

Abstract
The emergence of biochemical homochirality was a key step in the origin of life, yet prebiotic mechanisms for chiral separation are not well constrained. Here we demonstrate a geochemically plausible scenario for chiral separation of amino acids by adsorption on mineral surfaces. Crystals of the common rock-forming mineral calcite (CaCO3), when immersed in a racemic aspartic acid solution, display significant adsorption and chiral selectivity of d- and l-enantiomers on pairs of mirror-related crystal-growth surfaces. This selective adsorption is greater on crystals with terraced surface textures, which indicates that d- and l-aspartic acid concentrate along step-like linear growth features. Thus, selective adsorption of linear arrays of d- and l-amino acids on calcite, with subsequent condensation polymerization, represents a plausible geochemical mechanism for the production of homochiral polypeptides on the prebiotic Earth.
One of life's most distinctive biochemical signatures is its strong selectivity for chiral molecular species, notably l-amino acids and d-sugars. Prebiotic synthesis reactions, with the possible exception of some interstellar processes, yield essentially equal amounts of l- and d-enantiomers (1–4). A significant challenge in origin-of-life research, therefore, is to identify natural mechanisms for the homochiral selection, concentration, and polymerization of molecules from an initially racemic mixture (5, 6). Symmetry breaking on a chirally selective mineral surface offers a viable scenario for the origin of life; according to Lahav (5), “if a selective adsorption of chiral amino acids . . . on certain crystal faces were observed, then the problem of biological homochirality would be possible to comprehend.”
Effective chiral selectivity by a crystal requires at least three noncolinear configuration-dependent points of interaction between the molecule and an acentric mineral surface (7). Most previous experimental studies of adsorption of organic molecules on mineral surfaces have focused on the symmetry-breaking effects of right- vs. left-handed quartz, which is the most common acentric mineral (8). However, recent studies, which report minimal adsorption of organic molecules by quartz in an aqueous environment, cast doubt on any significant role of quartz in prebiotic chiral selectivity (6). Centric minerals, although seldom studied for their chiral selectivity, also have the potential for symmetry breaking (5, 9, 10). Indeed, any general crystal face with a surface structure that does not possess mirror symmetry has the potential for enantioselectivity. Crystal faces that meet these conditions are displayed by most common rock-forming minerals (11, 12).
We chose calcite (CaCO3; rhombohedral space group
R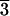 c) for adsorption experiments for
three reasons. First and foremost, calcite was one of the most abundant
marine minerals in the Archaean era. Thick limestone (calcium
carbonate) formations are common in Archaean terranes, including the
oldest known sedimentary formations (13). Sumner (14) has proposed that
before 2.5 billion years ago, massive calcite deposits “precipitated
as crystals directly on the sea floor.” Carbonate-rich formations,
furthermore, often partially dissolve and reprecipitate under mildly
acidic or hydrothermal conditions, and the [21
c) for adsorption experiments for
three reasons. First and foremost, calcite was one of the most abundant
marine minerals in the Archaean era. Thick limestone (calcium
carbonate) formations are common in Archaean terranes, including the
oldest known sedimentary formations (13). Sumner (14) has proposed that
before 2.5 billion years ago, massive calcite deposits “precipitated
as crystals directly on the sea floor.” Carbonate-rich formations,
furthermore, often partially dissolve and reprecipitate under mildly
acidic or hydrothermal conditions, and the [21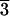 1]
trigonal scalenohedral is the most commonly observed crystal form in
vein and vug coatings. Calcite crystal surfaces would thus have been
widely present in prebiotic environments.
1]
trigonal scalenohedral is the most commonly observed crystal form in
vein and vug coatings. Calcite crystal surfaces would thus have been
widely present in prebiotic environments.
A second, more pragmatic reason for employing calcite is that it is
readily available in large (>10 cm) crystals with prominent
[21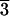 1] trigonal scalenohedral crystal forms, commonly
referred to as “dog-tooth” crystals (12). Pairs of adjacent
scalenohedral faces, for example (21
1] trigonal scalenohedral crystal forms, commonly
referred to as “dog-tooth” crystals (12). Pairs of adjacent
scalenohedral faces, for example (21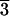 1) and
(3
1) and
(3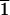
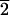 1), possess surface structures that are
related by mirror symmetry (Fig. 1) and
thus have the potential for chiral selectivity.
1), possess surface structures that are
related by mirror symmetry (Fig. 1) and
thus have the potential for chiral selectivity.
Figure 1.

(A) The common [21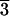 1] trigonal
scalenohedral (dog-tooth) form of calcite features adjacent crystal
faces with enantiomorphic surface structures [after Dana (12)]. The
markedly acentric surface structures of both the
(3
1] trigonal
scalenohedral (dog-tooth) form of calcite features adjacent crystal
faces with enantiomorphic surface structures [after Dana (12)]. The
markedly acentric surface structures of both the
(3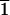
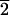 1) face (B) and the
(21
1) face (B) and the
(21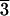 1) face (C) consist of corner-linked
chains of CaO6 octahedra, cross-linked by planar
CO3 groups, which are seen almost on edge. The
l-aspartic acid is observed to adsorb preferentially on the
(3
1) face (C) consist of corner-linked
chains of CaO6 octahedra, cross-linked by planar
CO3 groups, which are seen almost on edge. The
l-aspartic acid is observed to adsorb preferentially on the
(3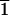
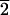 1) face, whereas d-aspartic
acid adsorbs preferentially on the (21
1) face, whereas d-aspartic
acid adsorbs preferentially on the (21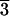 1) face.
1) face.
A third reason for using calcite in studies of chiral separation is its well documented tendency to adsorb amino acids. Biomineralized calcite is strongly bonded to proteins in the shells of many invertebrates (15, 16). Surface-growth topology of calcite, furthermore, may be affected strongly by the presence of L- vs. d-amino acids (17)—a phenomenon that underscores calcite's potential for chiral selectivity.
Experimental Methods
As a test of chirally selective adsorption, we immersed four
calcite crystals for 24 h in a 0.05 M solution of racemic aspartic
acid, which is strongly adsorbed onto calcite (14, 15). Crystal nos. 1,
3, and 4 are pale golden calcite from the Gordonville–Elmwood Mine
(Carthage, TN). Crystal no. 2 is a pale golden calcite from Joplin, MO.
Maximum crystal dimensions ranged from 11 to 18 cm, whereas surface
areas of crystal faces selected for study range from 6 to 36
cm2. Each crystal displays at least one
termination with no significant development of surfaces other than
faces of the [21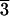 1] form, although not all faces are well
developed on all crystals. Three types of faces, R,
L, and C, were studied. The R and
L denote enantiomeric scalenohedral faces, which have the
potential for chiral selectivity (Fig. 1). The R-type faces
are equivalent to (21
1] form, although not all faces are well
developed on all crystals. Three types of faces, R,
L, and C, were studied. The R and
L denote enantiomeric scalenohedral faces, which have the
potential for chiral selectivity (Fig. 1). The R-type faces
are equivalent to (21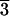 1), whereas L-type faces
are equivalent to (3
1), whereas L-type faces
are equivalent to (3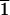
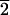 1). The C
denotes a rhombohedral cleavage face equivalent to (10
1). The C
denotes a rhombohedral cleavage face equivalent to (10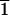 1);
these faces have a centric surface structure that should not
selectively adsorb l- or
d-amino acids, and thus can serve as an
experimental control. The initial assignment of an index to a calcite
face is somewhat arbitrary, owing to the rhombohedral symmetry. We
assign indices in the same sense as Dana (12). Note, however, that once
one face is assigned indices, the indices of all other faces are
defined completely.
1);
these faces have a centric surface structure that should not
selectively adsorb l- or
d-amino acids, and thus can serve as an
experimental control. The initial assignment of an index to a calcite
face is somewhat arbitrary, owing to the rhombohedral symmetry. We
assign indices in the same sense as Dana (12). Note, however, that once
one face is assigned indices, the indices of all other faces are
defined completely.
We cleaned each crystal successively with deionized water, methanol, methylene chloride, methanol, and deionized distilled water for two cycles. Each face was then etched for 20 seconds in 0.02 M HCl and rinsed in HPLC-grade water. After this thorough surface cleaning, we immersed each crystal in a separate 0.05 M solution of racemic aspartic acid. After 24 h, each crystal was removed from its aspartic acid solution and washed by immersion in HPLC-grade, distilled deionized water. We subsequently removed adsorbed aspartic acid from each individual crystal face by holding the face horizontally, pipetting 0.02 M HCl onto the face for 20 sec, and then collecting the acid wash in the same pipet. We repeated this procedure three times on 3 successive days for each face.
The d/l-aspartic acid values were measured by gas chromatography of N-trifluoroacetyl isopropyl ester derivatives, with a Chirasil-val 50-m × 0.25-mm i.d. column (Alltech Associates) and a nitrogen–phosphorus detector in a Hewlett–Packard 5890 series II gas chromatograph. Derivatization followed the methods of Goodfriend (18), except that the esterification step was carried out for 60 min and the fully derivatized samples were dried under a nitrogen stream rather than low vacuum. Maximum aspartic acid adsorption, estimated by comparison with GC peak areas of a concentration series of standard solutions, was ≈1 nmol/cm2. No other amino acids were detected in any of our GC analyses; therefore, contamination by environmental l-amino acids is not a significant factor in these experiments.
Results
The d/l values for three sets of
acid-wash experiments on 23 different crystal faces are recorded in
Table 1. The
d/l values for 12 acid-wash experiments on
four different (10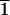 1)-type cleavage faces (denoted
C) range from 0.9889 ± 0.0051 to 1.0025 ±
0.0051, with a mean value of 0.9964—consistent with the 0.9948 ±
0.0015 d/l value observed
for the aspartic acid experimental solution.
1)-type cleavage faces (denoted
C) range from 0.9889 ± 0.0051 to 1.0025 ±
0.0051, with a mean value of 0.9964—consistent with the 0.9948 ±
0.0015 d/l value observed
for the aspartic acid experimental solution.
Table 1.
Observed d/l values for aspartic acid adsorbed on calcite crystal faces for three successive acid-wash experiments
| Crystal no. | Indices | Face type | d/l (1) | d/l (2) | d/l (3) |
|---|---|---|---|---|---|
| 1 | (2 1 3̄ 1) | R | 1.0020(25) | 1.0123(12) | 1.0365(19) |
| (3 1̄2̄ 1) | L | 0.9922(2) | 0.9978(15) | 0.9857(12) | |
| (1 3̄ 2 1) | R | 1.0053(20) | — | 1.0029(34) | |
| (1̄2̄ 3 1) | L | 0.9398(16) | — | 0.9991(34) | |
| (3̄ 2 1 1) | R | 0.9904(28) | 1.0062(45) | — | |
| (2̄ 3 1̄ 1) | L | 0.9894(10) | 0.9918(37) | 0.8960(49) | |
| 2 | (2 1 3̄ 1) | R | 1.0004(51) | 1.0042(51) | — |
| (3 1̄2̄ 1) | L | 0.9850(51) | 0.9908(51) | 0.9594(36) | |
| (1 3̄ 2 1) | R | 0.9957(51) | 0.9974(51) | 1.0113(27) | |
| (1̄2̄ 3 1) | L | 1.0036(51) | 0.9800(23) | 0.9930(51) | |
| (3̄ 2 1 1) | R | 0.9984(2) | 1.0001(51) | 0.9996(51) | |
| (2̄ 3 1̄ 1) | L | 0.9925(51) | 0.9695(23) | 0.9811(24) | |
| (0 1 1̄1̄) | C | 0.9889(51) | 1.0025(51) | 0.9972(25) | |
| 3 | (2 1 3̄ 1) | R | 1.0011(51) | 0.9986(51) | 0.9852(8) |
| (3 1̄2̄ 1) | L | — | 0.9979(51) | 0.9288(51) | |
| (1̄2̄ 3 1) | L | 0.9942(51) | 1.0028(51) | 0.9470(12) | |
| (2̄ 3 1̄ 1) | L | 1.0028(51) | 0.9993(51) | 0.9938(51) | |
| (0 1 1̄1̄) | C | 1.0024(51) | 0.9928(51) | 1.0002(51) | |
| (1 1̄ 0 1̄) | C | 0.9917(51) | 0.9988(51) | 0.9936(51) | |
| 4 | (2 1 3̄ 1) | R | 0.9955(51) | 0.9935(51) | 0.9860(29) |
| (1̄2̄ 3 1) | L | 1.0038(51) | — | 0.9952(51) | |
| (2̄ 3 1̄ 1) | L | 1.0026(51) | 1.0013(6) | 0.9675(31) | |
| (1 1̄ 0 1̄) | C | 0.9975(51) | 0.9972(51) | 0.9937(12) |
The analytical error of d/l-aspartic acid values, based on 3–6 (mean of 3.6) replicate injections of 15 samples, averaged 0.0051 or 0.52% of the d/l value. For samples analyzed only once, this value is taken as the error of the measurement. For samples analyzed two or more times, the standard error of the measurements is used as the measure of analytical error.
Two crystals (nos. 1 and 2) with finely terraced, chevron-like growth
textures on [21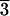 1]-type scalenohedral faces display
significant chiral selectivity (Fig. 2
and Table 1). Faces of the same handedness as (21
1]-type scalenohedral faces display
significant chiral selectivity (Fig. 2
and Table 1). Faces of the same handedness as (21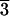 1), which
are designated R in Fig. 2 and Table 1, selectively adsorb
d-aspartic acid. The mean value of
d/l from 15 separate
acid-wash experiments (Table 1) is 1.0042, with a maximum observed
value in a single acid wash of 1.0365 ± 0.0019. Of these 15
d/l values, 14 are
greater than the 0.9948 value of the experimental solution.
1), which
are designated R in Fig. 2 and Table 1, selectively adsorb
d-aspartic acid. The mean value of
d/l from 15 separate
acid-wash experiments (Table 1) is 1.0042, with a maximum observed
value in a single acid wash of 1.0365 ± 0.0019. Of these 15
d/l values, 14 are
greater than the 0.9948 value of the experimental solution.
Figure 2.

The d/l values for aspartic acid adsorbed on
calcite crystals nos. 1 and 2, which display a finely terraced surface
structure, differ for different faces. The R-type faces
generally display d/l > 1, whereas L-type
faces display d/l < 1. Rhombohedral
(1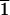 0
0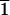 )-type cleavage faces (denoted C) show no
significant d or l selectivity. The horizontal
dashed line represents the observed 0.9948 ± 0.0015
d/l value for the aspartic acid experimental
solution, based on four separate derivatizations of a 0.05 M
solution.
)-type cleavage faces (denoted C) show no
significant d or l selectivity. The horizontal
dashed line represents the observed 0.9948 ± 0.0015
d/l value for the aspartic acid experimental
solution, based on four separate derivatizations of a 0.05 M
solution.
The opposite pattern is observed for mirror-related
(3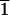
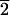 1)-type faces (designated L in
Fig. 2 and Table 1), which selectively adsorb
l-aspartic acid. The mean
d/l value for 17
acid-wash experiments range is 0.9792, with a minimum observed value of
0.8960 ± 0.0049. Of these 17
d/l values, 14 are less
than the 0.9948 value of the experimental solution.
1)-type faces (designated L in
Fig. 2 and Table 1), which selectively adsorb
l-aspartic acid. The mean
d/l value for 17
acid-wash experiments range is 0.9792, with a minimum observed value of
0.8960 ± 0.0049. Of these 17
d/l values, 14 are less
than the 0.9948 value of the experimental solution.
The maximum observed chiral excess of ≈10% is a lower limit for calcite chiral selectivity, because each acid wash may incorporate some of the 0.05 M racemic aspartic acid solution. This solution, trapped in microscopic cracks and pits on the calcite surface, cannot be removed fully by the water rinse, but it may be extracted in part during the acid-wash process. Less than a microliter of this racemic experimental solution would overwhelm the nanomolar amounts of adsorbed amino acid. Microscopically localized chiral excess of d- and l-aspartic acid may thus be many times greater than the bulk values reported here.
In contrast, adsorption of aspartic acid on the other two crystals (nos. 3 and 4 in Table 1), which possess mirror-smooth scalenohedral faces, is approximately racemic in 17 of 20 acid-wash experiments on scalenohedral faces. Three single experiments yielded l-aspartic acid excesses (d/l < 0.97) from L-type faces, but no other systematic trends are evident.
Conclusions
This study of calcite–aspartic acid interactions points to the need for additional experimental and theoretical studies. The contrast in behavior between crystals with different surface-growth textures underscores the need for these studies. Experiments with racemic mixtures of other amino acids, as well as sugars and nucleotides, will also be of importance in understanding the extent and relevance of these effects. Preliminary experiments reveal that d- and l-alanine display selective adsorption behavior similar to aspartic acid, whereas d- and l-valine and lysine may not be adsorbed selectively on calcite.
Many other mineral–molecule pairs also have the potential for chirally selective adsorption. Large natural crystals with enantiomeric faces occur commonly for the evaporite minerals gypsum (hydrous calcium sulfate) and barite (barium sulfate), as well as apatite (calcium phosphate). Significantly, chiral amino acids affect the growth morphology of gypsum crystals (19), so chiral selectivity by gypsum may be expected also. All of the major rock-forming minerals in basalt, including pyroxenes, feldspars, and olivines, also may display enantiomorphic pairs of faces (12).
These experiments have implications for origin-of-life scenarios. The Archaean Earth had substantial, although presumably dilute, inventories of racemic amino acids, both from exogenous sources (20, 21) and from prebiotic synthesis (22, 23). This study demonstrates that calcite and other minerals may have provided a mechanism for concentrating and chirally selecting these amino acids.
These results may also point to a possible mechanism for prebiotic synthesis of homochiral polypeptides. Chirally selective adsorption occurs preferentially on calcite surfaces with terraced growth features. This phenomenon suggests that local, highly selective concentration of d- and l-enantiomers may arise along step-like features, with calcite serving as a template for linear arrays of homochiral amino acids. Such a configuration may promote homochiral polymerization (24–26), which is a key step in the synthesis of self-replicating peptides (27, 28). We suggest that mineral-mediated chiral selectivity, in conjunction with homochiral polymerization, may thus provide a link between prebiotic synthesis and the RNA/protein world.
Acknowledgments
We thank L. Becker, G. Cody, J. Cronin, D. Deamer, R. Downs, W. Huntress, A. Knoll, M. McCarthy, M. Singer, J. Szostak, N. Tuross, P. Unrau, and H. Yang for useful discussions and constructive reviews of the manuscript. R. Filley, M. Grosso, and M. Horan contributed to the development of analytical procedures. This work was supported by the National Aeronautics and Space Administration Astrobiology Institute, the National Science Foundation, and the Carnegie Institution of Washington.
References
- 1.Cronin J R, Pizzarello S. Science. 1997;275:951–955. doi: 10.1126/science.275.5302.951. [DOI] [PubMed] [Google Scholar]
- 2.Pizzarello S, Cronin J R. Geochim Cosmochim Acta. 2000;64:329–338. doi: 10.1016/s0016-7037(99)00280-x. [DOI] [PubMed] [Google Scholar]
- 3.Bailey J, Chrysostomou A, Hough J H, Gledhill T M, McCall A, Clark S, Menard F, Tamura M. Science. 1998;281:672–674. [PubMed] [Google Scholar]
- 4.Mason S F. Origins Life Evol Biosphere. 2000;30:435–437. doi: 10.1023/a:1006765216225. [DOI] [PubMed] [Google Scholar]
- 5.Lahav N. Biogenesis: Theories of Life's Origin. New York: Oxford Univ. Press; 1999. p. 259. [Google Scholar]
- 6.Bonner W A. Origins Life Evol Biosphere. 1995;25:175–190. doi: 10.1007/BF01581581. [DOI] [PubMed] [Google Scholar]
- 7.Davankov V A. Chirality. 1997;9:99–102. [Google Scholar]
- 8.Soai K, Osanai S, Kadowaki K, Yonekubo S, Shibata T, Sato I. J Am Chem Soc. 1999;121:11235–11236. [Google Scholar]
- 9.McFadden C F, Cremer P S, Gellman A J. Langmuir. 1996;12:2483–2487. [Google Scholar]
- 10.Sholl D S. Langmuir. 1998;14:862–867. [Google Scholar]
- 11.Smyth J R, Bish D L. Crystal Structures and Cation Sites of the Rock-Forming Minerals. Boston: Allen & Unwin; 1988. [Google Scholar]
- 12.Dana E D. A Textbook of Mineralogy. 1958. , revised and enlarged by Ford, W. E. (Wiley, New York), 4th Ed., p. 125. [Google Scholar]
- 13.Bally A W, Palmer A R, editors. The Geology of North America: An Overview. Boulder, CO: Geol. Soc. Am.; 1989. [Google Scholar]
- 14.Sumner D W. Am J Sci. 1997;297:455–487. [Google Scholar]
- 15.Lowenstam H A, Weiner S. On Biomineralization. New York: Oxford Univ. Press; 1989. [Google Scholar]
- 16.Weiner S, Addadi L. J Mater Chem. 1997;7:689–702. [Google Scholar]
- 17.Teng H H, Dove P M, Orme C A, De Yoreo J J. Science. 1998;282:724–727. doi: 10.1126/science.282.5389.724. [DOI] [PubMed] [Google Scholar]
- 18.Goodfriend G A. Geochim Cosmochim Acta. 1991;55:293–302. [Google Scholar]
- 19.Cody A M, Cody R D. J Crystalogr Growth. 1991;113:508–519. [Google Scholar]
- 20.Chyba C, Sagan C. Nature (London) 1992;355:125–132. doi: 10.1038/355125a0. [DOI] [PubMed] [Google Scholar]
- 21.Glavin D P, Bada J L, Brinton K F, McDonald G D. Proc Natl Acad Sci USA. 1999;96:8835–8838. doi: 10.1073/pnas.96.16.8835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller S L. Science. 1953;117:528–529. doi: 10.1126/science.117.3046.528. [DOI] [PubMed] [Google Scholar]
- 23.Marshall W L. Geochim Cosmochim Acta. 1994;58:2099–2106. [Google Scholar]
- 24.Avetisov VA, Goldanskii V I, Kuz'min V V. Phys Today. 1991;44:33–41. doi: 10.1063/1.881264. [DOI] [PubMed] [Google Scholar]
- 25.Ertem G, Ferris J P. J Am Chem Soc. 1997;119:7197–7201. doi: 10.1021/ja970422h. [DOI] [PubMed] [Google Scholar]
- 26.Orgel L E. Origins Life Evol Biosphere. 1998;28:227–234. doi: 10.1023/a:1006595411403. [DOI] [PubMed] [Google Scholar]
- 27.Lee D H, Severin K, Yokobayashi Y, Ghadiri M R. Nature (London) 1997;390:591–594. doi: 10.1038/37569. [DOI] [PubMed] [Google Scholar]
- 28.Yao S, Ghosh I, Zutshi R, Chimielewski J. Nature (London) 1998;396:447–450. doi: 10.1038/24814. [DOI] [PubMed] [Google Scholar]