

Abstract
Recent biochemical studies have identified a cell surface receptor for thyroid and steroid hormones that bind near the arginine-glycine-aspartate (RGD) recognition site on the heterodimeric αvβ3 integrin. To further characterize the intermolecular interactions for a series of hormone analogues, combined quantum mechanical and molecular mechanical (QM/MM) methods were used to calculate their interaction energies. All calculations were performed in the presence of either calcium (Ca2+) or magnesium (Mg2+) ions. These data reveal that 3,5′-triiodothyronine (T3) and 3,5,3′,5′-tetraiodothyroacetic acid (T4ac) bound in two different modes, occupying two alternate sites, one of which is along the Arg side chain of the RGD cyclic peptide site. These orientations differ from those of the other ligands whose alternate binding modes placed the ligands deeper within the RGD binding pocket. These observations are consistent with biological data that indicate the presence of two discrete binding sites that control distinct downstream signal transduction pathways for T3.
1. Introduction
Integrins are plasma membrane proteins that generate complex and intracellular signals during morphogenesis, tissue remodeling, and repair [1]. Interactions of integrins with specific extracellular matrix proteins and growth factors or with certain small molecules generate ligand-specific signals. The integrin superfamily of heterodimeric glycoproteins consists of α- and β-monomers that associate in defined combinations that include more than twenty different mammalian subtypes, of which at least eight are characterized by the presence of an Arg-Gly-Asp (RGD) site used in the process of recognition of different protein ligands [1–3]. The combination of αvβ3 subunits is the most abundant integrin expressed in mammalian cells [1].
Structural data have revealed that the extracellular segment of integrin αvβ3 (i.e., lacking the transmembrane and cytoplasmic tails) is V shaped with the 4-domain αv subunit and the 8-domain β3 subunit bent by 135° [4–6]. Data for the RGD cyclic pentapeptide complex reveals that the RGD peptide occupies a shallow crevice between the propeller and βA domains in the integrin head.
The X-ray structure of apo αvβ3 has been determined in the presence of Ca2+ ions [6] and in the presence of Mn2+ as a complex with the cyclic pentapeptide Arg-Gly-Asp-{D-Phe}-{N-methyl-Val} (Figure 1) [4]. These data showed that replacement of Ca2+ with Mn2+ in the αvβ3 integrin did not result in any important structural changes to the protein. There are also relatively small changes in the αvβ3 structure with and without the cyclic pentapeptide bound in the RGD site of integrin.
Figure 1.
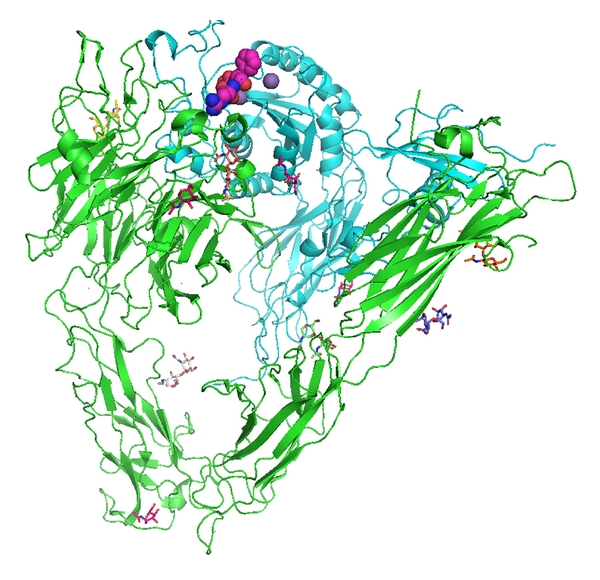
View of αvβ3 integrin from 1L5G.pdb (Xiong et al., 2001 [4]). RGD cyclic pentapeptide is shown as space filling atoms (violet), NAG sugars are stick figures in various colors, and the sphere (purple) is a Mn2+ ion. The αv domain is green and the β3 domain is cyan. Figure drawn with PyMol.
Ligand binding to integrins is dependent upon bivalent-cation interactions that are generally stimulated by Mg2+ or Mn2+ and inhibited by Ca2+. Competitive binding data revealed preferential binding sites for one or more cations that could also be displaced by other cations and that their specific occupancy can lead to enhanced ligand binding in an allosteric or synergistic manner [1, 7]. The central core of the β-propeller is one of the primary sites for cation binding. A common structural motif observed for integrins is the presence of a metal binding site, termed metal ion-dependent adhesion site (MIDAS), that is unique for Mg2+/Mn2+ and is located in the integrin α subunit I domain. Mutagenesis data have shown that the MIDAS motif and the nearby solvent exposed side chains are required for ligand binding [3].
The integrin protein family has been studied both experimentally and theoretically [8–15]. Studies using NMR and molecular dynamics methods to map the interaction of the cyclic β-peptide isoDGR, a competitive antagonist of RGD-containing ligands of αvβ3 integrin that inhibits endothelial cell adhesion, proliferation, and tumor growth, revealed a new recognition motif of the RGD binding pocket of αvβ3 integrin [9]. Docking studies for a series of cyclic peptides further defined the structure-function relationships in binding to the RGD recognition site of αvβ3 integrin.
Studies using a combination of experimental and computational approaches defined the role of divalent cations in mediating the binding of integrins to RGD peptides. Structural data revealed that the divalent cation at the MIDAS site in the βA domain of integrin contacts the Asp of the RGD peptide. This site is occupied by the divalent ion only in the RGD-liganded state [15]. The results of these molecular dynamics studies support the stability of the orientation of Asp residue of the RGD recognition site that interacts with the MIDAS which is essential for binding, whereas the orientation of Glu220 carboxyl oxygens of the β3 domain for the ligand associated metal binding sites (LIMBSs) was maintained throughout the molecular dynamics simulations [10, 15]. The steered molecular dynamics studies of the unbinding of RGD cyclic peptides from αvβ3 integrin revealed the importance of single water molecules tightly coordinated to the MIDAS ion. These data suggest that this process is a general strategy for the stabilization of protein-protein adhesion against cell-derived forces through divalent cations [15].
Recently, a cell surface receptor for thyroid hormone has been identified on the extracellular domain of the αvβ3 integrin [16] that is relevant to the proliferative [16–19] and proangiogenic activity of thyroid hormone [16, 20, 21]. Transduction of the hormone signal into proliferation and angiogenesis at the integrin is achieved by the mitogen-activated protein kinase (MAPK; extracellular regulated kinase 1/2, ERK1/2) pathway [16, 19, 22, 23] or by the phosphatidylinositol 3-kinase (P3K)/Akt pathway [24–26]. L-Thyroxine (T4) and 3,5,3′-triiodo-L-thyronine (T3) are agonist thyroid hormone analogues at the integrin receptor, whereas a deaminated derivative of T4, tetraiodothyroacetic acid (T4ac), is a nonagonist inhibitor of the binding of T4 and T3 to integrin. Competition data revealed that RGD peptides also block hormone binding by integrin, suggesting that the hormone-binding site is near the RGD recognition site on αvβ3. Further analysis revealed that RGD peptides could inhibit the binding of other biologically active small molecules with estrogen-like structures, such as cis- and trans-resveratrol and stilbene (Figure 2) [18, 27–29]. These data reveal that T4 promotes cell proliferation in cancer cells whereas resveratrol is proapoptotic and that further shows that T4 inhibits resveratrol-induced apoptosis [18]. This action is prevented by T4ac, shown to be an inhibitor of T4 binding to integrin. In the presence of T4, T4ac does not affect the induction of apoptosis by resveratrol [18, 30]. These observations suggest that there is a discrete binding site for resveratrol and thyroid hormone on integrin.
Figure 2.
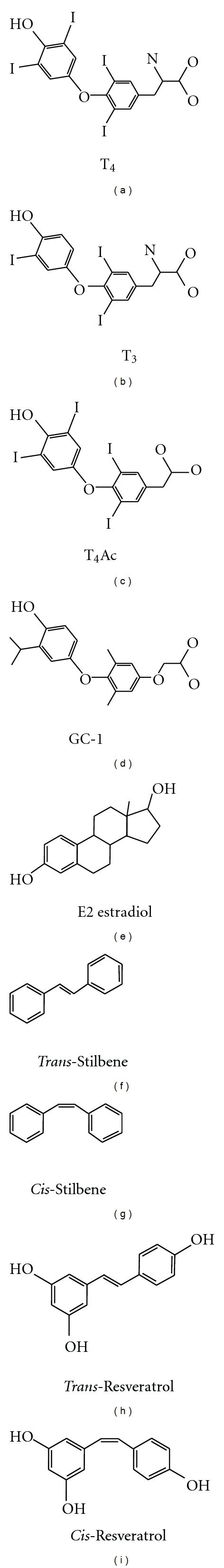
Schematic of ligands bound to αvβ3 integrin.
The noniodinated thyroid hormone analogue GC-1 lacks the amino group of the alanine side chain, lacks the ether linkage between phenyl rings, and has methyl groups at the 3,5-positions and an isopropyl group at the 3′-position, compared with the thyroid hormone T3 (Figure 2). This thyroid hormone analogue is a selective agonist for nuclear thyroid hormone receptor TRβ1 and has the desirable metabolic effect of lowering circulating cholesterol without inducing tachycardia [31, 32]. GC-1 was also demonstrated to be proangiogenic at the plasma membrane and require interaction with αvβ3 integrin [20].
To understand how these hormones and their analogues interact with integrin, modeling studies were carried out using the coordinates of the extracellular fragment of αvβ3 integrin (1L5G.pdb) [4, 5] as a starting model for their interactions near the RGD recognition site [27]. These preliminary models revealed that the hormone analogues shown in Figure 2 can bind in the RGD recognition site and that there may be two closely related binding pockets that can bind these hormone analogues. The smaller of these sites may be preferential for the more planar compounds such as estradiol, stilbene, or resveratrol.
2. Material and Methods
To provide more definitive details of the intermolecular interactions of these hormone analogues with the RGD recognition site in αvβ3 integrin, quantitative molecular modeling including molecular dynamics (MD) simulations and combined quantum mechanical—molecular mechanical (QM/MM) methods were employed. These calculations also incorporated the effects of solvent and divalent cations on ligand-binding interactions. In order to calculate the desolvation energies of these ligands, an explicit water environment based on molecular dynamics (MD) simulations was used in the QM/MM calculations.
In the present study we utilized our own method of combined QM/MM calculations which are based on MD. In this approach, standard MD of the protein solvated by a sphere of explicit water molecules was carried out. Then a randomly chosen set of snapshots was selected from the MD, and for each snapshot, full geometry optimization of the ligand (QM) in a fixed protein matrix (MM) was performed. In order to calculate the desolvation energy of the ligand, the same QM/MM was used to calculate the ligand surrounded by a sphere of explicit water molecules. The ligand-binding energy to the protein was calculated as a difference between the average ligand energy in the protein and the water environment. These computations were performed using the quantum-chemical program Q-Chem [33], which includes a modified version of an interface between QM and MM atoms [34]. Since the combined QM/MM calculations for each snapshot are independent of the other snapshots, this method referred to as the large-scale QM/MM method has the advantage of perfect parallel scalability [35]. All calculations were performed using a large 1.000 node Linus cluster where each node consists of two 3.2 GHz Xeon processors and 2 GB of shared memory.
3. Computational Details
Coordinates from the X-ray structure of the αvβ3 receptor (1L5G PDB) were used as a starting model [4]. The cyclic RGD pentapeptide was removed from the file and original Mn2+ ions were replaced by Mg2+ or Ca2+ ions. Parameters of the NAG residues observed in this structure were obtained from the ANTECHAMBER program of the AMBER programming suite [36]. All hydrogen atom positions were populated based on the AMBER force field. A total of 25 Na+ cations were used to neutralize the entire protein. The positions of these cations were obtained from grid calculations of the protein electrostatic potential [37, 38]. The ligand structures (Figure 2) were obtained using quantum mechanical (QM) calculations at the B3LYP/6-31+G* level of theory. The QM calculations consisted of full geometry optimization of the ligands performed in the gas phase. These calculations, including iodine atoms of the thyroid hormones (T4, T3, T4ac), were obtained using the lanl2dz effective core potential with a corresponding basis set [38].
Docking calculations were carried out using the Auto Dock 3 program [39]. Because of the lack of molecular mechanical parameters for iodine in the Auto Dock program, this atom was temporary replaced in the docking process by a carbon atom. The docking procedure was performed using a fixed internal geometry of these ligands. After docking, one snapshot of these compounds was randomly selected for subsequent study. The precise selection of a docked structure is not important because geometry and orientation of the molecule inside the protein active site were recalculated in the following MD simulations and in the QM/MM calculations. Once these ligands were docked to the protein near the RGD recognition site of αvβ3 integrin, another round of MD simulations was carried out. Parameters of the ligands were obtained using the ANTECHAMBER program in the AMBER suite.
The molecular system, including the protein, the ligand, and the metal ions, was placed into a center of TIP3P water sphere of radius 80 Å. The MD consisted of 100 ps heating dynamics from 0 to 300 K, followed by equilibration dynamics performed for 5 ns. The MD simulation was also performed at constant temperature and volume, with application of constrained harmonic potentials outside the water sphere and for the metal ions. The MD has been performed separately for each ligand (Figure 2) and separately for the protein system with Ca2+ and Mg2+ ions. Following the MD simulations, combined QM/MM calculations of the interaction between the ligand and the protein environment were carried out. First, a randomly selected protein snapshot from MD was chosen that was then divided into QM part and MM contributions. The QM part included the ligand and the MM part included the rest of the protein system with water molecules and the metal ions. Finally, full geometry optimization of the QM part inside the fixed positions of all atoms of the MM part was performed using the B3LYP/6-31+G*/AMBER method [34], as implemented in the Q-Chem program [33]. During geometry optimization, the ligands were allowed to change their internal geometry and position relative to the fixed protein matrix. The calculations of the ligands, including those with iodine atoms (T4, T3, T4ac), were performed using the lanl2dz effective core potential with a corresponding basis set.
The interaction energy between the ligand and the protein was calculated as an energy difference between the energy of the molecule inside the protein and the energy of the molecule in the gas phase which was obtained from the optimal geometry of the molecule in the protein and in the gas phase, respectively. The same procedure was then applied to 600 randomly selected protein snapshots from the last 200 ps of MD, and the results of the calculations are reported statistically based on the selected protein ensemble (Tables 1 and 2).
Table 1.
The interaction energy between the ligands (Figure 2) and the protein (or water). Numbers in parenthesis indicate the interaction energy of the ligand bound in the protein relative to the interaction energy of this molecule in water. The interaction energy is calculated as a difference between the energy of the ligand bound in the protein (or water) and the energy of this molecule in the gas phase, for the optimal geometry of this molecule bound in the protein (water) and the gas phase, respectively.
| Ligands | Interaction energy (kcal/mol) | ||
|---|---|---|---|
| Protein | Water | ||
| Mg2+ | Ca2+ | ||
| cis-Resveratrol | −49 (−6) | −63 (−19) | −44 |
| trans-Resveratrol | −59 (−14) | −65 (−20) | −45 |
| cis-Stilbene | −48 (−19) | −53 (−24) | −29 |
| trans-Stilbene | −45 (−12) | −48 (−20) | −32 |
| Estradiol | −124 (−72) | −60 (−8) | −52 |
| GC-1 | −77 (−13) | −96 (−33) | −63 |
| T3 | −109 (−60) | −84 (−35) | −49 |
| T4 | −75 (−27) | −62 (−14) | −48 |
| T4ac | −60 (−15) | −59 (−14) | −44 |
Table 2.
The induced dipole moment of the ligands (Figure 2) bound in the protein (water). Numbers in parenthesis indicate the induced dipole moment of the ligand in the protein relative to the induced dipole moment of this molecule in water. The induced dipole moment is calculated as a difference between the permanent dipole moment of the ligand in the protein (water) and the permanent dipole moment of this molecule in the gas phase, for the optimal geometry of this molecule in the protein (water) and the gas phase, respectively.
| Ligands | Induced dipole moment (debye) | ||
|---|---|---|---|
| Protein | Water | ||
| Mg2+ | Ca2+ | ||
| cis-Resveratrol | 1.4 (0.1) | 2.6 (1.3) | 1.3 |
| trans-Resveratrol | 3.4 (1.2) | 4.4 (2.3) | 2.1 |
| cis-Stilbene | 1.8 (1.0) | 1.8 (1.0) | 0.8 |
| trans-Stilbene | 2.3 (1.3) | 1.5 (0.5) | 1.0 |
| Estradiol | 6.2 (3.8) | 1.1 (−1.3) | 2.4 |
| GC-1 | 5.2 (2.3) | 3.6 (0.6) | 2.9 |
| T3 | 3.1 (0.2) | 5.5 (2.6) | 2.9 |
| T4 | 5.2 (3.8) | 1.3 (−0.2) | 1.4 |
| T4ac | 2.5 (2.5) | 1.5 (1.4) | 0.1 |
Calculations of an induced dipole moment of the ligands (Figure 2) bound to the protein and in water were also carried out (Table 2). The induced dipole moment is reported as a difference between the permanent dipole moment of the ligand in the protein (water) and the permanent dipole moment of the ligand in the gas phase, obtained from the optimal geometry of this molecule in the protein (water) and the gas phase, respectively. The calculated values of the interaction energies and induced dipole moments are reported as average values based on an ensemble of 600 protein (solvent) samples. Use of a larger number of samples in the ensemble would represent the protein (solvent) system much more realistically; however it requires more time-consuming calculations. Therefore the number of samples (snapshots) chosen in the calculations was a compromise between computational accuracy and efficiency.
4. Results and Discussion
4.1. Computational Models of Ligand Interactions
Structural data for the extracellular portion of αvβ3 integrin reveals a heterodimer with 12 domains assembled into an ovoid “head” and two “tails.” The αv and βA domains form a seven-bladed propeller and the β3 portion of the integrin “head” is composed of the βA and hybrid domains (Figure 1) [4, 5]. The RGD cyclic peptide containing the RGD sequence binds in a crevice between the propeller and the βA domains of the integrin head (Figure 1) [5]. These data reveal interactions of the RGD peptide Asp with the backbone of Tyr122 and Asn215 and the side chain atoms of Arg214 of the β3 domain of αvβ3 integrin and the Arg of RGD interacts with Asp150 and Asp218 of the αv domain (Figure 3). Also illustrated in Figure 3 is the binding orientation of T4ac (red) and estradiol (yellow) as docked in the RGD cyclic peptide binding pocket. Only small conformational changes were noted near the βA domain between the apo integrin structure and the RGD cyclic peptide complex [5]. These changes are also involved the metal (Ca2+ or Mn2+) binding site in this region.
Figure 3.
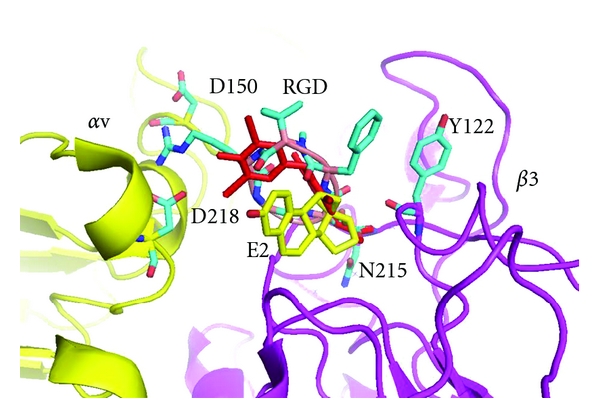
Crystal structure of αvβ3 integrin (yellow/violet) with RGD cyclic peptide (cyan) (1I5g.pdb) with T4Ac (red) and estradiol (E2, yellow) modeled into the RGD binding pocket. Residues Asp150 and Asp218 in the αv domain and Tyr122 and Asn215 in the β3 domain are labeled for orientation. Figure drawn with PyMol.
To model thyroid or steroid hormone analogue interactions near the RGD cyclic peptide binding site, molecular docking experiments using quantum chemical calculations (QM/MM) were carried out in the presence of Ca2+ or Mg2+ for each of the ligands shown in Figure 2. Furthermore, these calculations were carried out with the ligand in two different starting positions near the RGD binding site (indicated as Ca1 and Ca2) to remove modeling bias from the calculations. Similar calculations were made with the ligand starting position about 15 Å away from the RGD binding site. The results of the distant starting site calculations showed much smaller stabilization energies and suggest that the interaction between the ligand and metal cations is an important part of ligand binding (data not shown). The results of these quantitative calculations are compared with previously reported qualitative docking studies [27].
4.2. Thyroid Hormone Binding
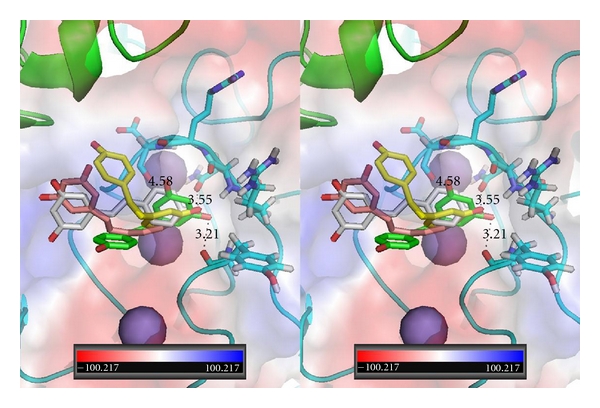
The results of the QM/MM modeling for the thyroid hormones, T4, T3, and T4ac, based on two starting orientations near the RGD binding site at the interface of the αv and β3 domains, show that the thyroid hormones can bind in a wide range of orientations. In the case of T4, the different starting positions result in binding modes in which the amino and hydroxyl ends of the molecule are flipped in the binding pocket. In one orientation, the 4′-hydroxyphenyl ring occupies a binding pocket deeper within the crevice between the αvβ3 domains, while in the other binding mode, the phenolic ring occupies the interface between the integrin αvβ3 domains (Figure 4). Consistent with pharmacokinetic/pharmacodynamic modeling reported elsewhere [26], the QM/MM modeling results for T3 differ significantly from those of T4 (Figure 5). In one mode, the tyrosyl moiety lies along the Arg side chain of the cyclic RGD peptide (Figure 6). The second binding mode for T3 places the 4′-phenolic ring in a different binding pocket than observed for T4 (Figure 5). The results for the two starting positions of T4ac reveal yet another variation in binding modes (Figure 7). In this case, one of the T4ac orientations aligns along the side chain of Arg in the RGD peptide binding site, as was observed for one of the orientations of T3 (Figure 6). Comparison of the other starting position for T3 and T4ac reveals a binding mode in which the phenolic ring of T4ac binds deeper within the RGD binding pocket, similar to that of T4. The phenolic ring of T3 occupies an alternate pocket not occupied by either T4 or T4ac (Figure 8). The one starting position calculated for the noniodinated thyroid analogue GC-1 binds in a similar manner to that of T3 (Ca2) (Figure 9).
Figure 4.
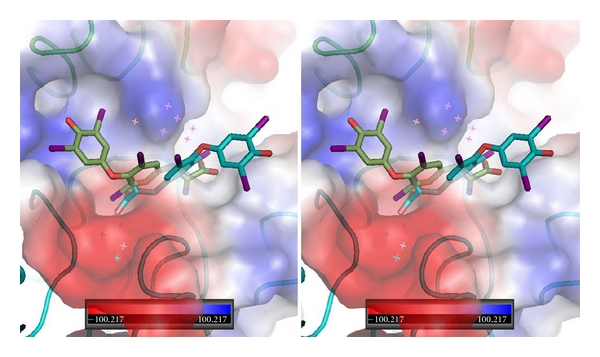
Stereoview of QM/MM model for T4 (green, Ca1) and T4 (cyan, Ca2) bound in αvβ3 integrin at the RGD binding site. Note that this energy minimum conformation has T4 bound in a “forward” and “reverse” orientation with the phenolic ring flipped in opposite directions. The electrostatic surface for integrin is shown. Red is negative and blue is positive surface. αv domain is green and β3 domain is cyan. Figure drawn with PyMol.
Figure 5.
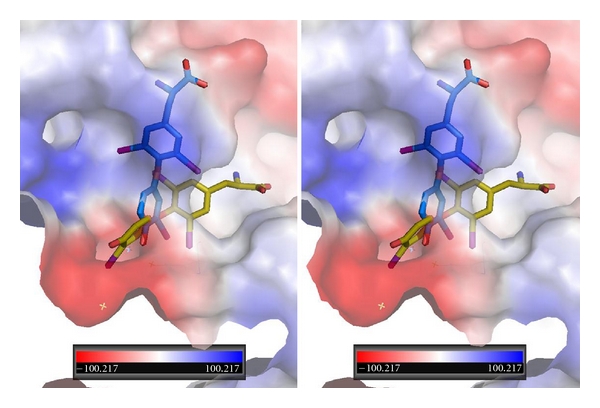
Stereoview of QM/MM model for T3 bound in αvβ3 integrin at the RGD binding site. T3 bound in two orientations: one orientation (yellow, Ca1) has the phenolic ring binding in a different pocket than that of T4 while the other orientation of T3 (blue, Ca2) binds in a mode that is perpendicular to the first.
Figure 6.
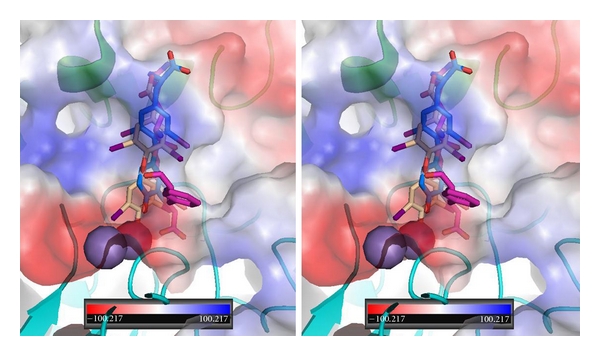
Stereoview of QM/MM model for T3 (blue-Ca2) and T4ac (beige) bound in αvβ3 integrin at the RGD peptapeptide (violet) binding site. The electrostatic surface for integrin is shown. Red is negative and blue is positive surface. αv domain is green and β3 domain is cyan. Spheres (purple/violet) are Ca2+/Mg2+. Figure drawn with PyMol.
Figure 7.
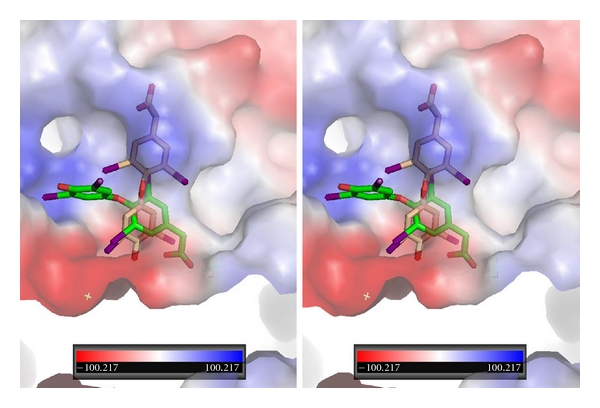
Stereoview of QM/MM model for T4ac bound in αvβ3 integrin at the RGD binding site. T4ac bound in two orientations: one orientation (green, Ca1) has the phenolic ring binding in a different pocket than that of T4 while the other orientation of T3 (beige, Ca2) binds in a mode that is perpendicular to the first.
Figure 8.
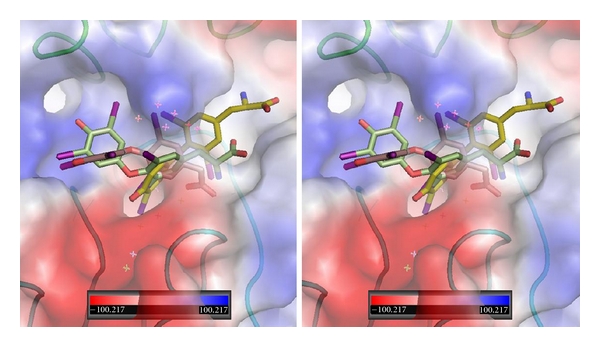
Stereoview of QM/MM model for T3 (gold, Ca2), T4ac (beige, Ca1), and T4 (green, Ca1) bound in αvβ3 integrin at the RGD binding site. The electrostatic surface for integrin is shown. Red is negative and blue is positive surface. αv domain is green and β3 domain is cyan. Figure drawn with PyMol.
Figure 9.
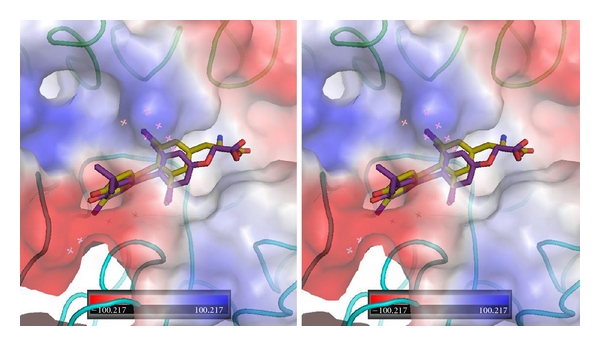
Stereoview of αvβ3 integrin RGD electrostatic surface showing GC-1 (violet) and T3 (gold, Ca2) near the RGD binding site.
The differences in binding modes possible for the thyroid hormones are in agreement with competition data that indicate that RGD peptides block hormone binding by αvβ3 integrin. These studies further show that T4ac is a nonagonist inhibitor of T4 and T3 binding to αvβ3 integrin.
4.3. Steroid-Like Ligands
The results of the QM/MM modeling of ligand interactions with cis- and trans-reservatrol based upon two starting positions show that these ligands have similar binding orientations near the RGD binding site (Figure 10). In the case of the cis-resveratrol, both starting positions placed the ligand in similar orientations such that the dihydroxyphenyl ring makes interactions with Asn215 of the β3 domain. In the case of trans-resveratrol, the two starting positions result in the molecule being flipped such that the dihydroxyphenyl ring is bound the opposite direction. The 4′-hydroxyphenyl ring still maintains contact to Asn215 of the β3 domain (Figure 10). These results are in agreement with biochemical data that indicate only the β3 monomer was essential for activation and induction of apoptosis by resveratrol and that the occlusion of the RGD binding site blocks the cellular actions of resveratrol [30].
Figure 10.
Stereoview of αvβ3 integrin RGD showing cis-resveratrol (green, Ca1), cis-resveratrol (yellow, Ca2), trans-resveratrol (beige, Ca1), and trans-resveratrol (grey, Ca2) and the residues that make contact with the RGD site. The purple spheres are Mg2+ ions.
The modeling results for the QM/MM binding modes of cis- and trans-stilbene, the nonhydroxylated parent compound of resveratrol, revealed that the energy minimum conformation for the two starting positions of cis-stilbene were perpendicular to each other with one conformer bound deeper in the binding pocket than the other cis-conformer (Figure 11). The results for the two starting positions of trans-stilbene show one molecule bound deeper in the RGD pocket than the other which is outside the binding pocket. The more hydrophobic nature of cis- and trans-stilbene compared with cis- and trans-resveratrol results in the greater variability of its binding modes as these molecules do not optimize binding involving specific hydrogen bonding interactions with residues in the RGD binding site.
Figure 11.
Stereoview of αvβ3 integrin RGD showing cis-stilbene (gold, Ca1), cis-stilbene (cyan, Ca2), trans-stilbene (green, Ca1), and trans-stilbene (purple, Ca2) and the residues that make contact with the RGD site.
The binding data for estradiol show that the 3-hydroxyl can interact with Asn215, as observed for resveratrol. An alternate orientation for estradiol binding shows the molecule perpendicular to that of the first and filling an alternate binding site (Figure 12).
Figure 12.
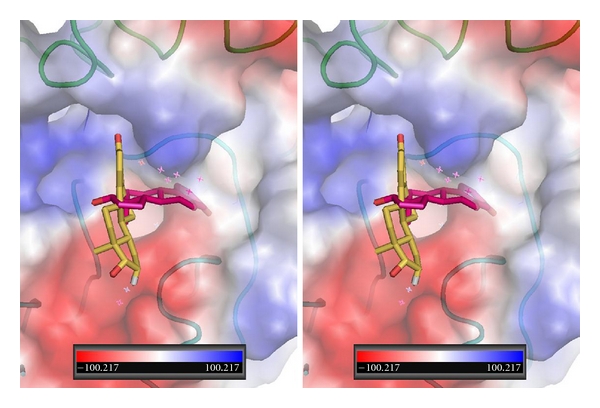
Stereoview of αvβ3 integrin RGD showing two orientations for estradiol (Ca2, (violet) and Ca2 (gold)).
4.4. Computational Models
The interaction energies between thyroid hormone analogues and the RGD recognition site of αvβ3 integrin are presented in Table 1 that also shows the interaction energies between these ligands and water. As illustrated in Table 1, all calculated interaction energies in the protein are more negative than the corresponding interaction energies of those molecules in the water environment, which indicates that these molecules are more strongly stabilized by the protein environment than by the water environment. Among these ligands, estradiol shows the strongest interaction energy with the protein containing Mg2+ ions, namely, −72 kcal/mol, while GC-1 shows the strongest interaction energy with the protein containing Ca2+, namely, −33 kcal/mol. Both values of the interaction energy include the calculated desolvation energies obtained in this study. T3 shows a relatively strong interaction with the αvβ3 integrin and the type of metal binding present; namely, the interaction energy with the protein containing the Mg2+ ions has a value of −60 kcal/mol, and the interaction energy with the protein containing the Ca2+ ions has a value of −35 kcal/mol. Similarly, both values of the interaction energy include the calculated desolvation energies obtained in this study.
It is interesting to note that the interaction energy of T3 with the protein containing both divalent metal ions is larger than the interaction energy of T4, even though both compounds have similar interaction energies with water. This observation indicates that an additional iodine atom in T4 actually decreases its interaction energy with the protein. Moreover, the interaction energy of T4 is larger than the interaction energy of T4ac indicating that the NH2-group which is present in T4 and is absent in T4ac increases the interaction energy between the ligand and protein. Generally, the protein environment with the Ca2+ ions stabilizes ligands such as resveratrol and stilbene, stronger than the protein environment with the Mg2+ ions. However, for the other ligands studied the protein environment with Mg2+ stabilizes the ligand more strongly than the protein environment with Ca2+. Resveratrol and stilbene both have less flexible geometries, where internal rotations around the single C–C bonds between phenyl rings and a C=C double bond are constrained by steric interactions of phenyl hydrogen atoms. Less flexible geometries prevent those molecules from penetrating deeper into the protein active site and keeps them constrained on the surface of the binding pocket. The other ligands show much more flexible geometries, giving greater opportunity to penetrate deeper into the protein active site. Deeper penetration of the protein active site by estradiol, GC-1, T3, T4, and T4ac, increases the interaction between the ligand and the divalent cations, particular for Mg2+, and this effect is observed in these calculations.
Table 2 shows the values of the induced dipole moment of the ligands, obtained in the protein and water environments. The largest induced dipole moment is observed for estradiol bound in the presence of Mg2+, which has a value of 3.8 debye relative to the induced dipole moment of this ligand in water. The similar increased induced dipole moment in the protein with Mg2+ is also observed in these calculations for T4.
Although it is not systematically observed for all ligands, there is a correlation between a value of the induced dipole moment of the ligand and its interaction energy with the surrounding environment that is caused by two effects. The first is related to the change of the molecular geometry by steric interactions between atoms of the ligand and atoms of the active site pocket, or atoms of a first solvation shell of water. The second effect is related to polarization of the electronic density of the molecule by an electrostatic field of the protein or water surroundings. These calculations show the second effect is more important. The change of the electronic density of the ligand inside the protein or water, observed as an induced dipole moment, generates a stronger attraction between polarized electrons of the ligand and the electrostatic field of the protein or water. As a consequence, the stronger attraction increases the total interaction energy between the ligand and the protein or water. The interaction energies show a strong stabilization effect of the bound ligand to integrin. However, these QM/MM calculations do not include polarization effects of the protein or water by the ligand that is inside these two environments. The QM/MM calculations have been performed using an approach in which all atoms of the protein or water are represented by molecular mechanical atoms, and therefore they cannot be polarized. Consequently, if polarization of the protein (or water) is included in the QM/MM calculation, the calculated interaction energy between the ligand and the surrounding environments is expected to be even larger.
These studies reveal that there are two important effects responsible for the stabilization of the small molecules inside the active site of integrin binding domain. The first effect is related to internal flexibility of the molecule which yields better penetration of the molecule into the protein active site, and consequently it increases its stabilization. The second effect is related to polarization of electronic density of the molecule, by the electrostatic field of the protein (or water). Greater molecular polarization is responsible for a larger induced dipole moment, which consequently interacts more strongly with the surrounding protein (or water) environments. Therefore, it can be finally concluded that both molecular flexibility and molecular polarizability of the ligands are the main forces responsible for strength of the binding process of the small molecules to αvβ3 integrin.
5. Conclusions
Recent data have shown that the hormone receptor on αvβ3 integrin enables the thyroid hormones T4 and T3 to stimulate cancer cell proliferation and angiogenesis and to regulate the activity of certain membrane pumps [28]. The results of these QM/MM modeling studies for a series of thyroid or steroid-like ligands (Figure 2) bound to the αvβ3 integrin reveal that these ligands have multiple modes of binding near the RGD peptide binding site that was shown to occupy a shallow crevice between the propeller and βA domains of the integrin head [4] (Figure 1). The most notable observation from these studies is that the thyroid hormone analogues probe significantly different conformational space near the RGD recognition site and that these modes can be correlated with their biologic response to signaling via the integrin. Furthermore, the binding modes for T3 and T4ac are consistent with pharmacodynamic data that indicate the presence of two discrete binding sites for T3 that control distinct downstream signal transduction pathways [28, 40]. Comparison of the low-energy binding modes for thyroid hormones suggest that the positions of T4 and T4ac illustrated in Figure 8 are consistent with data that shows T4ac blocks T4 binding to integrin. The different binding positions of T3 (Figure 5) are also consistent with data that suggest two discrete, but close, binding sites that activate different downstream signaling pathways [28].
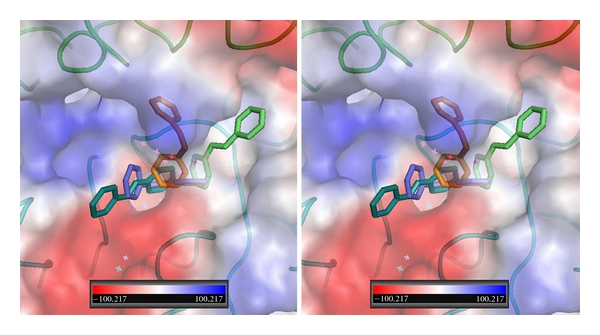
The binding of resveratrol has been shown to interact primarily with the β3 monomer of integrin, activating the ERK1/2 pathway [18, 30]. These QM/MM modeling data for cis- and trans-resveratrol indicate that both conformers have intermolecular interactions with the side chain residues of the β3 domain, although the cis-conformer which is flipped end to end with those of the trans conformers can potentially be involved in more hydrogen bond interactions (Figure 10). Furthermore, comparison of the binding of resveratrol with T4 and T4ac (Figure 13) suggests a mechanism of ligand recognition that is compatible with the observed downstream biological responses.
Figure 13.
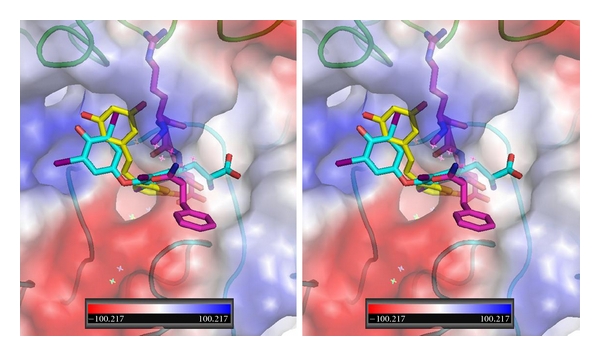
Stereoview of QM/MM model for T4 (cyan, Ca1), trans-resveratrol (yellow, Ca1), and RGD peptapeptide (violet) bound in αvβ3 integrin at the RGD binding site. The electrostatic surface for integrin is shown. Red is negative and blue is positive surface. αv domain is green and β3 domain is cyan. Figure drawn with PyMol.
The QM/MM modeling data presented here show that these ligands have flexible binding modes and that some ligands can bind in either a forward or reverse direction near the RGD recognition site. This flexibility in their low energy conformations suggests that each ligand can selectively take advantage of specific interactions within the local environment and with the metal binding sites. The observation of multiple binding pockets near the RGD recognition site also indicates that there may be subtle changes in the integrin binding site that can dictate binding to alternate sites that influence downstream signaling pathways.
Acknowledgment
The authors gratefully acknowledge support of NIH grants R44GM65617 and R44GM073408.
References
- 1.Humphries MJ. Integrin structure. Biochemical Society Transactions. 2000;28(4):311–340. [PubMed] [Google Scholar]
- 2.Baldwin ET, Sarver RW, Bryant GL, et al. Cation binding to the integrin CD11b I domain and activation model assessment. Structure. 1998;6(7):923–935. doi: 10.1016/s0969-2126(98)00093-8. [DOI] [PubMed] [Google Scholar]
- 3.Leitinger B, McDowall A, Stanley P, Hogg N. The regulation of integrin function by Ca. Biochimica et Biophysica Acta. 2000;1498(2-3):91–98. doi: 10.1016/s0167-4889(00)00086-0. [DOI] [PubMed] [Google Scholar]
- 4.Xiong J-P, Stehle T, Diefenbach B, et al. Crystal structure of the extracettular segment of integrin αvβ3. Science. 2001;294(5541):339–345. doi: 10.1126/science.1064535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xiong JP, Stehle T, Zhang R, et al. Crystal structure of the extracellular segment of integrin αvβ3 in complex with an Arg-Gly-Asp ligand. Science. 2002;296(5565):151–155. doi: 10.1126/science.1069040. [DOI] [PubMed] [Google Scholar]
- 6.Arnaout MA, Mahalingam B, Xiong JP. Integrin structure, allostery, and bidirectional signaling. Annual Review of Cell and Developmental Biology. 2005;21:381–410. doi: 10.1146/annurev.cellbio.21.090704.151217. [DOI] [PubMed] [Google Scholar]
- 7.Plow EF, Haas TA, Zhang L, Loftus J, Smith JW. Ligand binding to integrins. Journal of Biological Chemistry. 2000;275(29):21785–21788. doi: 10.1074/jbc.R000003200. [DOI] [PubMed] [Google Scholar]
- 8.Irvine DJ, Hue KA, Mayes AM, Griffith LG. Simulations of cell-surface integrin binding to nanoscale-clustered adhesion ligands. Biophysical Journal. 2002;82(1):120–132. doi: 10.1016/S0006-3495(02)75379-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spitaleri A, Mari S, Curnis F, et al. Structural basis for the interaction of isoDGR with the RGD-binding site of αvβ3 integrin. Journal of Biological Chemistry. 2008;283(28):19757–19768. doi: 10.1074/jbc.M710273200. [DOI] [PubMed] [Google Scholar]
- 10.Murcia M, Jirouskova M, Li J, Coller BS, Filizola M. Functional and computational studies of the ligand-associated metal binding site of β3 integrins. Proteins. 2008;71(4):1779–1791. doi: 10.1002/prot.21859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jin M, Andricioaei I, Springer TA. Conversion between three conformational states of integrin I domains with a C-terminal pull spring studied with molecular dynamics. Structure. 2004;12(12):2137–2147. doi: 10.1016/j.str.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 12.Gottschalk KE, Günther R, Kessler H. A three-state mechanism of integrin activation and signal transduction for integrin αvβ3. ChemBioChem. 2002;3(5):470–473. doi: 10.1002/1439-7633(20020503)3:5<470::AID-CBIC470>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 13.Gottschalk K-E, Kessler H. The structures of integrins and integrin-ligand complexes: implications for drug design and signal transduction. Angewandte Chemie—International Edition. 2002;41(20):3767–3774. doi: 10.1002/1521-3773(20021018)41:20<3767::AID-ANIE3767>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 14.Gottschalk KE, Kessler H. A computational model of transmembrane integrin clustering. Structure. 2004;12(6):1109–1116. doi: 10.1016/j.str.2004.02.039. [DOI] [PubMed] [Google Scholar]
- 15.Craig D, Gao M, Schulten K, Vogel V. Structural insights into how the MIDAS ion stabilizes integrin binding to an RGD peptide under force. Structure. 2004;12(11):2049–2058. doi: 10.1016/j.str.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 16.Bergh JJ, Lin H-Y, Lansing L, et al. Integrin αvβ3 contains a cell surface receptor site for thyroid hormone that is linked to activation of mitogen-activated protein kinase and induction of angiogenesis. Endocrinology. 2005;146(7):2864–2871. doi: 10.1210/en.2005-0102. [DOI] [PubMed] [Google Scholar]
- 17.Davis FB, Tang HY, Shih A, et al. Acting via a cell surface receptor, thyroid hormone is a growth factor for glioma cells. Cancer Research. 2006;66(14):7270–7275. doi: 10.1158/0008-5472.CAN-05-4365. [DOI] [PubMed] [Google Scholar]
- 18.Lin HY, Tang HY, Keating T, et al. Resveratrol is pro-apoptotic and thyroid hormone is anti-apoptotic in glioma cells: both actions are integrin and ERK mediated. Carcinogenesis. 2008;29(1):62–69. doi: 10.1093/carcin/bgm239. [DOI] [PubMed] [Google Scholar]
- 19.Lin HY, Tang HY, Shih A, et al. Thyroid hormone is a MAPK-dependent growth factor for thyroid cancer cells and is anti-apoptotic. Steroids. 2007;72(2):180–187. doi: 10.1016/j.steroids.2006.11.014. [DOI] [PubMed] [Google Scholar]
- 20.Mousa SA, O’Connor LJ, Bergh JJ, Davis FB, Scanlan TS, Davis PJ. The proangiogenic action of thyroid hormone analogue GC-1 is initiated at an integrin. Journal of Cardiovascular Pharmacology. 2005;46(3):356–360. doi: 10.1097/01.fjc.0000175438.94906.a0. [DOI] [PubMed] [Google Scholar]
- 21.Mousa SA, O’Connor L, Davis FB, Davis PJ. Proangiogenesis action of the thyroid hormone analog 3,5-diiodothyropropionic acid (DITPA) is initiated at the cell surface and is integrin mediated. Endocrinology. 2006;147(4):1602–1607. doi: 10.1210/en.2005-1390. [DOI] [PubMed] [Google Scholar]
- 22.Cheng SY, Leonard JL, Davis PJ. Molecular aspects of thyroid hormone actions. Endocrine Reviews. 2010;31(2):139–170. doi: 10.1210/er.2009-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davis PJ, Davis FB, Cody V. Membrane receptors mediating thyroid hormone action. Trends in Endocrinology and Metabolism. 2005;16(9):429–435. doi: 10.1016/j.tem.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 24.Moeller LC, Dumitrescu AM, Refetoff S. Cytosolic action of thyroid hormone leads to induction of hypoxia-inducible factor-1α and glycolytic genes. Molecular Endocrinology. 2005;19(12):2955–2963. doi: 10.1210/me.2004-0542. [DOI] [PubMed] [Google Scholar]
- 25.Lei J, Mariash CN, Ingbar DH. 3,3′,5-Triiodo-L-thyronine up-regulation of Na,K-ATPase activity and cell surface expression in alveolar epithelial cells is Src kinase- and phosphoinositide 3-kinase-dependent. Journal of Biological Chemistry. 2004;279(46):47589–47600. doi: 10.1074/jbc.M405497200. [DOI] [PubMed] [Google Scholar]
- 26.Lin HY, Sun M, Tang HY, et al. L-thyroxine vs. 3,5,3′-triiodo-L-thyronine and cell proliferation: activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase. American Journal of Physiology. 2009;296(5):C980–C991. doi: 10.1152/ajpcell.00305.2008. [DOI] [PubMed] [Google Scholar]
- 27.Cody V, Davis PJ, Davis FB. Molecular modeling of the thyroid hormone interactions with αvβ3 integrin. Steroids. 2007;72(2):165–170. doi: 10.1016/j.steroids.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 28.Lin HY, Cody V, Davis FB, et al. Identification and functions of the plasma membrane receptor for thyroid hormone analogues. Discovery Medicine. 2011;11(59):337–347. [PubMed] [Google Scholar]
- 29.Lin HY, Sun M, Lin C, et al. Androgen-induced human breast cancer cell proliferation is mediated by discrete mechanisms in estrogen receptor-α-positive and -negative breast cancer cells. Journal of Steroid Biochemistry and Molecular Biology. 2009;113(3–5):182–188. doi: 10.1016/j.jsbmb.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 30.Lin HY, Lansing L, Merillon JM, et al. Integrin αVβ3 contains a receptor site for resveratrol. The FASEB Journal. 2006;20(10):1742–1744. doi: 10.1096/fj.06-5743fje. [DOI] [PubMed] [Google Scholar]
- 31.Baxter JD, Webb P, Grover G, Scanlan TS. Selective activation of thyroid hormone signaling pathways by GC-1: a new approach to controlling cholesterol and body weight. Trends in Endocrinology and Metabolism. 2004;15(4):154–157. doi: 10.1016/j.tem.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 32.Chiellini G, Apriletti JW, Yoshihara HA, Baxter JD, Ribeiro RCJ, Scanlan TS. A high-affinity subtype-selective agonist ligand for the thyroid hormone receptor. Chemistry and Biology. 1998;5(6):299–306. doi: 10.1016/s1074-5521(98)90168-5. [DOI] [PubMed] [Google Scholar]
- 33.Shao Y, Molnar LF, Jung Y, et al. Advances in methods and algorithms in a modern quantum chemistry program package. Physical Chemistry Chemical Physics. 2006;8(27):3172–3191. doi: 10.1039/b517914a. [DOI] [PubMed] [Google Scholar]
- 34.Freindorf M, Shao Y, Furlani TR, Kong J. Lennard-Jones parameters for the combined QM/MM method using the B3LYP/6-31+G*/AMBER potential. Journal of Computational Chemistry. 2005;26(12):1270–1278. doi: 10.1002/jcc.20264. [DOI] [PubMed] [Google Scholar]
- 35.Freindorf M, Jones MD, Shao Y, Kong J, Furlani TR. Large-scale QM/MM calculations of electronic excitations in yellow protein: toward petascale level of protein calculations. In: 7th IEEE International Conference on Bioinformatics and Bioengineering, BIBE; January 2007; USA. pp. 614–619. [Google Scholar]
- 36.Case DA, Darden TA, Cheatham TE, III, et al. AMBER 8. University of California; 2004. [Google Scholar]
- 37.Hay PJ, Wadt WR. Ab initio effective core potentials for molecular calculations: potentials for K to Au including the outermost core orbitale. The Journal of Chemical Physics. 1985;82(1):299–310. [Google Scholar]
- 38.Wadt WR, Hay PJ. Ab initio effective core potentials for molecular calculations: potentials for main group elements Na to Bi. The Journal of Chemical Physics. 1985;82(1):284–298. [Google Scholar]
- 39.Morris GM, Goodsell DS, Halliday RS, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. Journal of Computational Chemistry. 1998;19(14):1639–1662. [Google Scholar]
- 40.Davis PJ, Davis FB, Mousa SA, Luidens MK, Lin HY. Membrane receptor for thyroid hormone: physiologic and pharmacologic implications. Annual Review of Pharmacology and Toxicology. 2011;51:99–115. doi: 10.1146/annurev-pharmtox-010510-100512. [DOI] [PubMed] [Google Scholar]