

Abstract
Soluble oligomers of the amyloid-β (Aβ) peptide are thought to play a key role in the pathophysiology of Alzheimer's disease (AD). Recently, we reported that synthetic Aβ oligomers bind to cellular prion protein (PrPC) and that this interaction is required for suppression of synaptic plasticity in hippocampal slices by oligomeric Aβ peptide. We hypothesized that PrPC is essential for the ability of brain-derived Aβ to suppress cognitive function. Here, we crossed familial AD transgenes encoding APPswe and PSen1ΔE9 into Prnp−/− mice to examine the necessity of PrPC for AD-related phenotypes. Neither APP expression nor Aβ level is altered by PrPC absence in this transgenic AD model, and astrogliosis is unchanged. However, deletion of PrPC expression rescues 5-HT axonal degeneration, loss of synaptic markers, and early death in APPswe/PSen1ΔE9 transgenic mice. The AD transgenic mice with intact PrPC expression exhibit deficits in spatial learning and memory. Mice lacking PrPC, but containing Aβ plaque derived from APPswe/PSen1ΔE9 transgenes, show no detectable impairment of spatial learning and memory. Thus, deletion of PrPC expression dissociates Aβ accumulation from behavioral impairment in these AD mice, with the cognitive deficits selectively requiring PrPC.
Introduction
In Alzheimer's disease (AD), the amyloid-β peptide accumulates in plaques while synapses are lost and the brain becomes dysfunctional. Soluble Aβ oligomer species have been implicated in the synaptic loss and the dementia of AD (Hsia et al., 1999; Mucke et al., 2000b). Aβ oligomers with deleterious effects on synaptic function have been prepared from synthetic peptide, from cell culture supernatants, and from brain extracts (Lambert et al., 1998; Walsh et al., 2002; Cleary et al., 2005; Lesné et al., 2006; Shankar et al., 2008). We searched for high-affinity oligomeric-specific Aβ-binding sites and identified cellular prion protein, PrPC (Laurén et al., 2009).
Our previous demonstration of Aβ oligomer binding to PrPC used synthetic peptide (Laurén et al., 2009), but did not explore whether the action of brain-derived Aβ species interact with PrPC. Earlier work also demonstrated a requirement of PrPC for acute Aβ oligomer suppression of synaptic plasticity in hippocampal slices (Laurén et al., 2009), leaving open the possibility that Aβ oligomer action to disrupt memory uses different or multiple pathways. A recent paper confirmed that PrPC is a high-affinity binding site for Aβ oligomers, but suggested that memory impairment by acute Aβ injection does not require PrPC (Balducci et al., 2010).
To extend analysis of PrPC function in AD models, we examined spatial learning and memory in transgenic APPswe/PSen1ΔE9 mice (Jankowsky et al., 2004) with and without PrPC (Manson et al., 1994). Since the manifestation of several other neurodegenerative conditions in mice is not altered by loss of PrPC expression (Steele et al., 2009), this paradigm assesses the role of PrPC in pathways specific to the APPswe/PSen1ΔE9 condition. Our hypothesis predicts that PrPC functions downstream of Aβ production but upstream of intracellular toxicity within neurons, and for this reason, we did not examine transgenic Tau-induced mouse phenotypes.
Here, we report that AD transgenic mice lacking PrPC accumulate Aβ, but show normal survival and no loss of spatial learning and memory.
Materials and Methods
Mice.
The APPswe/PSen1ΔE9-coinjected transgenic mice (Jankowsky et al., 2004) were obtained from The Jackson Laboratory, and Prnp−/− mice (Edinburgh strain) (Manson et al., 1994) were obtained from Dr. Chesebro of the Rocky Mountain Laboratories. Both strains were extensively backcrossed (>10 generations) onto the C57BL/6J background and were maintained on this strain background.
Brain tissue collection.
Mice were killed and immediately perfused with ice-cold 0.9% NaCl for 2 min. The brains were then dissected out and placed in ice-cold 0.9% NaCl. For biochemical analysis, the right posterior part of the brain extending to the hippocampus was weighed and immediately frozen in −80°. To extract the soluble/cytosolic fraction, the brains were homogenized in three volumes (w/v) of 50 mm Tris-HCl, 150 mm NaCl, pH 7.6 (TBS) containing a protease inhibitor cocktail (Roche Complete #10745000), 1 mm sodium orthovanadate, and 50 mm sodium fluoride. Tissue was homogenized using an ultrasonic cell disruptor (Ultrasonic Power), and ultracentrifuged at 100,000 × g for 20 min at 4°. The supernatant was mixed with 4× SDS-PAGE loading buffer, boiled for 5 min, and stored for subsequent analysis. The pellet was then resuspended to the same volume as the original homogenate in TBS with 2% Triton X-100 (American Bioanalytical), 0.1% SDS (American Bioanalytical), a protease inhibitor cocktail (Roche Complete #10745000), 1 mm sodium orthovanadate, and 50 mm sodium fluoride. Tissue was homogenized and ultracentrifuged at 100,000 × g for 20 min. The supernatant was mixed with 4× SDS-PAGE loading buffer, boiled for 5 min, and stored for subsequent analysis. The remaining pellet was then resuspended to the same volume in 0.1 m formic acid, homogenized, and ultracentrifuged, and the supernatant was frozen after neutralizing pH with 50 μl of 1 m Trizma-base. The remaining pellet was then resuspended to the same volume in 6N guanidine-HCl (American Bioanalytical), homogenized, and ultracentrifuged, and the supernatant was frozen for subsequent analysis.
Immunohistochemistry.
One hemisection containing the frontal cortex was immersed in fresh 4% paraformaldehyde (PFA) overnight. After the brains were fixed, they were embedded in 10% gelatin and placed in 4% PFA for 20 h at 4°C. Parasagittal sections (30 μm) were then cut using a Leica WT1000S vibratome. For immunohistochemistry, sections were blocked in 10% donkey serum for 1 h, followed by incubation with primary antibody overnight at 23°. Primary antibodies were diluted in PBS with 0.2% Triton X-100 (American Bioanalytical). The following antibodies were used: β-amyloid antibody (Cell Signaling Technology #2454, and Millipore 6E10 MAB 1560, both 1:250), 5-HT (Immunostar #20080, 1:16,000), PSD-95 (Invitrogen #51-6900, 1:250), GFAP (Sigma #C9205, 1:2500), and synaptophysin (Millipore MAB5258, 1:1000). Following incubation, the sections were washed three times with PBS, and incubated in secondary fluorescent antibody (Invitrogen Alexa Fluor donkey anti-rabbit or anti-mouse, all 1:500) for 2 h at room temperature (RT). The slices were then washed three times and transferred to PBS. Each free-floating section was mounted on a microscope slide (Fisherbrand Superfrost/Plus) and coverslipped using fluorescent mounting medium (Vector Laboratories #H-1000).
Imaging and analysis.
All images and analyses were generated by personnel without knowledge of the mouse genotype. β-Amyloid images were obtained using a Zeiss AxioImager Z1 fluorescent microscope with a 10× objective. Mosaic images of the entire frontal cortex from each animal were obtained and analyzed, and plaque burden was calculated using ImageJ. The fractional area occupied by GFAP-positive astrocytes from the frontal cortex was measured by computer-assisted analysis of images obtained at 20× objective magnification.
Z-stack images of 5-HT-labeled slices (30 μm stack of 15 images at 2 μm spacing) were obtained using an UltraView VoX spinning disc confocal microscope (PerkinElmer). Optical stacks from two sites in the frontal cortex were obtained using a 20× objective, and images were projected using Adobe Photoshop. All images were in the sagittal plane and were carefully matched for location in the rostral–caudal (1 mm posterior to bregma) and medial–lateral (1 mm lateral from midline) directions. All individual serotonin fibers within each image were traced and the total unit length of axon per area in the sagittal plane is reported, as described previously in our spinal cord studies (GrandPré et al., 2002; Kim et al., 2004; Li et al., 2004, 2005; Wang et al., 2006, 2009; Duffy et al., 2009), and in studies of cerebral cortex 5-HT innervation (Mamounas et al., 2000; Austin et al., 2002; Grider et al., 2006).
Using the UltraView VoX spinning disc confocal microscope (PerkinElmer), synaptophysin and PSD-95-immunoreactive puncta were imaged with a 100× objective in sections of the frontal lobe of cerebral cortex. Four to eight parasagittal images were obtained from two slices from each of 9–10 mice per genotype. Thus, the measurements for each genotype derived from 60 images with 100–200 synaptic puncta per image, or ∼10,000 synaptic puncta per genotype. The fractional area occupied by immunoreactive puncta from frontal cortex was measured as described (Masliah et al., 1992; Buttini et al., 1999; Mucke et al., 2000a; Chin et al., 2004; Buttini et al., 2005; Linhoff et al., 2009) using ImageJ, excluding cell somata.
Immunoblots.
Precast 10% tris-glycine or 10–20% tris-tricine gels were used (Bio-Rad). After transfer, the PVDF membranes (Bio-Rad #162-0174) were incubated in blocking buffer for 1 h at RT (Licor Odyssey blocking buffer #927-4000). Membranes were then washed five times in TBST, and incubated overnight in primary antibodies. The following antibodies were used: 6E10 (Millipore MAB 1560 1:1000), 22C11 (Millipore MAB 348, 1:100), 6D11 (Covance 39810-500, 1:500), and actin (Santa Cruz Biotechnology #SC1616, 1:200). All antibodies were diluted in Odyssey blocking buffer, and membranes were incubated overnight at 4°. Following primary antibody incubation, the membranes were washed five times with TBST, and secondary antibodies were applied for 1 h at 23° (Odyssey donkey anti-mouse or anti-goat IRDye 680 or 800). Membranes were then washed and proteins visualized using a Licor Odyssey Infrared imaging system. Blots were analyzed using ImageJ, and normalized to actin optical density.
Behavioral studies.
Animals were randomized and the experimenter blinded to genotype for the duration of behavioral testing.
Morris Water Maze testing (Vorhees and Williams, 2006) was performed over the course of 6 d and consisted of a 3 d learning trial and a 3 d reversal trial. Both trials were performed in an open-water pool ∼1 m in diameter and used a submerged, nonvisible escape platform located in the center of one of the pool's four quadrants. This location remained constant for each 3 d trial; for the reversal trial, the platform was placed, for the duration of the trial, in the quadrant diagonally across from its original location in the learning trial. Over the course of each testing day, an animal swam a total of eight times—four times in the morning, constituting one “block” of swims, and four times in an afternoon block. Over the course of a block, each mouse would begin its swim in each of four distinct locations around the wall of the pool, and was timed for its latency to reach the escape platform for a maximum time of 1 min. If the mouse did not find the submerged platform within 1 min, it was removed from the water and placed on the platform for ∼10 s before being taken from the pool.
The water maze probe trial was performed 2 d following the third and last day of the reversal trial, and in the same 1 m pool described above. For the purposes of the probe trial, the platform was removed from the pool; all mice were started from a location along the pool wall diagonally opposed to the location of the platform in the reversal trial and permitted to swim for 1 min. The probe trials were recorded on a JVC Everio G-series camcorder and analyzed using Panlab's Smart tracking and analysis program, v2.5.
A block of five swims to a visible platform was conducted after the probe trial.
Passive avoidance testing (King et al., 2003) was performed over the course of 3 d after water maze testing was complete. On the first day of testing, each subject was permitted 5 min to explore the passive avoidance apparatus, which consisted of two chambers, one well lit and one dark, connected by a door and sharing a metal grid floor through which current might be passed. The door between the light and dark boxes remained open for the duration of the 5 min, and no shock was administered. The second day of testing consisted of a single “shock trial.” Each animal was placed in the light box to begin and, upon entry into the dark box, the dividing door closed, and the mouse was subjected to a shock of 0.5 mA, duration 2 s; time to entry was recorded. On the third day of testing, each animal was placed in the light box to start, and its time to enter the dark box was recorded. No shock was administered. The following were constant settings throughout the 3 d experiment: door delay = 1 s, cutoff time = 5 min. Passive avoidance testing was performed using Ugo Basile's 7550 line passive avoidance set-up (step-through method) with model 7553 (mouse-specific apparatus).
Statistical analysis.
Repeated-measures two-way and one-way ANOVA tests were prepared using SPSS software.
Results
Mice with one of four genotypes were generated on an inbred C57BL/6J background, with and without the APPswe/PSen1ΔE9 transgene on the wild-type or Prnp−/− background. At 12 months of age, these mice were killed and the brain tissue was examined histochemically and biochemically. Since the reported interactions of Aβ and PrPC are posttranslational, the overall expression of APP and PrPC is not predicted to be altered by the status of the other gene. Indeed, levels of sAPP (22C11) and sAPPα (6E10) in the transgene-positive animals are indistinguishable between the wild-type and Prnp−/− backgrounds (Fig. 1a,b,g). Similarly, PrPC levels are not different in brain extracts of mice with and without the APPswe/PSen1ΔE9 transgene (Fig. 1f).
Figure 1.

APP and Aβ levels are not dependent on PrPC. a–f, Immunoblot analysis of soluble brain extracts (a–c, f) or detergent-extracted particulate fractions (d) or guanidinium-solubilized material (e) with anti-Aβ 6E10 antibody (a, c–e), anti-APP 22C11 antibody (b), or anti-PrPC 6D11 antibody (f). Genotype is indicated at the top of each blot for separate mice. The migration of MW standards is shown at the left. g, Densitometric analysis of immunoblot signals from experiments as in a–f. Data are mean ± SEM for n = 5–12 mice per genotype. h, i, Immunofluorescent detection of Aβ in a parasagittal section of the anterior half of a APPswe/PSen1ΔE9 transgenic mouse brain at 12 months of age of the Prnp+/+ (h) or Prnp−/− (i) genotype.
The processing of APP produces membrane-associated C-terminal fragments and soluble Aβ species. Immunoblot of detergent extracts of the particulate phase reveals no effect of Prnp genotype on β-CTF detected with 6E10 antibody (Fig. 1d,g). Soluble monomeric Aβ levels in brain homogenates prepared under non-denaturing conditions show no correlation with PrPC level (Fig. 1c,g). Thus, APP metabolism is independent of PrPC.
A role for PrPC in mediating Aβ oligomer synaptotoxicity is downstream of Aβ production and is not required for Aβ fibrillization, so Aβ plaque load is not predicted to be altered by the status of PrPC expression. The level of Aβ extracted from APPswe/PSen1ΔE9 brain homogenates by formic acid or guanidinium-HCl from the particulate phase is not altered by status of the Prnp gene (Fig. 1e,g). Similarly, the burden of Aβ-immunoreactive plaque is altered minimally in APPswe/PSen1ΔE9 transgenic brain without PrPC as compared to that with PrPC (Fig. 1h,i), 8.3 ± 0.5 versus 10.4 ± 0.7 as a percentage of brain area. Minimal effects of PrPC on Aβ level are generally consistent with previous observations of a minor increase in Aβ plaque in PrPC-overexpressing APPswe transgenic mice (Schwarze-Eicker et al., 2005) and a slight increase of mouse Aβ levels in Prnp−/− mice (Parkin et al., 2007).
While the synaptic action of Aβ oligomer is hypothesized to be mediated by PrPC, the non-neuronal reaction to Aβ plaque deposition may occur through alternate cellular receptors. We assessed astrogliosis in the APPswe/PSen1ΔE9 brains histologically (Fig. 2a). Familial AD transgene expression increased GFAP-positive area, but deletion of PrPC expression did not alter the astrocyte reaction. Thus, APP expression, Aβ level, and gliosis are not altered by deletion of PrPC.
Figure 2.

Serotonin axons and synaptic markers are preserved in APPswe/PSen1ΔE9 mice lacking PrPC. a, Anti-GFAP immunohistology of parasagittal brain sections was scored for astrocytic area in the anterior half of the cerebral cortex. Data are mean ± SEM for n = 10 mice per genotype. b, Parasagittal sections of cerebral cortex from mice of the indicated genotypes were stained with anti-5-HT. Projections of 30 μm confocal Z-stacks are shown. Note the reduced fiber length in the APPswe/PSen1ΔE9 sample. Scale bar, 50 μm. c, The 5-HT-immunoreactive fiber length in the parasagittal plane from images as in b was measured. Data are mean ± SEM for n = 10–16 mice per genotype. *p < 0.05, one-way ANOVA with post hoc comparisons as indicated; n.s., no significant difference. d, Cerebral cortex from mice of the indicated genotypes was stained with anti-synaptophysin antibody and imaged by confocal microscopy with a 100× objective. Scale bar, 10 μm. e, f, The fractional area of immunoreactive puncta from images as in d is reported as a function of genotype. Sections were stained with anti-synaptophysin (e) or anti-PSD-95 (f). Data are mean ± SEM for n = 4–8 sites from each of 9–10 mice per genotype. *p < 0.05, one-way ANOVA with post hoc comparisons.
Having shown that PrPC has little effect on Aβ metabolism, we assessed its role in mediating the deleterious chronic effects of brain-derived Aβ in vivo on neuronal form and function, focusing on measures that cannot be modeled in vitro. Aβ-dependent neurodegeneration in transgenic mice is not as pronounced as it is in human AD, but restricted aspects of degeneration have been reported by assessment of monoaminergic pathways (Liu et al., 2008). An examination of serotonin-immunoreactive fibers in the cerebral cortex reveals significant deficits in the APPswe/PSen1ΔE9 mice at 12 months of age (Fig. 2b,c), matching the selective degeneration described previously (Liu et al., 2008). At this age, axonal degeneration occurs, but serotonergic cell loss is not yet detectable (Liu et al., 2008). The appearance of 5-HT-immunopositive axons in Prnp−/− mice is similar to that in wild-type mice. In the transgenic APPswe/PSen1ΔE9 mice lacking PrPC, 5-HT-immunoreactive fibers are indistinguishable from control mice and significantly preserved relative to APPswe/PSen1ΔE9 mice with PrPC. Thus, this transgenic AD-like axon degeneration phenotype is dependent on PrPC.
Synapse loss has been considered a hallmark of AD and been detected in transgenic models (Davies et al., 1987; Scheff et al., 1990; Hsia et al., 1999; Mucke et al., 2000b; Biscaro et al., 2009). We examined a presynaptic marker, anti-synaptophysin, and a postsynaptic marker, anti-PSD-95, in separate sections (Fig. 2d–f). The immunopositive area for either of these markers is slightly, but significantly, reduced in cortical sections from APPswe/PSen1ΔE9 mice. Nontransgenic brain lacking PrPC exhibits no alteration of these markers relative to wild type. In contrast, the APPswe/PSen1ΔE9, Prnp−/− brain shows an increased area occupied by both markers relative to APPswe/PSen1ΔE9 samples with PrPC, and is indistinguishable from wild-type brain by these measures. Thus, the loss of synaptic marker area caused by APPswe/PSen1ΔE9 expression is rescued by a lack of PrPC expression.
An obvious but incompletely understood phenotype of the AD transgenic mice is reduced survival, characterized by sudden death occurring in the adult period. This phenotype may result from hyperexcitability and status epilepticus (Palop et al., 2007; Minkeviciene et al., 2009). Mouse survival was monitored between 0 and 12 months of age, with 40% of APPswe/PSen1ΔE9 transgenic mice being lost (Fig. 3), similar to previous reports for this strain (Halford and Russell, 2009). In contrast, <4% of APPswe/PSen1ΔE9 mice lacking PrPC died during the same age period (p < 0.005). The rescue of survival in the Prnp−/− group demonstrates that death in the transgenic group requires endogenous PrPC.
Figure 3.

Survival of APP-PSen mice improved by absence of PrPC. Survival of wild-type and APPswe/PSen1ΔE9 transgenic mice with and without PrPC expression. Survival of the APPswe/PSen1ΔE9 mice is significantly shorter than each of the other three genotypes by the Wilcoxon statistic (p < 0.005).
Previously, we examined the in vitro requirement of PrPC for acute Aβ suppression of CA1-LTP in hippocampal slices. While some strains of FAD transgenic mice have been shown to exhibit spatial memory deficits (Chen et al., 2000), deficits of CA1-LTP are not uniform, being present in some cases (Nalbantoglu et al., 1997; Chapman et al., 1999; Moechars et al., 1999; Trinchese et al., 2004) but not in others (Hsia et al., 1999; Larson et al., 1999; Fitzjohn et al., 2001; Roder et al., 2003; Gureviciene et al., 2004; Volianskis et al., 2008). We observed no deficit in the magnitude of this form of LTP in hippocampal slices of 12-month-old plaque-bearing APPswe/PSen-1ΔE9 mice (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), consistent with previous studies of this strain (Volianskis et al., 2008). There was a slight reduction of hippocampal input–output curves for this pathway, but this did not reach statistical significance in our studies. Intact LTP may reflect homeostatic compensation in the chronic condition with reduced synaptic numbers, but it precluded an assessment of PrPC's in vivo role for CA1-LTP in this cohort of mice. Since some Aβ plaque-bearing mouse strains do exhibit reduced hippocampal LTP (Nalbantoglu et al., 1997; Chapman et al., 1999; Moechars et al., 1999; Trinchese et al., 2004), the degree of chronic compensation may vary by age, strain, pathway, and transgene.
The phenotype of APPswe/PSen1ΔE9 mice most directly related to Alzheimer's disease is age-dependent memory impairment, and this was our primary outcome measure. Spatial learning and memory were examined in the Morris water maze (Morris, 1984) for the four groups of mice at 3 and 12 months of age. Visually cued performance was rapid in all four genotypes at 12 months of age (Fig. 4a, right). For hidden platform tests, mice were trained to one platform location, and then the platform position was reversed and training continued for the new location. At 12 months of age, the APP-PSen and the Prnp genotypes altered performance, and there was a significant interaction of the two genotypes [two-way repeated-measures (RM) ANOVA: APP-PSen × Prnp interaction, p = 0.015; APP-PSen, p < 0.001; Prnp, p < 0.001]. Both wild-type and Prnp−/− mice learn the new location of the hidden platform over several trial blocks, and latencies to reach the platform approach those for visible platform locations (Fig. 4a). For APPswe/PSen1ΔE9 transgenic mice, learning is severely impaired with much longer latencies to reach a hidden platform. The learning of the APPswe/PSen1ΔE9 mice on the Prnp−/− background is indistinguishable from the control groups. By post hoc pairwise RM-ANOVA, the APPswe/PSen1ΔE9 mice differ (p < 0.001) from each of the other three groups, and none of the other groups differ significantly from one another. For the APPswe/PSen1ΔE9 strain, enhanced learning was observed for both males and females lacking PrPC as compared to those with PrPC expression (Fig. 4b).
Figure 4.
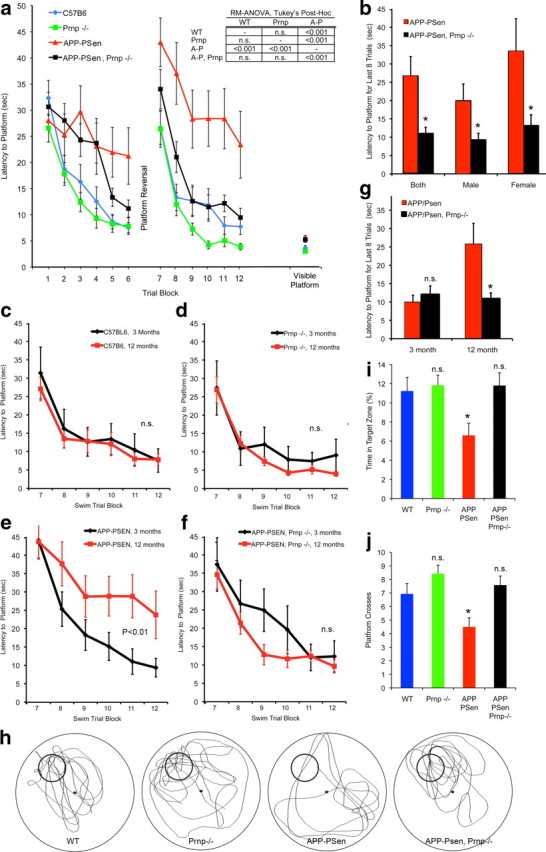
Deletion of PrPC expression rescues spatial learning and memory in APPswe-PSen1ΔE9 transgenic mice. a, Spatial learning is plotted as the latency to find a hidden platform in the Morris water maze at 12 months of age. After the first set of swim trials, the platform location was reversed for the second set of trials. At a later time, after the probe trial, latency to reach a visible platform was scored. Data are mean ± SEM for these groups: C57BL/6, n = 19 (8 male, 11 female); Prnp−/− n = 15 (7 male, 8 female); APP-PSen n = 11 (6 male, 5 female); APP-PSen, Prnp−/− n = 20 (11 male, 9 female). Performance differed by genotype across trial blocks 5–6 and 9–12 (two-way RM-ANOVA: APP-PSen by Prnp interaction, p = 0.015; APP-PSen, p < 0.001; Prnp, p < 0.001). For the indicated Tukey post hoc pairwise comparisons, the APP-PSen group differed from each of the other three groups (p < 0.001), but none of the other genotypes differed significantly from one another. b, The performance of the APPswe/PSen1ΔE9 transgenic mice with and without PrPC expression from trial blocks 11–12 of a is separated by sex. The latencies are reduced in the Prnp−/− groups compared to the Prnp+/+ groups, *p < 0.05, two-tailed Student's t test. c–f, Mice of each genotype were tested for spatial learning at 3 months, and the data are compared to the 12 month performance curves replotted from a. By RM-ANOVA, only the APP-PSen group differed significantly (p < 0.05) between age groups. Data are mean ± SEM for n = 11–20 mice per group. g, The performance of the APPswe/PSen1ΔE9 transgenic mice with and without PrPC expression from trial blocks 11–12 of d–f is separated by age. The latencies are reduced in the Prnp−/− groups compared to the Prnp+/+ at 12 months but not 3 months, *p < 0.05, two-tailed Student's t test. h, Two days after the second set of hidden platform learning trials from a, a 60 s probe trial was conducted. The swim track of one representative mouse from each of the four genotypes is illustrated. A small circle indicates the learned platform location. i, j, The time spent at the site of the previous platform location (i) and crosses of the previous platform location (j) are shown. Data are mean ± SEM for n = 11–20 mice per group. *p < 0.05, one-way ANOVA with post hoc comparison to the WT group.
As an AD model, the PrP-dependent learning deficit of APPswe/PSen1ΔE9 mice is predicted to be an age-dependent phenomenon. Previously, we have shown that APPswe/PSen1ΔE9 mice learn normally while young adults, but progressively exhibit learning deficits in a radial arm water maze from 6 to 12 months of age as Aβ plaque develops (Park et al., 2006). Here, we examined Morris maze learning for each of the four genotypes at 3 months and 12 months (Fig. 4c–g). The APPswe/PSen1ΔE9 mice show a significant decline in learning over this period (Fig. 4e) (RM-ANOVA, p < 0.01). None of the other strains exhibit an age-dependent decline in learning for this task. Thus, the Prnp−/− genotype interacts with APPswe/PSen1ΔE9 transgene to prevent the expression of an age-dependent learning deficit.
For the 12-month-old mice, the memory of learned platform locations was examined in a 60 s probe trial 2 d after the last learning trial (Fig. 4h–j). The wild-type and Prnp−/− mice prefer the location of the previously learned hidden platform, making significantly more platform crosses and spending more time in the platform location. In contrast, the APPswe/PSen1ΔE9 mice show no preference for the hidden platform location, and spend significantly less time in the target zone. The transgenic memory deficit is alleviated on the Prnp−/− background, demonstrating that PrPC is essential for impairment of memory by these AD transgenes in mice.
A second, but less specific, test of learning is passive avoidance. In this paradigm, wild-type mice exhibit an increased latency to enter a preferred dark chamber after receiving a footshock in that location (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). The APPswe/PSen1ΔE9 mice enter the dark chamber significantly sooner, consistent with reduced learning or memory at 24 h. In the absence of PrPC, postshock latencies are statistically indistinguishable from wild-type mice regardless of the presence of the APPswe/PSen1ΔE9 transgenic allele. Thus, on the Prnp−/− background, the APPswe/PSen1ΔE9 passive avoidance deficit is not detectable.
Discussion
The major finding of this study is that memory deficits in AD transgenic mice require the presence of PrPC. While previous studies had shown that synthetic Aβ oligomers bind to PrPC and depend on PrPC to suppress LTP in slices acutely (Laurén et al., 2009), the role of PrPC for in vivo toxicity of brain-derived Aβ had not been tested. The data here demonstrate a requirement of endogenous PrPC for the ability of familial AD transgenes to induce several deficits. In addition to rescuing impairments in spatial memory, deletion of Prnp expression improved survival, and prevented loss of serotonin axons and synaptic markers. It is striking that APP and Aβ levels are not altered in APPswe/PSen1ΔE9 mice by the lack of PrPC. In this regard, the effect of deleting PrPC on mouse transgene phenotypes is similar to that of deleting Tau expression (Roberson et al., 2007). The dissociation of Aβ accumulation and dysfunction is consistent with PrPC functioning immediately downstream of Aβ oligomers in a pathological cascade. To the extent that certain deficits might be derived from the PSen1ΔE9 transgene independently of APPswe (Zhang et al., 2009), they still require PrPC to be manifest.
Recently, Balducci et al. (2010) confirmed the direct binding of Aβ oligomers to PrPC but reported that PrPC is not essential for Aβ-induced memory deficits. This study used distinct methods from those used here. In place of AD transgenesis and spatial memory tests in the Morris water maze, the authors injected mice intracerebroventricularly with synthetic Aβ oligomer and examined object recognition memory. It may be significant that they synthesized a depsipeptide preparation and alkalinized it to induce conversion to the Aβ itself. Some studies of transgenic APP mice have shown impairments of spatial learning but not object recognition (Chen et al., 2000), suggesting that the Balducci experiment may score Aβ actions distinct from, and less sensitive than, the genetic model. In their study, mice lacking PrPC show a typical preference for novelty, but after Aβ oligomer injection they show reversed behavior [reversed discrimination = negative 0.22 ± 0.8 in the study by Balducci et al. (2010), their Fig. 4C]. The reversal is not an absence of object memory, as in wild-type mice injected with Aβ oligomer. The Aβ-oligomer-injected PrPC-null mice actively avoid novel objects and prefer familiar objects. Since novelty seeking is reversed, an assessment of memory function is tenuous but, at face value, object memory is detectable in the Aβ-oligomer-injected PrPC-null mice. Thus, their method might be construed to support, rather than refute, the hypothesis that PrPC is required for memory impairment by Aβ. Clearly, Aβ has an unexplained action in this experiment that alters preference for novelty versus familiarity, possibly via arousal or anxiety (Berlyne, 1966; Berlyne et al., 1966; Mumby, 2001). Regardless of these uncertainties regarding the acute synthetic Aβ injection model, our studies demonstrate that PrPC is essential for APP/PSen transgenic memory and survival deficits.
The molecular events that link Aβ oligomer binding to PrPC with subsequent loss of synaptic markers, 5-HT axon degeneration, survival, and memory function are not yet defined. Several other neurodegenerative conditions are not dependent on PrPC (Steele et al., 2009), so there is specificity for APP/Aβ-dependent phenotypes. Similar to Aβ oligomers, misfolded infectious PrPSc requires PrPC to induce neurotoxicity. The requirement of PrPC for degeneration as opposed to infection has been demonstrated by the infection of grafted wild-type brain tissue in Prnp−/− hosts (Brandner et al., 1996), and the requirement for neuronal PrPC (Mallucci et al., 2003) and membrane-anchored PrPC (Chesebro et al., 2005) for neurotoxicity. Since PrPC is a lipid-anchored cell surface protein, it is presumably coupled to transmembrane proteins and intracellular signal transduction, but which associated proteins are relevant for these degenerative phenotypes is not known. The proteome of PrPC-associated proteins (Schmitt-Ulms et al., 2004) and the neurodegeneration of mice expressing truncated PrPC (Baumann et al., 2007; Li et al., 2007) may facilitate future mechanistic studies.
In the constitutive Prnp mutant, loss of PrPC expression throughout life rescues memory deficits in the transgenic mice. It is not clear which phenotypes might be reversible if PrPC expression or Aβ-oligomer binding were blocked at some point after the onset of AD-related deficits. However, further molecular and pharmacological analysis should define the potential utility of PrPC blockade in alleviating AD onset, progression, and symptoms.
Footnotes
H.B.N. is a Brown-Coxe Postdoctoral Fellow and S.M.S. is a member of the Kavli Institute for Neuroscience at Yale University. We acknowledge support from the National Institutes of Health, the Falk Medical Research Trust, an anonymous donor, the Alzheimer's Association, and the Cure Alzheimer's Fund to S.M.S.
References
- Austin MC, Whitehead RE, Edgar CL, Janosky JE, Lewis DA. Localized decrease in serotonin transporter-immunoreactive axons in the prefrontal cortex of depressed subjects committing suicide. Neuroscience. 2002;114:807–815. doi: 10.1016/s0306-4522(02)00289-0. [DOI] [PubMed] [Google Scholar]
- Balducci C, Beeg M, Stravalaci M, Bastone A, Sclip A, Biasini E, Tapella L, Colombo L, Manzoni C, Borsello T, Chiesa R, Gobbi M, Salmona M, Forloni G. Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc Natl Acad Sci U S A. 2010;107:2295–2300. doi: 10.1073/pnas.0911829107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann F, Tolnay M, Brabeck C, Pahnke J, Kloz U, Niemann HH, Heikenwalder M, Rülicke T, Bürkle A, Aguzzi A. Lethal recessive myelin toxicity of prion protein lacking its central domain. EMBO J. 2007;26:538–547. doi: 10.1038/sj.emboj.7601510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlyne DE. Curiosity and exploration. Science. 1966;153:25–33. doi: 10.1126/science.153.3731.25. [DOI] [PubMed] [Google Scholar]
- Berlyne DE, Koenig ID, Hirota T. Novelty, arousal, and the reinforcement of diversive exploration in the rat. J Comp Physiol Psychol. 1966;62:222–226. doi: 10.1037/h0023681. [DOI] [PubMed] [Google Scholar]
- Biscaro B, Lindvall O, Hock C, Ekdahl CT, Nitsch RM. Aβ immunotherapy protects morphology and survival of adult-born neurons in doubly transgenic APP/PS1 mice. J Neurosci. 2009;29:14108–14119. doi: 10.1523/JNEUROSCI.2055-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, Kobayashi Y, Marino S, Weissmann C, Aguzzi A. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature. 1996;379:339–343. doi: 10.1038/379339a0. [DOI] [PubMed] [Google Scholar]
- Buttini M, Orth M, Bellosta S, Akeefe H, Pitas RE, Wyss-Coray T, Mucke L, Mahley RW. Expression of human apolipoprotein E3 or E4 in the brains of Apoe−/− mice: isoform-specific effects on neurodegeneration. J Neurosci. 1999;19:4867–4880. doi: 10.1523/JNEUROSCI.19-12-04867.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttini M, Masliah E, Barbour R, Grajeda H, Motter R, Johnson-Wood K, Khan K, Seubert P, Freedman S, Schenk D, Games D. β-Amyloid immunotherapy prevents synaptic degeneration in a mouse model of Alzheimer's disease. J Neurosci. 2005;25:9096–9101. doi: 10.1523/JNEUROSCI.1697-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, Younkin L, Good MA, Bliss TV, Hyman BT, Younkin SG, Hsiao KK. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci. 1999;2:271–276. doi: 10.1038/6374. [DOI] [PubMed] [Google Scholar]
- Chen G, Chen KS, Knox J, Inglis J, Bernard A, Martin SJ, Justice A, McConlogue L, Games D, Freedman SB, Morris RG. A learning deficit related to age and beta-amyloid plaques in a mouse model of Alzheimer's disease. Nature. 2000;408:975–979. doi: 10.1038/35050103. [DOI] [PubMed] [Google Scholar]
- Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, Raymond L, Favara C, Baron G, Priola S, Caughey B, Masliah E, Oldstone M. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science. 2005;308:1435–1439. doi: 10.1126/science.1110837. [DOI] [PubMed] [Google Scholar]
- Chin J, Palop JJ, Yu GQ, Kojima N, Masliah E, Mucke L. Fyn kinase modulates synaptotoxicity, but not aberrant sprouting, in human amyloid precursor protein transgenic mice. J Neurosci. 2004;24:4692–4697. doi: 10.1523/JNEUROSCI.0277-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- Davies CA, Mann DM, Sumpter PQ, Yates PO. A quantitative morphometric analysis of the neuronal and synaptic content of the frontal and temporal cortex in patients with Alzheimer's disease. J Neurol Sci. 1987;78:151–164. doi: 10.1016/0022-510x(87)90057-8. [DOI] [PubMed] [Google Scholar]
- Duffy P, Schmandke A, Schmandke A, Sigworth J, Narumiya S, Cafferty WB, Strittmatter SM. Rho-associated kinase II (ROCKII) limits axonal growth after trauma within the adult mouse spinal cord. J Neurosci. 2009;29:15266–15276. doi: 10.1523/JNEUROSCI.4650-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzjohn SM, Morton RA, Kuenzi F, Rosahl TW, Shearman M, Lewis H, Smith D, Reynolds DS, Davies CH, Collingridge GL, Seabrook GR. Age-related impairment of synaptic transmission but normal long-term potentiation in transgenic mice that overexpress the human APP695SWE mutant form of amyloid precursor protein. J Neurosci. 2001;21:4691–4698. doi: 10.1523/JNEUROSCI.21-13-04691.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GrandPré T, Li S, Strittmatter SM. Nogo-66 receptor antagonist peptide promotes axonal regeneration. Nature. 2002;417:547–551. doi: 10.1038/417547a. [DOI] [PubMed] [Google Scholar]
- Grider MH, Chen Q, Shine HD. Semi-automated quantification of axonal densities in labeled CNS tissue. J Neurosci Methods. 2006;155:172–179. doi: 10.1016/j.jneumeth.2005.12.021. [DOI] [PubMed] [Google Scholar]
- Gureviciene I, Ikonen S, Gurevicius K, Sarkaki A, van Groen T, Pussinen R, Ylinen A, Tanila H. Normal induction but accelerated decay of LTP in APP + PS1 transgenic mice. Neurobiol Dis. 2004;15:188–195. doi: 10.1016/j.nbd.2003.11.011. [DOI] [PubMed] [Google Scholar]
- Halford RW, Russell DW. Reduction of cholesterol synthesis in the mouse brain does not affect amyloid formation in Alzheimer's disease, but does extend lifespan. Proc Natl Acad Sci U S A. 2009;106:3502–3506. doi: 10.1073/pnas.0813349106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci U S A. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, Copeland NG, Lee MK, Younkin LH, Wagner SL, Younkin SG, Borchelt DR. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet. 2004;13:159–170. doi: 10.1093/hmg/ddh019. [DOI] [PubMed] [Google Scholar]
- Kim JE, Liu BP, Park JH, Strittmatter SM. Nogo-66 receptor prevents raphespinal and rubrospinal axon regeneration and limits functional recovery from spinal cord injury. Neuron. 2004;44:439–451. doi: 10.1016/j.neuron.2004.10.015. [DOI] [PubMed] [Google Scholar]
- King SL, Marks MJ, Grady SR, Caldarone BJ, Koren AO, Mukhin AG, Collins AC, Picciotto MR. Conditional expression in corticothalamic efferents reveals a developmental role for nicotinic acetylcholine receptors in modulation of passive avoidance behavior. J Neurosci. 2003;23:3837–3843. doi: 10.1523/JNEUROSCI.23-09-03837.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson J, Lynch G, Games D, Seubert P. Alterations in synaptic transmission and long-term potentiation in hippocampal slices from young and aged PDAPP mice. Brain Res. 1999;840:23–35. doi: 10.1016/s0006-8993(99)01698-4. [DOI] [PubMed] [Google Scholar]
- Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009;457:1128–1132. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Li A, Christensen HM, Stewart LR, Roth KA, Chiesa R, Harris DA. Neonatal lethality in transgenic mice expressing prion protein with a deletion of residues 105–125. EMBO J. 2007;26:548–558. doi: 10.1038/sj.emboj.7601507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Liu BP, Budel S, Li M, Ji B, Walus L, Li W, Jirik A, Rabacchi S, Choi E, Worley D, Sah DW, Pepinsky B, Lee D, Relton J, Strittmatter SM. Blockade of nogo-66, myelin-associated glycoprotein, and oligodendrocyte myelin glycoprotein by soluble nogo-66 receptor promotes axonal sprouting and recovery after spinal injury. J Neurosci. 2004;24:10511–10520. doi: 10.1523/JNEUROSCI.2828-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Kim JE, Budel S, Hampton TG, Strittmatter SM. Transgenic inhibition of Nogo-66 receptor function allows axonal sprouting and improved locomotion after spinal injury. Mol Cell Neurosci. 2005;29:26–39. doi: 10.1016/j.mcn.2004.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linhoff MW, Laurén J, Cassidy RM, Dobie FA, Takahashi H, Nygaard HB, Airaksinen MS, Strittmatter SM, Craig AM. An unbiased expression screen for synaptogenic proteins identifies the LRRTM protein family as synaptic organizers. Neuron. 2009;61:734–749. doi: 10.1016/j.neuron.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Yoo MJ, Savonenko A, Stirling W, Price DL, Borchelt DR, Mamounas L, Lyons WE, Blue ME, Lee MK. Amyloid pathology is associated with progressive monoaminergic neurodegeneration in a transgenic mouse model of Alzheimer's disease. J Neurosci. 2008;28:13805–13814. doi: 10.1523/JNEUROSCI.4218-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallucci G, Dickinson A, Linehan J, Klöhn PC, Brandner S, Collinge J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science. 2003;302:871–874. doi: 10.1126/science.1090187. [DOI] [PubMed] [Google Scholar]
- Mamounas LA, Altar CA, Blue ME, Kaplan DR, Tessarollo L, Lyons WE. BDNF promotes the regenerative sprouting, but not survival, of injured serotonergic axons in the adult rat brain. J Neurosci. 2000;20:771–782. doi: 10.1523/JNEUROSCI.20-02-00771.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manson JC, Clarke AR, Hooper ML, Aitchison L, McConnell I, Hope J. 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol Neurobiol. 1994;8:121–127. doi: 10.1007/BF02780662. [DOI] [PubMed] [Google Scholar]
- Masliah E, Ellisman M, Carragher B, Mallory M, Young S, Hansen L, DeTeresa R, Terry RD. Three-dimensional analysis of the relationship between synaptic pathology and neuropil threads in Alzheimer disease. J Neuropathol Exp Neurol. 1992;51:404–414. doi: 10.1097/00005072-199207000-00003. [DOI] [PubMed] [Google Scholar]
- Minkeviciene R, Rheims S, Dobszay MB, Zilberter M, Hartikainen J, Fülöp L, Penke B, Zilberter Y, Harkany T, Pitkänen A, Tanila H. Amyloid β-induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci. 2009;29:3453–3462. doi: 10.1523/JNEUROSCI.5215-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moechars D, Dewachter I, Lorent K, Reversé D, Baekelandt V, Naidu A, Tesseur I, Spittaels K, Haute CV, Checler F, Godaux E, Cordell B, Van Leuven F. Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J Biol Chem. 1999;274:6483–6492. doi: 10.1074/jbc.274.10.6483. [DOI] [PubMed] [Google Scholar]
- Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Mucke L, Yu GQ, McConlogue L, Rockenstein EM, Abraham CR, Masliah E. Astroglial expression of human alpha(1)-antichymotrypsin enhances Alzheimer-like pathology in amyloid protein precursor transgenic mice. Am J Pathol. 2000a;157:2003–2010. doi: 10.1016/s0002-9440(10)64839-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of Aβ1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000b;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumby DG. Perspectives on object-recognition memory following hippocampal damage: lessons from studies in rats. Behav Brain Res. 2001;127:159–181. doi: 10.1016/s0166-4328(01)00367-9. [DOI] [PubMed] [Google Scholar]
- Nalbantoglu J, Tirado-Santiago G, Lahsaïni A, Poirier J, Goncalves O, Verge G, Momoli F, Welner SA, Massicotte G, Julien JP, Shapiro ML. Impaired learning and LTP in mice expressing the carboxy terminus of the Alzheimer amyloid precursor protein. Nature. 1997;387:500–505. doi: 10.1038/387500a0. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Widi GA, Gimbel DA, Harel NY, Lee DH, Strittmatter SM. Subcutaneous Nogo receptor removes brain amyloid-β and improves spatial memory in Alzheimer's transgenic mice. J Neurosci. 2006;26:13279–13286. doi: 10.1523/JNEUROSCI.4504-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkin ET, Watt NT, Hussain I, Eckman EA, Eckman CB, Manson JC, Baybutt HN, Turner AJ, Hooper NM. Cellular prion protein regulates beta-secretase cleavage of the Alzheimer's amyloid precursor protein. Proc Natl Acad Sci U S A. 2007;104:11062–11067. doi: 10.1073/pnas.0609621104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Roder S, Danober L, Pozza MF, Lingenhoehl K, Wiederhold KH, Olpe HR. Electrophysiological studies on the hippocampus and prefrontal cortex assessing the effects of amyloidosis in amyloid precursor protein 23 transgenic mice. Neuroscience. 2003;120:705–720. doi: 10.1016/s0306-4522(03)00381-6. [DOI] [PubMed] [Google Scholar]
- Scheff SW, DeKosky ST, Price DA. Quantitative assessment of cortical synaptic density in Alzheimer's disease. Neurobiol Aging. 1990;11:29–37. doi: 10.1016/0197-4580(90)90059-9. [DOI] [PubMed] [Google Scholar]
- Schmitt-Ulms G, Hansen K, Liu J, Cowdrey C, Yang J, DeArmond SJ, Cohen FE, Prusiner SB, Baldwin MA. Time-controlled transcardiac perfusion cross-linking for the study of protein interactions in complex tissues. Nat Biotechnol. 2004;22:724–731. doi: 10.1038/nbt969. [DOI] [PubMed] [Google Scholar]
- Schwarze-Eicker K, Keyvani K, Görtz N, Westaway D, Sachser N, Paulus W. Prion protein (PrPc) promotes beta-amyloid plaque formation. Neurobiol Aging. 2005;26:1177–1182. doi: 10.1016/j.neurobiolaging.2004.10.004. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele AD, Zhou Z, Jackson WS, Zhu C, Auluck P, Moskowitz MA, Chesselet MF, Lindquist S. Context dependent neuroprotective properties of prion protein (PrP) Prion. 2009;3:240–249. doi: 10.4161/pri.3.4.10135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinchese F, Liu S, Battaglia F, Walter S, Mathews PM, Arancio O. Progressive age-related development of Alzheimer-like pathology in APP/PS1 mice. Ann Neurol. 2004;55:801–814. doi: 10.1002/ana.20101. [DOI] [PubMed] [Google Scholar]
- Volianskis A, Kostner R, Molgaard M, Hass S, Jensen MS. Episodic memory deficits are not related to altered glutamatergic synaptic transmission and plasticity in the CA1 hippocampus of the APPswe/PS1DeltaE9-deleted transgenic mice model of beta-amyloidosis. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2008.08.005. [DOI] [PubMed] [Google Scholar]
- Vorhees CV, Williams MT. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc. 2006;1:848–858. doi: 10.1038/nprot.2006.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Wang X, Baughman KW, Basso DM, Strittmatter SM. Delayed Nogo receptor therapy improves recovery from spinal cord contusion. Ann Neurol. 2006;60:540–549. doi: 10.1002/ana.20953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Budel S, Baughman K, Gould G, Song KH, Strittmatter SM. Ibuprofen enhances recovery from spinal cord injury by limiting tissue loss and stimulating axonal growth. J Neurotrauma. 2009;26:81–95. doi: 10.1089/neu.2007.0464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Wu B, Beglopoulos V, Wines-Samuelson M, Zhang D, Dragatsis I, Südhof TC, Shen J. Presenilins are essential for regulating neurotransmitter release. Nature. 2009;460:632–636. doi: 10.1038/nature08177. [DOI] [PMC free article] [PubMed] [Google Scholar]