

Abstract
The chromosomal protein passenger complex, a key mitotic regulator, consists of at least four proteins, INCENP, Aurora B, Survivin and Borealin. Survivin, in contrast to the other members of the chromosomal protein passenger complex (CPC), is mobile at metaphase. This protein is also phosphorylated by Aurora B at Threonine 117. In this work we have studied the role of the phosphorylation of Survivin in mitotosis by using non phosphorylable T117A and phosphomimic T117E silent resistant Survivin mutants, inducible cell lines expressing these mutants and a combination of siRNA, time-lapse microscopy and FRAP analysis. Time lapse microscopy and FRAP analysis show that Survivin T117A mutant is very stably associated with centromeres and its expression induces a prometaphasic arrest in endogenous survivin depleted cells. In addition, Survivin T117A was unable to rescue the phenotypes of the endogenous survivin depleted cells. Expressed in these cells, the phosphomimic Survivin T117E mutant exhibits a very weak interaction with the centromeres and behaves as a dominant negative mutant inducing severe mitotic defects. Our data suggest that the Aurora B generated phosphorylation/dephosphorylation cycle of Survivin is required for proper proceeding of mitosis.
Keywords: Centromere; genetics; HeLa Cells; Humans; Metaphase; Microtubule-Associated Proteins; metabolism; Mitosis; Mutation; genetics; Phosphorylation; Protein-Serine-Threonine Kinases; genetics; metabolism; RNA, Small Interfering; genetics; Thymidine; genetics
Keywords: inactive X chromosome, histone variant, macroH2A distribution
Introduction
The chromosomal passenger protein complex (CPC) plays key roles in mitotic events. [1] In early mitosis, CPC promotes chromosome alignment and bi-orientation by correcting mis-attachments of microtubules to the kinetochores. [2, 3] CPC is also responsible for the phosphorylation of Histone H3 and in turn, for the displacement of HP1 (Heterochromatin protein 1) from condensed chromatin. [4, 5] Furthermore, CPC is an up-stream actor of the mitotic spindle control, orchestrating mitotic spindle assembly and cytokinesis. [6]
The Chromosomal passenger complex is composed of at least four proteins: INCENP, Survivin, Aurora B kinase and Borealin. [7] Among the members of CPC, Aurora B plays a key role, since it is the only passenger protein, which exhibits enzymatic activity. [8] INCENP, the first identified member of CPC, binds to Aurora B through its C-terminus and stimulates its kinase activity. [2] Survivin also directly interacts with Aurora B and regulates the activity of the kinase. [9]
The passenger proteins show specific localization pattern during mitosis. At metaphase they are localized at the inner centromeres and, as mitosis proceeds, they are transferred to the central spindle at anaphase and finally to the midbody at cytokinesis. [1] Depletion of any one of the passenger proteins resulted in very similar mitotic defects, including redistribution of the other members of the CPC, perturbations in mitotic progression, kinetochore/spindle misattachments and the generation of polyploid cells.[1, 2]
Survivin and Aurora B exhibit distinct dynamics during mitosis. [10] Both Survivin and Aurora B are immobile at telophase and cell cleavage, and Survivin, but not Aurora B, is highly mobile at prometaphase and metaphase. The mobility of Survivin is dependent on the presence of Aurora B, since the ablation of the kinase by siRNA treatment results in dramatic decrease of the mobility of Survivin. [10, 11] The reported data show, that Survivin, in contrast to Aurora B, is weakly associated with centromeric chromatin at prometaphase and metaphase [10]. Detailed studies on the mobility of the other passenger proteins during mitosis are not available in the literature. In addition, Survivin is phosphorylated at Threonine 117 by Aurora B. The role of this phosphorylation of Survivin is unknown.[12]
In this work we have studied the function of the Threonie 117 phosphorylation of Survivin during mitosis by expressing Threonine 117 non-phosphorylable or phosphomimic silent resistant Survivin mutants in endogenous Survivin depleted cells. Our immunofluorescence, time-lapse microscopy and FRAP data evidence that the Aurora B dependent phosphorylation/dephosphorylation cycle of Survivin is required for proper proceeding of mitosis.
Results
Survivin exhibits distinct behaviour at metaphase
We have recently reported that Aurora B and Survivin showed distinct mobility at mitosis: both proteins were found immobile on microtubules and Survivin, but not Aurora B, was found mobile at centromeres at both prometaphase and metaphase.[10] The arising question was if the two other members of the CPC, INCENP and Borealin, behaved like Survivin or like Aurora B. To address this question we have carried out FRAP studies by using HeLa cell lines expressing GFP-fusions of either INCENP or Borealin or Aurora B or Survivin. All these GFP-fusions showed the typical localization pattern of the passenger proteins during mitosis. They localized at the centromeres at metaphaphase and were transferred to the central spindle and the midbody as mitosis proceeds [10] and data not shown). In agreement with our previous data, we observed that Aurora B-GFP was immobile during all mitotic phases, while Survivin-GFP was mobile at metaphase, but not at anaphase and cytokinesis (Figure 1A, the upper two panels and [10]). Both Borealin-GFP and GFP-INCENP behaved like Aurora B-GFP, i.e. they exhibited a very restricted mobility during all phases of mitosis (Figure 1A, the two lower panels). We conclude that Survivin shows a distinct behaviour compared to the other CPC members, being the only CPC member mobile at metaphase.
Figure 1. FRAP analysis of the passenger proteins mobility during mitosis.
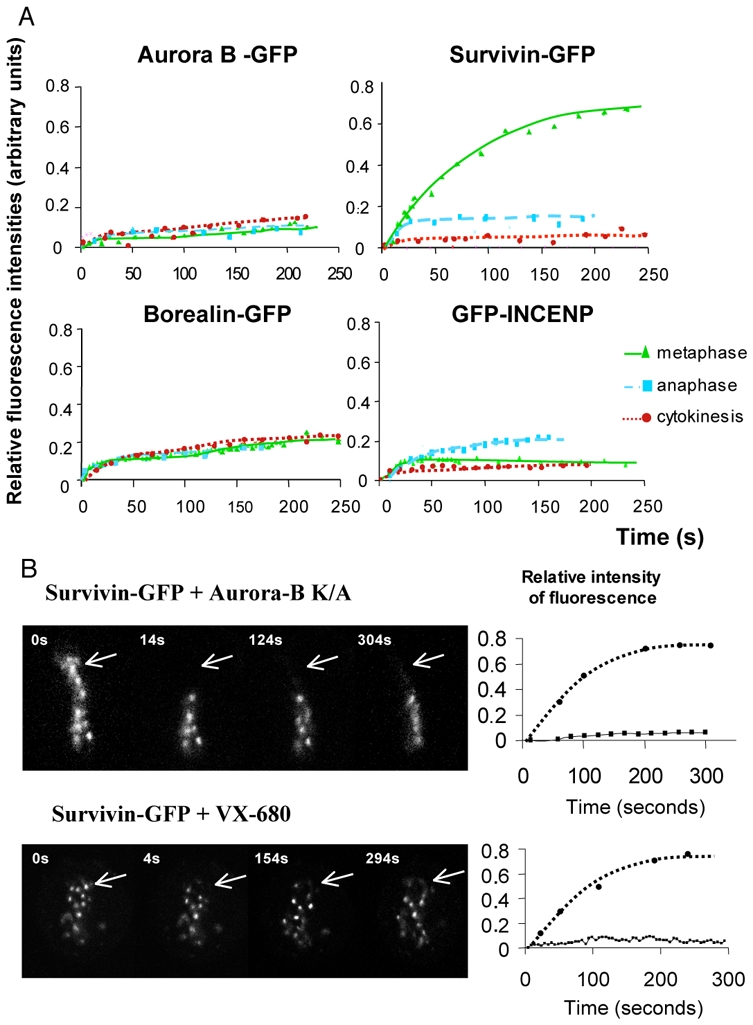
(A) HeLa cells were stably transfected with either Aurora B-GFP or Survivin–GFP or transitory transfected with either Borealin-GFP or GFP-INCENP. Either few centromeres at metaphase or a restricted area of the midzone at anaphase and cytokinesis were photobleached. The recovery of fluorescence was measured on the sequence of images acquired at the indicated intervals postbleaching. Note that Survivin, in contrast to the remaining passenger proteins, is highly mobile at metaphase.
(B) The mobility of Survivin is dependent on an active Aurora kinase.
HeLa cells expressing stably Survivin-GFP were either transfected with an inactive Aurora B kinase (Aurora B K109A) or treated overnight with the specific Aurora B kinase inhibitor, VX-680 at a 300 nM concentration. Few centromeres, indicated by an arrow, were photobleached. The kinetics of recovery of fluorescence are shown in the right part of the figure.
The presence of Aurora B was required for the higher mobility of Survivin at metaphase.[10] To further test if the mobility of Survivin depended on the enzymatic activity of Aurora B, we have either expressed the dominant negative dead Aurora B K106A mutant [13] in stable Survivin-GFP cell lines or treated these cells with VX-680, a specific inhibitor for Aurora kinases. These procedures led to abolishment of the activity of Aurora B, as judged by the lack of phosphorylation of histone H3 at mitosis ([13, 14] and data not shown). Then we have carried out FRAP experiments. The data show very clearly that in both cases Survivin-GFP became very stably associated with centromeres at metaphase (Figure 1B). Therefore, the enzymatic activity of Aurora B is essential for maintaining the higher mobility of Survivin at metaphase.
Survivin T117 mutant was unable to rescue Survivin function in cells in which Survivin has been ablated by siRNA
Aurora B phosphorylates Survivin at Threonine 117.[12] Since the enzymatic activity of Aurora B is required for the mitotic behaviour of Survivin, this suggests that the phosphorylation of Survivin by Aurora B would affect the mitotic properties of Survivin and consequently the fate of the cells. To address this we have performed a series of experiments by using a non phosphorylable Survivin T117A-GFP mutant in which threonine 117 was substituted for alanine. In order to study the behaviour of this mutant in the absence of endogenous Survivin we have taken advantage of the pseudogenetic approach described previously[10]. Briefly, the endogenous Survivin was ablated by siRNA treatment in HeLa T-rex cell lines containing stably integrated expression vector for either silent resistant SurvivinT117A-GFP mutant (SurvivinSRT117A-GFP) or silent resistant wild type Survivin (SurvivinSRWT-GFP). Both chimera were under the control of the tetracycline operator and the protein expression was induced by tetracycline (we have used HeLa T-rex cells since we expected this to allow a quick induction of the SurvivinSRT117A-GFP by tetracycline addition and the study of its behaviour in a relatively safe conditions for the cell, see below). Indeed, treatment with tetracycline resulted in a rapid induction of the expression of both proteins as judged by GFP fluorescence visualization and Western blotting (Figure 2 and data not shown).
Figure 2. Ectopically expressed Survivin T117A mutant did not restore wild type Survivin function.
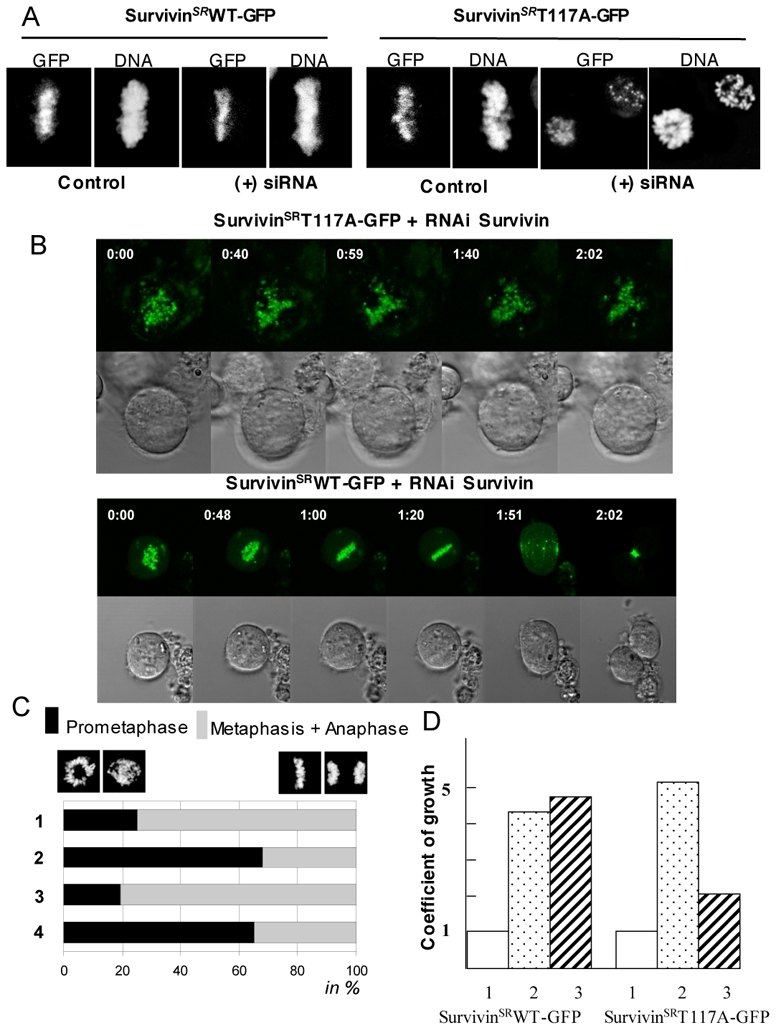
A) HeLa T-rex cells containing stably integrated tetracyclin inducible expression vectors for either silent resistant wild type Survivin (SurvivinSRWT-GFP, left panel) or silent resistant mutant Survivin (SurvivinSRT117A-GFP, right panel) were treated with Survivin siRNA for 48 hours and the expression of the exogenous Survivin proteins was induced by addition of tetracycline. 16 hours after the tetracycline addition, cells were analysed by microscopy. DNA was stained with Hoechst 33342. Note that the siRNA treated cells expressing SurvivingSRT117A-GFP were arrested at prometaphase.
B) GFP-fluorescence and transmission time-lapses imaging of endogenous Survivin depleted cells expressing either SurvivinSRT117A-GFP (top) or SurvivinSRWT-GFP (bottom). Note that the cells expressing SurvivinSRWT-GFP proceed normally in mitosis while the SurvivingSRT117A-GFP are arrested at prometaphase.
C) Quantifications of the data. 1, control HeLa T-rex cells; 2, Survivin siRNA treated HeLa T-rex cells; 3, Survivin siRNA treated HeLa T-rex cells expressing SurvivinSRWT-GFP; 4, Survivin siRNA treated HeLa T-rex cells expressing SurvivinSRT117A-GFP. The data represent the average of three independent experiments 100 mitotic cells were scored in each experiment. The percentage of prometaphase (black) and this of the sum of metaphase and anaphase (grey) cells are shown.
D) The expression of SurvivinSRT117A-GFP in endogenous Survivin-depleted cells affects cell growth. The same number of cells treated as described in (A) was seeded and the growth rate was estimated 84 hours post seeding. Punctuated rectangles (2) and striated rectangles (3) represent the coefficient of growth (the fold increase of the number of initially seeded cells) for control and Survivin siRNA treated cells, expressing either SurvivinSRWT-GFP (left) or SurvivinSRT117A-GFP (right), respectively. The initial amount of cells was set as one and presented as white rectangles (1).
Both SurvivinSRWT-GFP and SurvivingSRT117A-GFP were recruited to centromeres in the presence of endogenous Survivin (Figure 2 A, B), a result in agreement with the previously reported data.[12] The centromeric localization of both proteins was not affected by the absence of endogenous Survivin (Figure 2A, B). The suppression of the expression of the endogenous Survivin resulted, as previously reported[2], in a prometaphase arrest (Figure 2 A–C). The ectopic expression of SurvivinSRWT-GFP, but not SurvivinSRT117A-GFP, was sufficient, however, to restore the proper proceeding of mitosis (Figure 2C). In agreement with this, the growth rate of cells, in which the expression of endogenous Survivin was compromised by siRNA treatment, was rescued by the expression of SurvivinSRWT-GFP (Figure 2D). Conversely, no growth rate cell rescue was observed following SurvivingSRT117A-GFP expression (Figure 2D). Consequently, the substitution of the phosphorylable threonine 117 residue for alanine resulted in non-functional Survivin mutant, suggesting that the phosphorylation of threonine 117 by Aurora B is essential for the function of Survivin at mitosis.
Survivin T117A mutant remains stably associated with centromeres at metaphase
Since Survivin was found highly mobile on centromeres, we next asked if the T117A mutation, which creates non-phosphorylatable mutant, affects Survivin mobility. FRAP experiments were conducted on both control and Survivin siRNA treated HeLa T-rex, in which the expression of either SurvivinSRWT-GFP or SurvivingSRT117A-GFP was induced by tetracycline. The absence of endogenous Survivin did not affect the recovery of fluorescence of SurvivinSRWT-GFP (compare the upper and lower panels of Figure 3A; to note is that these recovery kinetics are more rapid than those for Survivin-GFP presented on Figure 1A; we attribute this to the higher expression of SurvivinSRWT-GFP induced by tetracycline in the HeLa T-rex cells). The picture was, however, completely different for SurvivinSRT117A-GFP. Indeed, while in the presence of endogenous Survivin, the fluorescence recovery curves for SurvivingSRT117A-GFP were very similar to those for SurvivinSRWT-GFP (compare Figure 3A with the upper panel of Figure 3B), the absence of endogenous Survivin resulted in an impressive decrease in the mobility of SurvivinSRT117A-GFP (Figure 3B, lower panel and Figure 3C). It is noteworthy that we have observed upon knock in of endogenous Survivin some variations in the mobility of SurvivinSRT117A-GFP within the individual cells (Figure 3C). We attribute these differences mainly to different efficiencies of endogenous Survivin suppression by the siRNA treatment.
Figure 3. The substitution of threonine 117 for an alanine residue results in a strong decrease of the mobility of the mutant SurvivinSRT117A-GFP at metaphase.
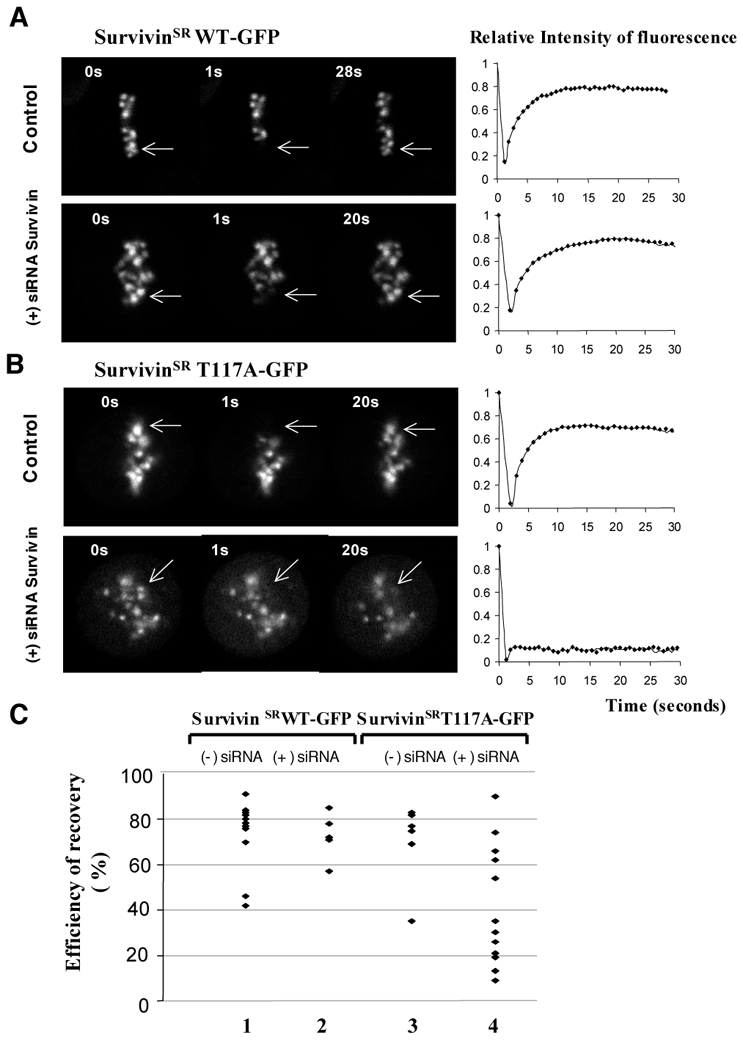
The expression of either SurvivinSRWT-GFP (A) or SurvivinSRT117A-GFP (B) was induced by addition of tetracyclin in either control or Survivin siRNA treated HeLa T-rex cells. The efficiency of expression of both mutatnts was very similar as judged by Western Blotting (data not shown). Few centromeres (indicated by an arrow) of the mitotic cells were photobleached and the recovery of fluorescence was measured at different times postbleaching. The quantification of the FRAP data is shown at the right of each panel. To note is that the kinetics of fluorescence recovery for SurvivingSRT117A-GFP in the siRNA treated cells was decreased to different extent (see panel C) and an extreme example of a quasi-total lost of mobility of SurvivinSRT117A-GFP is shown.
C) Summary of the variation of the efficiency of fluorescence recovery (the saturation level of fluorescence recovery measured at 15 seconds postbleaching) in the different FRAP experiments for SurvivinSRWT-GFP and SurvivinSRT117A-GFP in control and Survivin siRNA treated cells. 1 and 2, the efficiency of SurvivinSRWT-GFP fluorescence recovery in control and siRNA treated cells, respectively; 3 and 4, same as 1 and 2, but for the efficiency of SurvivinSRT117A-GFP fluorescence recovery. Each diamond accounts for one cell. Note that SurvivinSRT117A-GFP exhibits very low mobility in a large number of individual Survivin siRNA treated cells.
Therefore, the substitution of threonine 117 for alanine creates a mutant Survivin protein, which remains stably associated with centromeres at metaphase. This, in turn, suggests that the phosphorylation of Survivin might be essential for its increased mobility at metaphase. To test this we have used a SurvivinSRT117E-GFP mutant, which mimics the phosphorylated Survivin at threonine 117.
Localisation and behaviour of Survivin T117E mutant
In spite of several attempts, we were unable to establish HeLa T-rex cell lines containing stably integrated tetracycline inducible expression vector for SurvivinSRT117E-GFP. All experiments were conducted on transiently transfected HeLa T-rex cells with the expression vector for SurvivinSRT117E-GFP and compared to HeLa T-rex cells expressing SurvivinSRWT-GFP under the same conditions.
Initially, we have carried out experiments on paraformaldehyde fixed cells that expressed SurvivinSRT117E-GFP and we have co-detected SurvivinSRT117E-GFP and the other passenger proteins. Although Aurora B and INCENP decorated the centromeres, SurvivinSRT117E-GFP, in agreement with the previously reported data[12], was found excluded from chromatin in metaphasic cells (Figure 4A). However, when live cells were imaged, SurvivinSRT117E-GFP was found localized as endogenous Survivin on centromeres (Figure 4B, t0). The time-lapse studies performed on these cells revealed that SurvivingSRT117E-GFP was released from chromatin at the anaphase onset (Figure 4B, 9min). In late anaphase, SurvivinSRT117E-GFP was present in the whole cytoplasm (Figure 4B, 19 min) and no midbodies were decorated by SurvivinSRT117E-GFP at cell cleavage (Figure 4B, 70 minutes). We further confirmed the early mitotic chromosomal localization of SurvivinSRT117E-GFP by adding Hoechst to the culture medium. GFP and DNA were immediately imaged. SurvivinSRT117E-GFP was detected on the centromeres in all imaged metaphasic cells (Figure 4C).
Figure 4. Localisation and mobility of SurvivinSRT117E-GFP mutant.
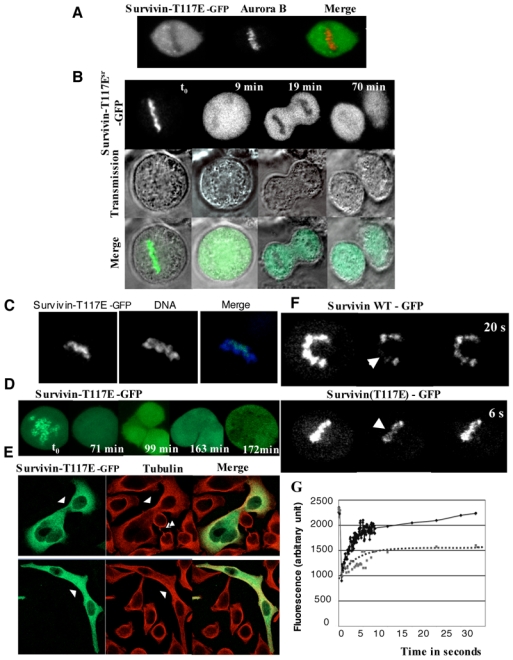
HeLa T-rex cells were transiently transfected with SurvivinSRT117E-GFP expression vector and the expression of SurvivinSRT117E-GFP was induced by tetracyclin.
A) SurvivinSRT117E-GFP loose its centromeric localization in paraformaldehyde fixed cells. HeLa T-rex cells expressing SurvivinSRT117E-GFP were fixed with parafolmaldehyde and the localization of SurvivinSRT117E-GFP was visualized by the fluorescence of its GFP. Aurora B was detected by immunofluorescence by using specific antibodies. DNA was stained with Hoechst 33242. Typical example of a metaphasic cell is shown.
B) GFP-fluorescence and transmission time lapse imaging of SurvivinSRT117E-GFP.
The merge of both signals is also presented. Note that SurvivinSRT117E-GFP is localized at the chromosomes in metaphase, but is not transferred to the central spindle and the midbody as mitosis proceeds.
C) SurvivinSRT117E-GFP is associated with mitotic chromosomes. SurvivinSRT117E-GFP was visualized by the GFP-fluorescence, while DNA was stained with Hoechst 33242. Hoechst 33242 was added for 10 min in the culture medium. Following a quick wash with culture medium, the cells were immediately imaged at 488 nm (GFP) and 720 nm (Hoechst). The merge of both signals is also shown.
D) Expression of SurvivinSRT117E-GFP results in a complex cell phenotype. A Hela T-rex cell expressing SurvivinSRT117E-GFP was continuously imaged. This time-lapse reveals the formation of a polyploidy cell within 3 hours.
E) Immunofluorescence microscopy of fixed cells expressing SurvivinSRT117E-GFP. Cells were fixed and submitted to an IF. Green, SurvivinSRT117E-GFP; red, tubulin, detected with a specific anti-tubulin antibody. Single arrowheads point abnormal connection between cells, whereas double arrowhead point a normal mid-body in non transfected cells.
F) SurvivinSRT117E-GFP is highly mobile at metaphase. HeLa T-rex cells were transiently transfected with either SurvivinSRWT-GFP or SurvivinSRT117E-GFP expression vectors and the protein expression was induced by tetracyclin. Few centromeres were bleached and fluorescence recovery was imaged.
G) FRAP quantifications. FRAP were performed as in F except that a rapid scanning of fluorescence recovery was performed by imaging a Region Of Interest (ROI) including the bleached region. Times between two scanning was 60 ms. The squares (dotted line) represented the chimera SurvivinSR-GFP and the diamonds (continuous line) the SurvivinSRT117E-GFP.
Such a discrepancy between the observations in fixed and alive cells indicated that although localized at the centromeres, SurvivinSRT117E-GFP was poorly bound and dissociated easily during cell fixation. To further show that SurvivinSRT117E-GFP was loosely associated with centromeres we have carried out FRAP experiments (Figure 4 F, G). As seen, the kinetics of fluorescence recovery of SurvivinSRT117E-GFP was much more rapid compared to SurvivinSRWT-GFP and the saturation level of the fluorescence recovery was also higher (Figures 4G). Taken collectively these data strongly suggest that the phosphorylation of Survivin at threonine 117 by Aurora B results in perturbations of its interactions with the centromeres at metaphase.
Moreover, SurvivinSRT117E-GFP behaved as a dominant negative mutant since its expression led to very severe cell phenotypes, mainly reflecting cytokinesis defects (Figure 4 D, E). Cells, expressing SurvivinSRT117E-GFP, were unable to cleave properly and polyploid cells were observed by both IF (figure 4 E) and time-lapse microscopy (figure 4 D).
Discussion
In this work we have studied the role of the phosphorylation of Survivin at threonine 117 by Aurora B. First, we showed that Survivin exhibited unique property among the remaining passenger proteins: it was loosely associated with centromeres at both promethaphase and metaphase. We further presented evidence that the enzymatic activity of Aurora B was required for the generation of this peculiar property of Survivin. To test whether the phosphorylation status of Survivin itself was important for the mobility of Survivin at mitosis, we have expressed either a non-phosphorylable silent resistant Survivin (SurvivinSRT117A-GFP) or a phosphomimic silent resistant SurvivinSRT117E-GFP mutant, in both control and Survivin siRNA treated cells. We found that the substitution of threonine for alanine at a position 117 resulted in a marked decrease in the mobility of Survivin in cells in which the expression of endogenous Survivin was suppressed by the siRNA treatment, but not in control cells. This evidences that the non-phosphorylable Survivin, in contrast to the wild type one, associated in a stable manner with the centromeres at metaphase. We attributed the lack of change in the mobility of SurvivinSRT117A-GFP observed in the presence of endogenous Survivin in control (non-siRNA) treated cells to the dimeric structure of Survivin.[15, 16] Survivin could form a “heterotypic dimer”, consisting of one wild type protein and one SurvivinSRT117A-GFP mutant, which might be sufficient to preserve the mobility of the protein at the centromeres at metaphase.
The phosphomimic SurvivinSRT117E-GFP mutant, in contrast to the non-phosphorylable mutant SurvivinSRT117A-GFP, exhibited a much higher mobility when compared to the wild type SurvivinSR-GFP in control, non siRNA treated cells. This suggests that the structure of the “heterotypic” SurvivinSRT117E-GFP-endogenous Survivin dimer would be strongly perturbed, affecting in turn its association with the centromeres at metaphase.
Interestingly, the expression of each Survivin mutant, either the non-phosphorylable or the phosphorylable one, had deleterious consequences for the cell. In the case of SurvivinSRT117A-GFP, the knock in of the endogenous protein was required for anaphase onset, whereas SurvivinSRT117E-GFP behaved as dominant negative and its expression even in the presence of endogenous Survivin led to strong mitotic defects and polyploidy.
These data taken together could shed light on the role of the phosphorylation of Survivin at mitosis. We hypothesize that endogenous Survivin is subjected to a phosphorylation/dephosphorylation cycling at Threonine 117 and this dynamic equilibrium of Survivin phosphorylation determined by Aurora B would be required for its function at mitosis. The CPC complex is a “sensor” for the proper attachment of the microtubules to the kinetochores and it is implicated in the corrections of the improper microtubules-kinetochores attachment. The reported data suggest that the suppression of the enzymatic activity of Aurora B, which appeared to happen at improper attached kinetochores and thus, in the absence of tension, is associated with the degradation of the K-mitotic microtubule formed fibers and the subsequent correction of the attachment. In addition, it is well established that the presence of Survivin is essential for a functional CPC complex.[17] Our data suggest that Survivin phosphorylation would release it from the centromeres and the remaining incomplete CPC complex would be no longer functional. A signal for Survivin phosphorylation could be associated with the lack of tension at improper attached kinetochores. The generation of non-functional CPC complex (resulting from the release of the phosphorylated Survivin) and in particular, from a non-functional Aurora B, would lead to corrections of the attachment according to the above-described scenario. Once the phosphorylated Survivin is released from the kinetochores, it would be dephosphorylated and then it would be able to associate again with the centromeres and to fulfill its function. This model is in good agreement with our data and explains why the presence of only the non-phosphorylable SurvivinSRT117A-GFP or the phosphomimic SurvivinSRT117E-GFP affects severely the fate of the cells.
Material and Methods
SiRNA experiments and reagents
Double stranded RNAi for Survivin suppressions were purchased from Eurogentec. They recognized the AAAGAACUGGCCCUUCUUGGA sequence. Single stranded sense and antisense strands, used as controls and siRNA duplexes were transfected into cells using oligofectamine (Invitrogen) according to the manufacturer’s protocols.
VX-680 was purchased from Kava technology, Inc, Blasticidin from Invitrogen and Nocodazole from Sigma.
Cell culture, transfection, immunofluorescence microscopy
Hela T-Rex™ cells were grown on Dulbecco’s modified Eagle’s (BioWhittaker, Europe) supplemented with 10 % foetal bovine serum (BioWhittaker, Europe) and blasticidin (5 mg/mL). Hela were grown under similar conditions but blasticidin was omitted.
Survivin silent resistant cDNA and Hela cells stably expressing Survivin-GFP or Aurora B-GFP were already described and characterised in Delacour et al. [10] Borealin was amplified from testis Marathon library (Clontech) and then cloned in pEGFP-N1 (Clontech) in order to express the fusion Borealin-GFP. GFP-INCENP was a gift from Takeshi Urano (Japan). Mutations were introduced in Survivin and Aurora B by using the QuickChange Site-Directed Mutagenesis kit (Quiagen) under conditions suggested by the manufacturer. SurvivinSR-GFP, SurvivinSR-T117A-GFP and SurvivinSR-T117E-GFP were cloned in pcDNA5/TO plasmid (invitrogen). Their expression was under the control of the tetracycline promoter, the induction was performed by the addition of tetracyclin (250 ng/ml), overnight. All plasmids were transfected into cells using lipofectamin (Invitrogen) under conditions suggested by the manufacturer. 48H after transfection cells were transferred in selecting medium (either geneticin (750 mg/ml) or hygromycin (200 mg/ml)).
For immunofluorescence experiments, cells grown on glass coverslips for 24 hours were fixed, for 15 min, at 37°C, in a solution of PBS, 4% paraformaldehyde and 2% sucrose. Cells were permeabilised in PBS containing 0.2% Triton X-100 for 10 minutes. Free binding sites were blocked with 0.5 mg/ml BSA and specific antibodies were then incubated for at least 30 min in PBS supplemented with 10 % bovine serum, 0.2 % Tween-20 and 0.02 % NaN3. Aurora B was detected by mouse monoclonal AIM- (1/100, transduction Laboratories). Unbound antibodies were removed by washing with PBS, 0.2 % Tween-20 and specific staining was revealed with Hylite Fluor™ 546-conjugated secondary antibodies (Euromedex, France). DNA was visualised with 0.1 mM Hoechst 33342 (Sigma). Images were collected with a Zeiss 510 laser scanning confocal apparatus with a 63X oil immersion objective.
Ex vivo microscopy
Ex vivo experiments were conducted on cells grown on Lab-Tek chambered coverglass (Nalge Nunc International and maintained under standard culture conditions. RNAi were transfected since 48 hours. Images were acquired on a Zeiss LSM510 system using a Planapochromat 40 X water immersion objective. GFP was excited with a 488 nm Argon 2 laser (power varying from 0.1 to 2%). FRAP (“Fluorescent Recovery After Photobleaching”) were conducted as already described in[10]. Briefly, an outlined region was bleached with a full power laser and recovery was monitored repetitively during approximately 4 minutes. For rapid scanning, the registered region was restricted to a circle ROI of 40 mm of diameter.
Abreviations used
- CPC
Chromosomal Passenger protein Complex
- IF
immuno fluorescence
References
- 1.Adams RR, Carmena M, Earnshaw WC. Chromosomal passengers and the (aurora) ABCs of mitosis. Trends Cell Biol. 2001;11:49–54. doi: 10.1016/s0962-8924(00)01880-8. [DOI] [PubMed] [Google Scholar]
- 2.Honda R, Korner R, Nigg EA. Exploring the functional interactions between Aurora B, INCENP, and survivin in mitosis. Mol Biol Cell. 2003;14:3325–3341. doi: 10.1091/mbc.E02-11-0769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andrews PD, Ovechkina Y, Morrice N, Wagenbach M, Duncan K, Wordeman L, Swedlow JR. Aurora B regulates MCAK at the mitotic centromere. Dev Cell. 2004;6:253–268. doi: 10.1016/s1534-5807(04)00025-5. [DOI] [PubMed] [Google Scholar]
- 4.Hirota T, Lipp JJ, Toh BH, Peters JM. Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature. 2005;438:1176–1180. doi: 10.1038/nature04254. [DOI] [PubMed] [Google Scholar]
- 5.Nowak SJ, Corces VG. Phosphorylation of histone H3: a balancing act between chromosome condensation and transcriptional activation. Trends Genet. 2004;20:214–220. doi: 10.1016/j.tig.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 6.Lens SM, Medema RH. The survivin/Aurora B complex: its role in coordinating tension and attachment. Cell Cycle. 2003;2:507–510. doi: 10.4161/cc.2.6.559. [DOI] [PubMed] [Google Scholar]
- 7.Vader G, Medema RH, Lens SM. The chromosomal passenger complex: guiding Aurora-B through mitosis. J Cell Biol. 2006;173:833–837. doi: 10.1083/jcb.200604032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andrews PD, Knatko E, Moore WJ, Swedlow JR. Mitotic mechanics: the auroras come into view. Curr Opin Cell Biol. 2003;15:672–683. doi: 10.1016/j.ceb.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 9.Carvalho A, Carmena M, Sambade C, Earnshaw WC, Wheatley SP. Survivin is required for stable checkpoint activation in taxol-treated HeLa cells. J Cell Sci. 2003;116:2987–2998. doi: 10.1242/jcs.00612. [DOI] [PubMed] [Google Scholar]
- 10.Delacour-Larose M, Molla A, Skoufias DA, Margolis RL, Dimitrov S. Distinct dynamics of Aurora B and Survivin during mitosis. Cell Cycle. 2004;3:1418–1426. doi: 10.4161/cc.3.11.1203. [DOI] [PubMed] [Google Scholar]
- 11.Beardmore VA, Ahonen LJ, Gorbsky GJ, Kallio MJ. Survivin dynamics increases at centromeres during G2/M phase transition and is regulated by microtubule-attachment and Aurora B kinase activity. J Cell Sci. 2004;117:4033–4042. doi: 10.1242/jcs.01242. [DOI] [PubMed] [Google Scholar]
- 12.Wheatley SP, Henzing AJ, Dodson H, Khaled W, Earnshaw WC. Aurora-B phosphorylation in vitro identifies a residue of survivin that is essential for its localization and binding to inner centromere protein (INCENP) in vivo. J Biol Chem. 2004;279:5655–5660. doi: 10.1074/jbc.M311299200. [DOI] [PubMed] [Google Scholar]
- 13.Scrittori L, Skoufias DA, Hans F, Gerson V, Sassone-Corsi P, Dimitrov S, Margolis RL. A small C-terminal sequence of Aurora B is responsible for localization and function. Mol Biol Cell. 2005;16:292–305. doi: 10.1091/mbc.E04-06-0447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harrington EA, Bebbington D, Moore J, Rasmussen RK, Ajose-Adeogun AO, Nakayama T, Graham JA, Demur C, Hercend T, Diu-Hercend A, Su M, Golec JM, Miller KM. VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat Med. 2004;10:262–267. doi: 10.1038/nm1003. [DOI] [PubMed] [Google Scholar]
- 15.Chantalat L, Skoufias DA, Kleman JP, Jung B, Dideberg O, Margolis RL. Crystal structure of human survivin reveals a bow tie-shaped dimer with two unusual alpha-helical extensions. Mol Cell. 2000;6:183–189. [PubMed] [Google Scholar]
- 16.Muchmore SW, Chen J, Jakob C, Zakula D, Matayoshi ED, Wu W, Zhang H, Li F, Ng SC, Altieri DC. Crystal structure and mutagenic analysis of the inhibitor-of-apoptosis protein survivin. Mol Cell. 2000;6:173–182. [PubMed] [Google Scholar]
- 17.Lens SM, Vader G, Medema RH. The case for Survivin as mitotic regulator. Curr Opin Cell Biol. 2006;18:616–622. doi: 10.1016/j.ceb.2006.08.016. [DOI] [PubMed] [Google Scholar]