toxin receptor, CMG2, with protective antigen. J Biol Chem. 2004;279:23349–23356. doi: 10.1074/jbc.M401292200. [DOI] [PubMed] [Google Scholar]
- 39.Klimpel KR, Molloy SS, Thomas G, Leppla SH. Anthrax toxin protective antigen is activated by a cell surface protease with the sequence specificity and catalytic properties of furin. Proc Natl Acad Sci USA. 1992;89:10277–10281. doi: 10.1073/pnas.89.21.10277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mogridge J, Cunningham K, Collier RJ. Stoichiometry of anthrax toxin complexes. Biochemistry. 2002;41:1079–1082. doi: 10.1021/bi015860m. [DOI] [PubMed] [Google Scholar]
- 41.Petosa C, Collier RJ, Klimpel KR, Leppla SH, Liddington RC. Crystal structure of the anthrax toxin protective antigen. Nature. 1997;385:833–838. doi: 10.1038/385833a0. [DOI] [PubMed] [Google Scholar]
- 42.Kintzer AF, Thoren KL, Sterling HJ, Dong KC, Feld GK, Tang II, Zhang TT, Williams ER, Berger JM, Krantz BA. The protective antigen component of anthrax toxin forms functional octameric complexes. J Mol Biol. 2009;392:614–629. doi: 10.1016/j.jmb.2009.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mogridge J, Cunningham K, Lacy DB, Mourez M, Collier RJ. The lethal and edema factors of anthrax toxin bind only to oligomeric forms of the protective antigen. Proc Natl Acad Sci USA. 2002;99:7045–7048. doi: 10.1073/pnas.052160199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Melnyk RA, Hewitt KM, Lacy DB, Lin HC, Gessner CR, Li S, Woods VL, Jr, Collier RJ. Structural determinants for the binding of anthrax lethal factor to oligomeric protective antigen. J Biol Chem. 2006;281:1630–1635. doi: 10.1074/jbc.M511164200. [DOI] [PubMed] [Google Scholar]
- 45.Ren G, Quispe J, Leppla SH, Mitra AK. Large-scale structural changes accompany binding of lethal factor to anthrax protective antigen: a cryo-electron microscopic study. Structure. 2004;12:2059–2066. doi: 10.1016/j.str.2004.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Feld GK, Thoren KL, Kintzer AF, Sterling HJ, Tang II, Greenberg SG, Williams ER, Krantz BA. Structural basis for the unfolding of anthrax lethal factor by protective antigen oligomers. Nat Struct Mol Biol. 2010;17:1383–1390. doi: 10.1038/nsmb.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ezzell JW, Jr, Abshire TG. Serum protease cleavage of Bacillus anthracis protective antigen. J Gen Microbiol. 1992;138:543–549. doi: 10.1099/00221287-138-3-543. [DOI] [PubMed] [Google Scholar]
- 48.Panchal RG, Halverson KM, Ribot W, Lane D, Kenny T, Abshire TG, Ezzell JW, Hoover TA, Powell B, Little S, Kasianowicz JJ, Bavari S. Purified Bacillus anthracis lethal toxin complex formed in vitro and during infection exhibits functional and biological activity. J Biol Chem. 2005;280:10834–10839. doi: 10.1074/jbc.M412210200. [DOI] [PubMed] [Google Scholar]
- 49.Moayeri M, Wiggins JF, Leppla SH. Anthrax protective antigen cleavage and clearance from the blood of mice and rats. Infect Immun. 2007;75:5175–5184. doi: 10.1128/IAI.00719-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miller CJ, Elliott JL, Collier RJ. Anthrax protective antigen: prepore-to-pore conversion. Biochemistry. 1999;38:10432–10441. doi: 10.1021/bi990792d. [DOI] [PubMed] [Google Scholar]
- 51.Milne JC, Furlong D, Hanna PC, Wall JS, Collier RJ. Anthrax protective antigen forms oligomers during intoxication of mammalian cells. J Biol Chem. 1994;269:20607–20612. [PubMed] [Google Scholar]
- 52.Kintzer AF, Sterling HJ, Tang II, Abdul-Gader A, Miles AJ, Wallace BA, Williams ER, Krantz BA. Role of the protective antigen octamer in the molecular mechanism of anthrax lethal toxin stabilization in plasma. J Mol Biol. 2010;399:741–758. doi: 10.1016/j.jmb.2010.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gawlik K, Remacle AG, Shiryaev SA, Golubkov VS, Ouyang M, Wang Y, Strongin AY. A femtomol range FRET biosensor reports exceedingly low levels of cell surface furin: implications for the processing of anthrax protective antigen. PLoS One. 2010;5:e11305. doi: 10.1371/journal.pone.0011305. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 54.Abrami L, Liu S, Cosson P, Leppla SH, van der Goot FG. Anthrax toxin triggers endocytosis of its receptor via a lipid raft-mediated clathrin-dependent process. J Cell Biol. 2003;160:321–328. doi: 10.1083/jcb.200211018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Abrami L, Leppla SH, van der Goot FG. Receptor palmitoylation and ubiquitination regulate anthrax toxin endocytosis. J Cell Biol. 2006;172:309–320. doi: 10.1083/jcb.200507067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Abrami L, Bischofberger M, Kunz B, Groux R, van der Goot FG. Endocytosis of the anthrax toxin is mediated by clathrin, actin and unconventional adaptors. PLoS Pathog. 2010;6:e1000792. doi: 10.1371/journal.ppat.1000792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brown MS, Goldstein JL. Receptor-mediated endocytosis: insights from the lipoprotein receptor system. Proc Natl Acad Sci USA. 1979;76:3330–3337. doi: 10.1073/pnas.76.7.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Beauregard KE, Collier RJ, Swanson JA. Proteolytic activation of receptor-bound anthrax protective antigen on macrophages promotes its internalization. Cell Microbiol. 2000;2:251–258. doi: 10.1046/j.1462-5822.2000.00052.x. [DOI] [PubMed] [Google Scholar]
- 59.Go MY, Chow EM, Mogridge J. The cytoplasmic domain of anthrax toxin receptor 1 affects binding of the protective antigen. Infect Immun. 2009;77:52–59. doi: 10.1128/IAI.01073-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zornetta I, Brandi L, Janowiak B, Dal Molin F, Tonello F, Collier RJ, Montecucco C. Imaging the cell entry of the anthrax oedema and lethal toxins with fluorescent protein chimeras. Cell Microbiol. 2010;12:1435–1445. doi: 10.1111/j.1462-5822.2010.01480.x. [DOI] [PubMed] [Google Scholar]
- 61.Friedlander AM. Macrophages are sensitive to anthrax lethal toxin through an acid-dependent process. J Biol Chem. 1986;261:7123–7126. [PubMed] [Google Scholar]
- 62.Song L, Hobaugh MR, Shustak C, Cheley S, Bayley H, Gouaux JE. Structure of staphylococcal alpha-hemolysin, a heptameric transmembrane pore. Science. 1996;274:1859–1866. doi: 10.1126/science.274.5294.1859. [DOI] [PubMed] [Google Scholar]
- 63.Benson EL, Huynh PD, Finkelstein A, Collier RJ. Identification of residues lining the anthrax protective antigen channel. Biochemistry. 1998;37:3941–3948. doi: 10.1021/bi972657b. [DOI] [PubMed] [Google Scholar]
- 64.Nassi S, Collier RJ, Finkelstein A. PA63 channel of anthrax toxin: an extended beta-barrel. Biochemistry. 2002;41:1445–1450. doi: 10.1021/bi0119518. [DOI] [PubMed] [Google Scholar]
- 65.Nguyen TL. Three-dimensional model of the pore form of anthrax protective antigen. Structure and biological implications. J Biomol Struct Dyn. 2004;22:253–265. doi: 10.1080/07391102.2004.10531226. [DOI] [PubMed] [Google Scholar]
- 66.Katayama H, Janowiak BE, Brzozowski M, Juryck J, Falke S, Gogol EP, Collier RJ, Fisher MT. GroEL as a molecular scaffold for structural analysis of the anthrax toxin pore. Nat Struct Mol Biol. 2008;15:754–760. doi: 10.1038/nsmb.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 68.Katayama H, Wang J, Tama F, Chollet L, Gogol EP, Collier RJ, Fisher MT. Three-dimensional structure of the anthrax toxin pore inserted into lipid nanodiscs and lipid vesicles. Proc Natl Acad Sci USA. 2010;107:3453–3457. doi: 10.1073/pnas.1000100107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Krantz BA, Finkelstein A, Collier RJ. Protein translocation through the anthrax toxin transmembrane pore is driven by a proton gradient. J Mol Biol. 2006;355:968–979. doi: 10.1016/j.jmb.2005.11.030. [DOI] [PubMed] [Google Scholar]
- 70.Krantz BA, Melnyk RA, Zhang S, Juris SJ, Lacy DB, Wu Z, Finkelstein A, Collier RJ. A phenylalanine clamp catalyzes protein translocation through the anthrax toxin pore. Science. 2005;309:777–781. doi: 10.1126/science.1113380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Blaustein RO, Koehler TM, Collier RJ, Finkelstein A. Anthrax toxin: channel-forming activity of protective antigen in planar phospholipid bilayers. Proc Natl Acad Sci USA. 1989;86:2209–2213. doi: 10.1073/pnas.86.7.2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yeh HJC, Kirk KL, Cohen LA, Cohen JS. 19F and 1H nuclear magnetic resonance studies of ring-fluorinated imidazoles and histidines. J Chem Soc Perkin Trans. 1975;2:928–934. [Google Scholar]
- 73.Wimalasena DS, Cramer JC, Janowiak BE, Juris SJ, Melnyk RA, Anderson DE, Kirk KL, Collier RJ, Bann JG. Effect of 2-fluorohistidine labeling of the anthrax protective antigen on stability, pore formation, and translocation. Biochemistry. 2007;46:14928–14936. doi: 10.1021/bi701763z. [DOI] [PubMed] [Google Scholar]
- 74.Mourez M, Yan M, Lacy DB, Dillon L, Bentsen L, Marpoe A, Maurin C, Hotze E, Wigelsworth D, Pimental RA, Ballard JD, Collier RJ, Tweten RK. Mapping dominant-negative mutations of anthrax protective antigen by scanning mutagenesis. Proc Natl Acad Sci USA. 2003;100:13803–13808. doi: 10.1073/pnas.2436299100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sellman BR, Mourez M, Collier RJ. Dominant-negative mutants of a toxin subunit: an approach to therapy of anthrax. Science. 2001;292:695–697. doi: 10.1126/science.109563. [DOI] [PubMed] [Google Scholar]
- 76.Sellman BR, Nassi S, Collier RJ. Point mutations in anthrax protective antigen that block translocation. J Biol Chem. 2001:2768371–8376. doi: 10.1074/jbc.M008309200. [DOI] [PubMed] [Google Scholar]
- 77.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Cryst. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Janowiak BE, Finkelstein A, Collier RJ. An approach to characterizing single-subunit mutations in multimeric prepores and pores of anthrax protective antigen. Protein Sci. 2009;18:348–358. doi: 10.1002/pro.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sun J, Lang AE, Aktories K, Collier RJ. Phenylalanine-427 of anthrax protective antigen functions in both pore formation and protein translocation. Proc Natl Acad Sci USA. 2008;105:4346–4351. doi: 10.1073/pnas.0800701105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Melnyk RA, Collier RJ. A loop network within the anthrax toxin pore positions the phenylalanine clamp in an active conformation. Proc Natl Acad Sci USA. 2006;103:9802–9807. doi: 10.1073/pnas.0604000103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schleberger C, Hochmann H, Barth H, Aktories K, Schulz GE. Structure and action of the binary C2 toxin from Clostridium botulinum. J Mol Biol. 2006;364:705–715. doi: 10.1016/j.jmb.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 82.Scobie HM, Marlett JM, Rainey GJ, Lacy DB, Collier RJ, Young JA. Anthrax toxin receptor 2 determinants that dictate the pH threshold of toxin pore formation. PLoS One. 2007;2:e329. doi: 10.1371/journal.pone.0000329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wimalasena DS, Janowiak BE, Lovell S, Miyagi M, Sun J, Zhou H, Hajduch J, Pooput C, Kirk KL, Battaile KP, Bann JG. Evidence that histidine protonation of receptor-bound anthrax protective antigen is a trigger for pore formation. Biochemistry. 2010;49:6973–6983. doi: 10.1021/bi100647z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rainey GJ, Wigelsworth DJ, Ryan PL, Scobie HM, Collier RJ, Young JA. Receptor-specific requirements for anthrax toxin delivery into cells. Proc Natl Acad Sci USA. 2005;102:13278–13283. doi: 10.1073/pnas.0505865102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rajapaksha M, Eichler JF, Hajduch J, Anderson DE, Kirk KL, Bann JG. Monitoring anthrax toxin receptor dissociation from the protective antigen by NMR. Protein Sci. 2009;18:17–23. doi: 10.1002/pro.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Williams DH, O'Brien DP, Sandercock AM, Stephens E. Order changes within receptor systems upon ligand binding: receptor tightening/oligomerisation and the interpretation of binding parameters. J Mol Biol. 2004;340:373–383. doi: 10.1016/j.jmb.2004.04.056. [DOI] [PubMed] [Google Scholar]
- 87.Pentelute BL, Sharma O, Collier RJ. Chemical dissection of protein translocation through the anthrax toxin pore. Angew Chem Int Ed Engl. 2011;50:2294–2296. doi: 10.1002/anie.201006460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gajewski C, Dagcan A, Roux B, Deutsch C. Biogenesis of the pore architecture of a voltage-gated potassium channel. Proc Natl Acad Sci USA. 2011;108:3240–3245. doi: 10.1073/pnas.1017097108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Krantz BA, Trivedi AD, Cunningham K, Christensen KA, Collier RJ. Acid-induced unfolding of the amino-terminal domains of the lethal and edema factors of anthrax toxin. J Mol Biol. 2004;344:739–756. doi: 10.1016/j.jmb.2004.09.067. [DOI] [PubMed] [Google Scholar]
- 90.Backer MV, Patel V, Jehning BT, Claffey KP, Karginov VA, Backer JM. Inhibition of anthrax protective antigen outside and inside the cell. Antimicrob Agents Chemother. 2007;51:245–251. doi: 10.1128/AAC.00983-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Salles II, Voth DE, Ward SC, Averette KM, Tweten RK, Bradley KA, Ballard JD. Cytotoxic activity of Bacillus anthracis protective antigen observed in a macrophage cell line overexpressing ANTXR1. Cell Microbiol. 2006;8:1272–1281. doi: 10.1111/j.1462-5822.2006.00708.x. [DOI] [PubMed] [Google Scholar]
- 92.Wesche J, Elliott JL, Falnes PO, Olsnes S, Collier RJ. Characterization of membrane translocation by anthrax protective antigen. Biochemistry. 1998;37:15737–15746. doi: 10.1021/bi981436i. [DOI] [PubMed] [Google Scholar]
- 93.Thoren KL, Krantz BA. The unfolding story of anthrax toxin translocation. Mol Microbiol. 2011;80:588–595. doi: 10.1111/j.1365-2958.2011.07614.x. [DOI] [PMC free article] [PubMed] [Google Scholar]