

Abstract
Although a series of melanoma differentiation antigens for immunotherapeutic targeting has been described, heterogeneous expression of antigens such as Melan-A/MART-1 and gp100 results from a loss of antigenic expression in many late stage tumors. Antigen loss can represent a means for tumor escape from immune recognition, and a barrier to immunotherapy. However, since antigen-negative tumor phenotypes frequently result from reversible gene regulatory events, antigen enhancement represents a potential therapeutic opportunity. Accordingly, we have developed a cell-based assay to screen for compounds with the ability to enhance T-cell recognition of melanoma cells. This assay is dependent on augmentation of MelanA/MART-1 antigen presentation by a melanoma cell line (MU89). T-cell recognition is detected as interleukin-2 production by a Jurkat T cell transduced to express a T-cell receptor specific for an HLA-A2 restricted epitope of the Melan-A/MART-1 protein. This cellular assay was used to perform a pilot screen by using 480 compounds of known biological activity. From the initial proof-of-principle primary screen, eight compounds were identified as positive hits. A panel of secondary screens, including orthogonal assays, was used to validate the primary hits and eliminate false positives, and also to measure the comparative efficacy of the identified compounds. This cell-based assay, thus, yields consistent results applicable to the screening of larger libraries of compounds that can potentially reveal novel molecules which allow better recognition of treated tumors by T cells.
Introduction
Malignant melanoma represents one of the more aggressive and therapeutically resistant tumors once it has progressed beyond a surgically resectable stage.1,2 Due to the refractory responses to conventional cancer therapies, considerable energy has been devoted toward developing effective melanoma immunotherapeutic strategies. There is ample evidence that many melanoma tumor structures are immunogenic, and a growing number of melanoma-associated antigens are known targets for T-cell–mediated cytotoxicity.3,4 Also, although the presence of lymphocytes in and around melanomas is associated with an improved prognosis,5 there remain numerous obstacles to the successful elimination of clinically apparent tumors.5 Regrettably, tumors can escape from immune destruction through a variety of mechanisms, including suppressive effects on the host immune response6,7 as well as reduced antigen expression.8,9
Although the majority of malignant melanomas express differentiation antigens such as Melan-A/MART-1, in many tumor deposits, expression of this antigen is heterogeneous.10–12 Further, in late-stage disease, this antigen is lost with increasing frequency.13 Some groups have reported anecdotal cases of antigen loss with tumor progression,11 and Jager showed progressive loss of Melan-A/MART-1 in metastases over time in 4 out of 5 patients.13 Although it is possible to enhance cell mediated immunity in vivo by vaccination with several melanocyte antigens, including Melan-A/MART-1, gp100, and tyrosinase, several reports indicate that melanoma tumors that grow in these vaccinated patients show an increasing frequency of tumor cells that have lost the antigen against which the patient was vaccinated. This finding suggests that antigen loss by immune selection plays a role in immunotherapy outcomes.14–17
Tumor cells will escape recognition and killing if they undergo reduced expression of such differentiation antigens, or lose the ability to process and present them through the restricting HLA antigen. We have referred to this phenomenon as “antigen silencing,”18,19 and as noted above, it is exacerbated by the immune system itself through “immunoselection”13,20–22 or “immune editing”23,24 for antigen-negative variants. Since only a minority of the patients respond in most clinical trials, and many clinical responses are partial, the finding that tumor antigen expression is altered after immunotherapy indicates the importance of proactively retaining antigen expression to overcome the outgrowth of low-antigen-expressing tumor cells.
We have previously demonstrated that loss of antigen expression need not arise from irreversible processes (such as gene mutation or deletion), but can (and often does) come about through alterations in gene regulatory pathways.18,19 Such changes leave the relevant coding sequences intact and are, in principle, reversible. We, and others, have identified small molecules (such as MEK and B-raf inhibitors25) and cytokines (such as interferon-beta [IFN-beta]26) that can upregulate melanoma antigen expression. Likewise, chemotherapeutic agents such as topoisomerase inhibitors27 enhance antigen expression in both melanomas and gliomas. The plastic nature of antigen expression is further indicated by the fact that cytokines such as Oncostatin M,19 interleukin 1α (IL-1α), and IL-1β28 can diminish antigen expression in melanomas.
Here, we report the successful development and validation of a robust cellular assay for screening compounds that are able to increase recognition of melanoma tumors by T cells. The primary focus of this screen is to identify novel compounds that can fulfill the twofold goal of both increasing differentiation antigen expression in tumor cells (which may be the result of enhancing tumor cell differentiation itself) and simultaneously allowing T-cell recognition of the treated tumor cells. Thus, the screen allows identification of biologically relevant compounds that not only improve differentiation antigen expression, but also allow normal T-cell function.
For the purposes of the assay, we have focused on Melan-A/MART-1, toward which well-characterized cytotoxic T-cell responses have been demonstrated.29–32 Moreover, Melan-A/MART-1 is coordinately regulated with a panel of additional melanocytic antigens.33–37 We identified and validated a melanoma cell line with desired characteristics for use in the screen as a tumor line for Melan-A/MART-1 expression. Since this cell line acts to stimulate the T-cell responders in the assay, we refer to these Melan-A/MART-1 expressors as having the general functional role of “stimulators.” In parallel, we sub-cloned an HLA-A2-restricted Melan-A/MART-1 specific T-cell receptor (TCR) into a lentiviral vector for use in generating a stable Jurkat T-cell responder line with defined specificity against a Melan-A/MART-1 peptide displayed in the context of Class I MHC. The Jurkat cells produce readily detectable IL-2 in a quantitative enzyme-linked immunosorbent assay (ELISA) when exposed to tumor cells expressing HLA-A2 and Melan-A/MART-1.
Using this assay, we successfully performed a screen of 480 compounds. Confirmatory assays and orthogonal secondary screens were then used to evaluate the hits from the primary screen, which allows the identification of novel agents that increase expression of tumor antigens. Moreover, the assay read-out itself is the most immunologically relevant parameter: enhanced T-cell activation through specific recognition of tumor-displayed antigen, as a direct consequence of augmented antigen expression. Systematic application of the same screening procedure toward large compound libraries will allow identification of additional compounds with the ability to enhance T-cell recognition of treated tumors as a means of improving the outcome of immunotherapy of melanoma. The principles developed in this screening assay are also amenable to development of screens for additional cell-based assays for drugs with the ability to induce expression of other antigens recognized by additional known T-cell specificities.
Materials and Methods
Cell Culture
The conditions for cell culture and the origins of most of the melanoma cell lines have been previously described.18,19 Melanoma cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS). The TCR-negative Jurkat T-cell–line derivative J.RT3-T3.5 was obtained from American type culture collection, and these T cells were cultured in RPMI with 10% FBS. IFN-beta-1a (Avonex) was obtained from Biogen-Idec (Cambridge, MA) and reconstituted according to the manufacturer's recommendations.
Subcloning of the Melan-A/MART-1 Specific TCR
A Melan-A/MART-1 specific TCR was produced and inserted into a lentiviral vector as previously described.27 Briefly, a TCR specific for the MART-1 peptide EAAGIGILTV presented in HLA-A2 MHC Class I molecules (1D338,39) was synthesized by GeneArt (Burlingame, CA) and placed into a third-generation lentiviral transfer vector.40 The full-length insert (1,820 base pair) containing both chains of the TCR was then subcloned into the lentiviral vector using the restriction sites NheI and SalI. A silent mutation was made in the seventh amino acid (P139) of the alpha chain constant region of the TCR to generate an EagI restriction site. Two silent mutations were also made in the eighth (P141) and ninth amino acid (P142) of the beta chain constant region of the TCR to generate a BspeI restriction site. The construction of the vector with these additional restriction enzyme sites facilitates the subcloning of other TCRs by creating a way to readily synthesize and subclone the 450 base pair variable regions of each chain of the TCR.
Characteristics of the Melan-A/MART-1 Specific TCR
The sequence of the alpha and beta chains of a TCR specific for the MART-1 peptide EAAGIGILTV presented in HLA-A2 MHC Class I molecules was codon-optimized to increase expression as previously described.41 The TCRα and TCRβ sequences were separated by a 2A sequence, as previously described, to facilitate stoichiometric coordinate expression under control of the EF-1α promoter.42 The alpha and beta chains each contained a mutation (T183C and S190C, respectively) adding a cysteine to facilitate pairing and surface expression of the two chains of the TCR, by forming an additional inter-chain disulfide bond as previously described.43,44
Generation of TCR-Encoding Lentiviral Particles
Lentiviral vectors containing supernatants were prepared as previously described.27,45
J-TCR-M1 Cell Line Derivation
The TCR-minus cell line J.RT3-T3.546–48 was infected with lentiviral particles containing the Melan-A/MART-1 specific TCR as previously described.27 For transduction of the Melan-A/MART-1 specific TCR, 1×105 J.RT3-T3.5 cells were incubated with lentiviral vector at a MOI of 10. The surface expression and peptide specificity of the transduced TCR was established by using tetramer staining. The function of the transduced TCR was shown after coculture with peptide pulsed tumor cells and cytokine ELISA. A pool of transduced J.RT3 cells stably expressing the exogenous Melan-A/MART-1 specific TCR at >95% efficiency was expanded for further use.
Chemicals
A chemical library of 480 compounds was obtained from Enzo Life Sciences (Plymouth Meeting, PA) and is referred to as the “ICCB Known Bioactives Library.” The compounds within this library include a variety of receptor agonists, ion channel functional modulators, and kinase and enzyme inhibitors. Compounds for use in secondary screens were purchased and resuspended at 10 mg/mL following the manufacturer's recommendations. 17-allylaminogeldanamycin (17-AAG) was purchased from Sigma Aldrich (St. Louis, MO). Phorbol-12-myristate-13 acetate (PMA), flunarizine, 3-(4-octadecyl)benzoylacrylic acid (OBAA), dantrolene, damnacanthal, aphidicolin, and glyburide were obtained from Enzo Life Sciences.
Cell-Based Screening Assay Procedure
The protocol for the cell-based assay is described in greater detail in Table 1 and has been previously reported.27 The 480 compounds were tested in six parallel 96-well plates. The first two columns (16 wells) of each plate were used for standards (8 wells), untreated controls (4 wells), and positive controls (4 wells). The other ten columns (80 wells) were used for testing compounds in separate wells. A 100 μL aliquot containing 5×104 MU89 cells was added to the wells that contained 0.1 μL of each test drug diluted in 50 μL of RPMI media. The plates were incubated with drug for 72 h at 37° C in a humidified incubator with 5% CO2. After 72 h, each well received 2.5×104 J-TCR-M1 cells in 50 μL medium, and was then incubated for a further 24 h. At the conclusion of the 24-h coculture period, each plate was centrifuged to pellet the cells, and 100 μL supernatant fluid was collected from each well and plated in a parallel 96-well flat bottom well for IL-2 ELISA assay.
Table 1.
Cell-Based Assay Screening Protocol
| Step | Parameter | Description |
|---|---|---|
| 1 | Media | 50 μL DMEM with 10% serum plated in 384-well plate Corning (cat no 3712) |
| 2 | Add compounds | Use pin transfer to add 0.1 μL of compounds (5 mg/mL). Final concentration for most compounds about 10 μM |
| 3 | Plate tumor cells | 96-well plate, 5×104 cells per well in 100 μL. Falcon (cat no 353072) tissue culture treated plates |
| 4 | Add compounds to 96-well plate with tumor cells | 80 wells contain test compounds. 8 wells are established to generate a standard curve of known IL-2 pg/mL. 4 wells contain IFN-beta, 4 wells are untreated controls |
| 5 | Incubate | 72 h in 37°C tissue culture incubator containing 5% CO2 |
| 6 | Add T cells | 2.5×104 cells in 50 μL per well. Spin plates gently to pellet cells and facilitate interaction |
| 7 | Incubate | 24 h in 37°C tissue culture incubator containing 5% CO2 |
| 8 | Take supernatant | 100 μL aliquots from each well |
| 9 | IL-2 ELISA | IL-2 ELISA kit from BD Biosciences (cat no 555190). Microtest ELISA plates BD Bioscience (cat no 353279). Read OD in plate reader BioRad 3550 450–595 nm |
| 10 | Calculations | Use standard curve to calculate IL-2 pg/ml for each plate. Calculate fold increase over average of untreated control. Establish efficacy of positive control (IFN-beta) (>2-fold over control) |
DMEM, Dulbecco's modified Eagle's medium; ELISA, enzyme-linked immunosorbent assay; IFN-beta, interferon-beta; IL-2, interleukin 2; OD, optical density.
Confirmation Assays: Repeat Cellular Assay
Melanoma cells were treated with hits from the primary screen for 3 days before counting and plating for the coculture assay (cells were washed and resuspended in fresh medium that no longer contained the treatment agents during the coculture). A 100 μL volume of 5×105 cells/mL tumor cells was mixed with 50 μL of 2.5×105 cells/mL T cells in a 96-well V-bottom plate and incubated overnight (16–24 h). Plates were centrifuged to pellet the cells, and 100 μL of supernatant fluid was removed from each well for cytokine assay. To assess antigen-specific T-cell responses, a final concentration of 3.33 μg/mL Melan-A/MART-1 peptide (26ELAGIGILTV35 [A27L from wild type]) was added to the coculture. MU89 tumor cells were also treated with IFN-beta (5,000 U/mL) for 3 days as a positive control.
Cytokine ELISA
The protocol for evaluation of the cytokine IL-2 was performed as previously described27 by using the BD OptEAI kit, human IL-2 ELISA set from BD Biosciences (San Diego, CA) following the manufacturer's recommendations. The absorbance at 450 nm was read in a BioRad 3550 plate reader. A standard curve using known concentrations of IL-2 (from 500 to 8 pg/mL) was included in each ELISA plate. IL-2 levels for experimental samples (pg/mL) were calculated from the standard curve.
EGFP Reporter Cell Line
The generation and application of EGFP reporter cells has been previously described.8 Briefly, a 1,200 base pair human genomic DNA segment encompassing the Melan-A/MART-1 promoter was used to generate a construct driving expression of the EGFP reporter gene. Stable transfectants with this construct were generated in the low-antigen cell line A375 and the high-antigen line MM96L+. The EGFP expression response patterns of these cells to IFN-beta and MAP kinase inhibitors recapitulates the antigen upregulation of endogenous Melan-A/MART-1 induced by these agents.8
Flow Cytometry
Staining and flow cytometric analyses of cytoplasmic Melan-A/MART-1 and gp100 expression were performed as previously described.18 Cells were fixed with 1% formaldehyde, permeabilized with 0.1% saponin, and stained with monoclonal antibodies to melanocyte antigens. Antibodies were obtained for: MART-1 from Vector Labs (Burlingame, CA); gp100 HMB45 from DakoCytomation (Carpinteria, CA) and then visualized with goat anti-mouse fluorescein isothiocyanate-conjugated secondary antibody from Invitrogen (Frederick, MD). The level of EGFP was determined by flow cytometry of unfixed cells that were washed and resuspended in 1× phosphate-buffered saline.
Results
Transduced Jurkat Responder T-Cell Line
The assay relies on the use of an antigen-specific responder T-cell line. To achieve this goal, we utilized a transduced TCR with specificity for a known Melan-A/MART-1 peptide in the context of a specific MHC molecule, with suitable binding affinity, and demonstrated biological signaling functionality. The chosen TCR reacts specifically to a Melan-A/MART-1 decapeptide (26EAAGIGILTV35) presented by HLA-A2,38 with appropriate triggering of T-cell activation.39 We used a lentiviral vector system for transduction of this specific TCR into the J.RT3-T3.5 T cell line, a Jurkat cell derivative that fails to express endogenous surface TCR through a mutation in its beta chain.47,48 The functional specificity of this transduced Jurkat cell line (J-TCR-M1) was established by its stimulation (as measured by IL-2 ELISA) only by HLA-A2+ tumor cells presenting the correct Melan-A/MART-1 peptide.27
Melanoma Stimulator Cell Line
The stimulator melanoma cell line for the assay was selected on the basis of its constitutive low, but perceptible, expression of Melan-A/MART-1 that can be consistently enhanced by treatment with IFN-beta, a cytokine previously shown to upregulate Melan-A/MART-1 and MHC Class I.26 Since the transduced TCR used in the responding J-TCR-M1 cells is restricted by HLA-A2, the tumor cells were of necessity HLA-A2+. We evaluated several candidate melanoma cell lines for the ability to stimulate the J-TCR-M1 cell line. Levels of Melan-A/MART-1 (determined by intracellular staining and flow cytometry) and corresponding levels of induced IL-2 (determined by coculture with J-TCR-M1 and IL-2 ELISA) were tested for different melanoma cell lines. A clear correlation was seen between Melan-A/MART-1 levels and IL-2 induction (Table 2). In most of the melanoma cell lines tested, IFN-beta increased Melan-A/MART-1 levels and IL-2 induction, with the greatest fold induction shown by MU89, possibly due to its lower initial baseline expression level of Melan-A/MART-1. The MU89 cell line was chosen for use in the cell-based assay because of these characteristics.
Table 2.
Correlation of MART-1 Intracellular Staining and Interleukin-2 Secretion After Coculture of T Cells and Tumors
| Cell linea | Treatmentb | MART-1c | IL-2d |
|---|---|---|---|
| MU89 | Control | 79 | 25 |
| IFN-beta | 153 | 96 | |
| 453A | Control | 99 | 114 |
| IFN-beta | 113 | 159 | |
| H59-44T | Control | 91 | 21 |
| IFN-beta | 103 | 23 | |
| MALME-3M | Control | 117 | 186 |
| IFN-beta | 130 | 289 | |
| MUX | Control | 13 | 10 |
| IFN-beta | 18 | 12 | |
| A375 | Control | 9 | 15 |
| IFN-beta | 9 | 7 |
All cell lines are HLA-A2.
IFN-beta treatment with 5,000 IU/mL for 3 days.
Geometric mean of intracellular staining using anti-MART-1.
IL-2 (pg/mL) after coculture with MART-1 TCR Jurkat cells.
TCR, T-cell receptor.
Assay Implementation
IFN-beta was chosen as a positive control to include in the assay because of its favorable induction of IL-2 by the MU89 melanoma cell line. The robustness of a high-volume screening assay is generally assessed by calculating the Z′ factor,49–51 defined from the mean (μ) and standard deviations (σ) of the positive (+) and negative (−) controls in a screen by the equation 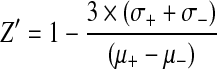 . Our positive and negative control data were accordingly used to derive a Z′ value for the screening assay (Fig. 1). Since a satisfactory Z′-factor is above 0.5,49–51 the calculated Z′-factor level (0.55 from an n=16) indicated that our assay showed sufficient robustness for its continued development.
. Our positive and negative control data were accordingly used to derive a Z′ value for the screening assay (Fig. 1). Since a satisfactory Z′-factor is above 0.5,49–51 the calculated Z′-factor level (0.55 from an n=16) indicated that our assay showed sufficient robustness for its continued development.
Fig. 1.

Z′ factor determination. The y-axis indicates levels of IL-2 measured in supernatants from coculture of 5×104 melanoma stimulator tumor cells (MU89) and 2.5×104 responder T cells (J-TCR-M1). IL-2 values are shown for 16 replicate determinations (wells 1–16 on x-axis) for both untreated negative control cells (squares), and positive control cells treated for 3 days with 5,000 U/mL IFN-beta (triangles). The solid line represents the average, and dashed lines represent plus and minus one standard deviation from the mean. IFN, interferon; IL, interleukin.
The use of IFN-beta as a positive control has an important role in determining whether a screened plate is of acceptable quality. Based on empirical testing, a screened plate cannot be considered valid unless the IFN-beta positive control shows a twofold increase over the untreated control value. The IFN-beta responses act as a quality control measure, flagging plates with poor performance in the assay that are necessarily rejected.
Previous experience has suggested that a 3 day treatment period would be suitable for evaluation of antigen-enhancing effects of either small molecules or protein mediators,25–27 and this time frame was also validated with the IL-2 assay coculture assay.27 We chose to use a concentration of 10 μM for library screening. Although most clinically relevant compounds work at nanomolar concentrations, by the use of a higher concentration, we aimed at avoiding excluding compounds with lower activity, whose potencies could be improved by structure-activity relationship studies and analog syntheses during subsequent lead development.
To determine optimal cell numbers per well for the indicator melanoma cell line MU89, it is necessary to account for assay sensitivity limits, cell growth limitations for assay scale-up (10 to 20 plates at a time), and allowance for the 3 day culture period before the addition of the transduced Jurkat responder line J-TCR-M1. During this 3 day incubation, cell growth will occur and may even be stimulated by certain screened library molecules, but the assay should also accommodate reduction in cell numbers through cytostatic or cytotoxic compounds. With these restrictions in mind, we chose to use 5×104 tumor cells per well of a 96-well plate for assay development.
Given the inherent variation in stimulator melanoma cell numbers after the initial 3 day treatment, it was important to determine the optimal ratio of stimulator cells to responder transduced T cells. We chose a 2: 1 stimulator to responder ratio as empirically giving reproducible results with tested positive controls. Since large excesses of stimulator cells alone (added at the beginning of the assay period) result in significant increases in IL-2 production from the responders (Fig. 2), in principle, some test compounds might give positive signals through differential augmentation of melanoma stimulator cell growth over the T-cell responders, rather than via enhancement of antigen expression per cell. In practice, there is no reason for suspecting such agents will be common, and culture conditions set physical limits to the maximal possible extent of stimulator cell proliferation. If such hypothetical agents were scored as primary hits, they will in any case be rapidly excluded through the first set of secondary screens, as detailed next.
Fig. 2.

IL-2 induction. IL-2 levels produced with a fixed number (2×104) of responder T cells (J-TCR-M1) while varying the number of untreated stimulator tumor cells (MU89). Number of tumor cells on x-axis plotted in log2. Mean and standard deviation of three replicates are shown.
Compound Library Screening
For evaluation of our cell-based assay, we screened 480 known bioactive compounds (Methods section), with results presented in Figure 3. Using the definition of a hit as a compound inducing a twofold signal above the untreated control, we found eight hits in the primary screen (Table 3). A twofold relative increase of IL-2 above the untreated control level is equivalent to 6 standard deviations (of the controls) from the control mean, a highly significant factor.
Fig. 3.

Primary screen results. Known bioactive library −480 compounds are shown as circles, triangles represent IFN-beta treated positive controls, and squares indicate untreated controls. Each point corresponds to a well that has been normalized to the average of the untreated controls for that plate. Dark circles are considered hits, and the IL-2 level relative to untreated controls is listed next to these points.
Table 3.
Hits from Pilot Screen
| Compound | Fold increase | Known inhibitory targets | References |
|---|---|---|---|
| OBAA | 5.8 | Phospholipase A2 | 52,53 |
| Flunarizine | 12.0 | Ca2+-channel blocker | 54 |
| 17-AAG | 7.7 | HSP90 | 55–57 |
| Aphidicolin | 1.9 | DNA polymerase | 58,59 |
| Damnacanthal | 2.1 | p56lck | 60 |
| PMA | 5.6 | PKC activator | 61,62 |
| Dantrolene | 2.3 | Ca2+ release from sarcoplama reticulum | 63 |
| Glyburide | 2.6 | ATP dependent K channels | 64 |
17-AAG, 17-allylaminogeldanamycin; OBAA, 3-(4-octadecyl)benzoylacrylic acid; PMA, phorbol-12-myristate-13 acetate.
Highly toxic compounds, to either cell type involved in the assay, will suppress the IL-2 read-out, sometimes to below baseline levels (as shown for some compounds plotted in Fig. 3). There is also the possibility that some molecules will block the signal transduction pathways involved in the activation of the T cell and subsequent IL-2 production. In turn, such toxic library members may fail to be scored as hits even if they possess inherent antigen upregulation potential. This issue is underscored by the observation that certain compounds within the ICCB known bioactives library were independently identified as upregulators of melanocyte gene expression including topoisomerase inhibitors (doxorubicin, etoposide, and camptothecin) and MEK inhibitors (U0126 and PD98059),25,27 which failed to be identified during our screening. In the case of daunorubicin (chemically very similar to doxorubicin), we have previously confirmed that this compound interferes with the direct coculture assay; however, an enhanced IL-2 signal is elicited if this drug is removed by washing after tumor cell treatment, before exposure of responder T cells.27 This interference could be due to direct T-cell toxicity. Both daunorubicin and doxorubicin are able to induce strong antigen upregulation. Thus, doxorubicin is a false negative, actually able to upregulate antigen expression, but not detected by our screen due to the ability of the compound to inhibit the assay read-out of T-cell IL-2 production. The failure of the MEK inhibitors U0126 and PD98059 to induce signals in our assay is attributable to similar toxic effects or to inhibition of signal transduction (data not shown).
The 480 compounds were screened in 6 plates, each of them containing 80 compounds and controls. The differences among the wells and plates assayed on the same day were minimal (Fig. 3). There is variability from day to day in absolute IL-2 induction; but after normalization (relative to untreated controls), there is very low day-to-day variation in the assay (Fig. 3). Thus, we observed that the assay had good reproducibility yielding similar results among different plates and different assay days.
Secondary Screens
Secondary screens were carried out on the 8 primary hits to further test performance robustness, specificity, and certain functional properties. Initially, for any previously described compound, extensive literature searches should be performed to look for all relevant known bioactivities, especially toward melanocytic cells or T cells. Experimental secondary screens were carried out on primary hits involved (in order of performance): (1) testing for nonspecific T-cell stimulation; (2) extensive dose-response testing in the coculture assay over broad concentration ranges; (3) performing intracellular staining testing multiple melanoma cell lines and using antibodies specific for an additional melanocyte-specific antigen; and (4) testing effects on a Melan-A/MART-1 promoter EGFP reporter cell line.
The published data bases allowed us to identify one of the hits, PMA, as a known T-cell activator that is capable of directly inducing T-cell IL-2 production.48,65 The results of the secondary screen for nonspecific T-cell activators are shown in Figure 4, showing that only PMA caused T-cells stimulation in the absence of tumor. This allowed us to exclude this agent as a “false positive.”
Fig. 4.

Nonspecific T-cell activation. Hits were examined in a secondary screen for nonspecific T-cell activation. Bars indicate the incubation of 2.5×104 T cells alone with the indicated compounds. The concentration of compounds used in this assay are 17-AAG 10 μg/mL, OBAA 25 μg/mL, Aphidicolin 4 μg/mL, Flunarizine 8 μg/mL, Dantrolene 2 μg/mL, Glyburide 4 μg/mL, PMA 2 μg/mL. 17-AAG, 17-allylaminogeldanamycin; OBAA, 3-(4-octadecyl)benzoylacrylic acid; PMA, phorbol-12-myristate-13 acetate.
From published information, none of the other hits were known to be capable of nonspecific T-cell stimulation, in accord with the data in Figure 4. Some of the compounds have previously been shown to affect melanoma cell biology. Flunarizine has been shown to affect melanoma cell motility.66–68 17-AAG has been reported to have clinical effects on B-RAF, an important mutated protein in a majority of melanomas.69–71 In a murine model, aphidicolin was effective against melanoma growth.58 Dantrolene has been shown to alter melanoma cell adhesion.72 The remaining hits were also further evaluated in secondary screens.
To confirm the activity of the hits from the primary screen, these seven hits passing the test for nonspecific T-cell effects were retested in the coculture assay over a wider dose range. Figure 5 shows IL-2 production induced by the various hits. We noted that 17-AAG and aphidicolin are compounds with large IL-2 stimulation effects, about seven- and fivefold, respectively. Two additional compounds flunarizine and OBAA gave more modest stimulation (1.5–2-fold) of IL-2 production by the responding T cells. Finally, dantrolene, damnacanthal, and glyburide did not stimulate significant IL-2 increases, thus failing to confirm the original screen results.
Fig. 5.

Repeat IL-2 ELISA. Evaluation of the hits in a repeat of the tumor T-cell coculture IL-2 ELISA. Note that the x-axes (drug concentrations) and y-axes (IL-2 production levels) cover different ranges to best indicate the activities of individual compounds. Data from a single determination, representative of at least three confirmatory experiments, are shown. ELISA, enzyme-linked immunosorbent assay.
We then tested the ability of the hit compounds to induce antigen increases in a more diverse set of tumors including melanomas and gliomas. In addition, the gp100 antigen that is expressed in melanocytes and gliomas was added to the Melan-A/MART-1 antigen expression tested in the screening cellular assay. Figure 6 illustrates examples of the flow histograms derived from the intracellular staining used to generate the data presented in Table 4. As can be seen by the ability of the compounds to stimulate antigen levels >1.5-fold, some hits clearly outperform others. Again, 17-AAG and aphidicolin are the best performers in this assay, inducing high levels (>2-fold) of antigen compared with untreated controls. Damnacanthal, OBAA, and flunarizine induced significant responses, increasing protein >1.5-fold in most cell lines. Glyburide and dantrolene showed minimal effects on protein expression and were only evaluated on a limited number of cell lines.
Fig. 6.

Intracytoplasmic antigen staining. Example of intracellular staining and flow histograms used to generate data for Table 4. All cells were stained with an antibody to gp100. Thin line represents untreated cells, bold line represents cells treated for 3 days with 17-AAG (1 μg/mL). Numbers represent geometric mean of flow histograms.
Table 4.
Intracellular Staining for Melanocyte Proteins Exposed to Hits from the Known Bioactives Library
| |
|
MU89 |
MALME-3M |
A375 |
MUX |
U-118 |
U-87 |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compounda | μg/mL | MART-1b | gp100 | MART-1 | gp100 | MART-1 | gp100 | MART-1 | gp100 | MART-1 | gp100 | MART-1 | gp100 |
| Control | 0 | 63 | 62 | 98 | 157 | 6 | 16 | 13 | 41 | 9 | 23 | 8 | 39 |
| 17-AAG | 0.5 | 203 | 108 | 138 | 458 | 8 | 37 | 21 | 75 | 16 | 60 | 16 | 90 |
| 5.0 | 142 | 112 | 165 | 540 | 6 | 16 | 19 | 45 | 14 | 53 | 14 | 81 | |
| Control | 0 | 60 | 80 | 65 | 113 | 6 | 16 | 13 | 41 | 9 | 23 | 8 | 39 |
| Aphidicolin | 0.5 | 113 | 159 | 131 | 307 | 22 | 83 | 27 | 86 | 18 | 43 | 19 | 72 |
| 5.0 | 99 | 127 | 19 | 429 | 25 | 145 | 27 | 108 | 17 | 53 | 15 | 71 | |
| Control | 0 | 63 | 71 | 59 | 140 | 4 | 10 | 9 | 21 | 7 | 16 | 5 | 17 |
| Damnacanthal | 3.0 | 22 | 34 | 67 | 133 | 5 | 9 | 10 | 22 | 9 | 19 | 7 | 19 |
| 6.0 | 19 | 43 | 74 | 170 | 8 | 15 | 13 | 31 | 11 | 25 | 6 | 16 | |
| 12.0 | 23 | 66 | 69 | 429 | 10 | 24 | 16 | 34 | 13 | 34 | 9 | 35 | |
| 24.0 | 30 | 109 | 33 | 343 | 8 | 14 | 15 | 12 | 28 | 9 | 29 | ||
| Control | 0 | 78 | 82 | 83 | 202 | 6 | 12 | ||||||
| Dantrolene | 3.0 | 63 | 64 | 85 | 170 | 5 | 10 | ||||||
| 6.0 | 65 | 74 | 90 | 210 | 6 | 13 | |||||||
| 12.0 | 65 | 84 | 90 | 172 | 6 | 14 | |||||||
| 24.0 | 58 | 87 | 85 | 190 | 7 | 18 | |||||||
| Control | 0 | 52 | 50 | 65 | 113 | 4 | 8 | 10 | 23 | 8 | 12 | 7 | 13 |
| Flunarizine | 2 | 57 | 80 | 79 | 141 | 5 | 8 | ||||||
| 4 | 49 | 75 | 73 | 132 | 4 | 9 | |||||||
| 8 | 66 | 107 | 77 | 138 | 6 | 11 | 11 | 29 | 11 | 21 | 7 | 16 | |
| 16 | 153 | 213 | 94 | 198 | 10 | 20 | 19 | 48 | 11 | 24 | 10 | 25 | |
| 32 | 39 | 25 | |||||||||||
| Control | 0 | 40 | 57 | 43 | 106 | 3 | 6 | ||||||
| Glyburide | 3 | 36 | 52 | 50 | 86 | 4 | 8 | ||||||
| 6 | 40 | 48 | 45 | 112 | 5 | 9 | |||||||
| 12 | 34 | 43 | 42 | 86 | 5 | 8 | |||||||
| 24 | 26 | 46 | 44 | 112 | 4 | 10 | |||||||
| Control | 0 | 47 | 64 | 65 | 113 | 4 | 7 | 8 | 16 | 17 | 16 | ||
| OBAA | 25 | 73 | 82 | 100 | 263 | 3 | 8 | 12 | 27 | 20 | 27 | ||
| 50 | 91 | 129 | 135 | 350 | 4 | 9 | 13 | 33 | 34 | 33 | |||
| 100 | 5 | 15 | 16 | 21 | 48 | 21 | |||||||
Bold values indicate an increase of ≥1.5-fold over control with compound treatment.
Treatment with indicated dose of compound was for 3 days.
Geometric mean of intracellular staining using antibodies to MART-1 or gp100 as indicated.
Assaying for stimulation of an EGFP reporter driven by the Melan-A/MART-1 promoter in a cellular system was used as the fourth criterion for demonstration of biological activity of the test compounds. Although some compounds might fail this test in principle, through differing mechanisms of action, this assay was considered useful as a lower-level secondary screen by virtue of its orthogonality with regard to the primary assay. Also, previous experience has shown that promoter enhancement is indeed a common feature of known antigen upregulators, from small molecules to proteins.25–27 The seven hits (excluding PMA) from the primary screen were evaluated over a dose range as shown in Figure 7. Again, the hits separated into three groups based on these results. As for the previous secondary screens, 17-AAG and aphidicolin had the largest impact on EGFP stimulation, inducing three- and fivefold increases, respectively. Four additional compounds (damnacanthal, flunarizine, OBAA, and dantrolene) stimulate EGFP at least twofold. Only glyburide is unable to stimulate significant EGFP increases. It is noteworthy that dantrolene and damnacanthal were active in this assay, but were poorly stimulatory in the IL-2 assay, whereas glyburide failed in both assays.
Fig. 7.

EGFP promoter assay. Evaluation of the ability of hits to upregulate a cell line containing a Melan-A/MART-1 promoter-driven EGFP reporter. Note that the x-axes (drug concentrations) and y-axes (EGFP fluorescent measurement) cover different ranges to best indicate the activities of individual compounds. Data from a single determination, representative of at least three confirmatory experiments, are shown.
Discussion
The identification of immune-targetable antigens in numerous tumors has encouraged more aggressive efforts to implement strategies for successful immunotherapy. Perhaps no tumor type has received more attention than melanoma, if for no other reason than its poor responsiveness to conventional chemotherapy and radiation. However, with each new advance comes recognition of new hurdles to therapeutic success. As our own group noted over a decade ago,73 one of the Achilles' Heels of targeted immunotherapy is the need for the tumor to express the target antigen. Indeed, we and others have shown that tumors have a survival advantage if they express lower levels of both target antigens and the presenting HLA alleles.19 These observations led to efforts to restore and enhance antigen expression of tumors, resulting in the identification of several different drugs that indeed improve immune recognition of melanoma cells, including MEK inhibitors,25 biologically distinct entities such as the cytokine IFN-beta,26 and topoisomerase inhibitors.27 IFN-beta signaling and topoisomerase inhibition can also enhance tumor expression of differentiation antigens and HLA molecules.26,27
These observations led us to design and implement a cell-based screening assay that would allow us reasonable throughput with the potential for discovery of additional biologically active compounds which could be therapeutically used. We have successfully constructed a responder T-cell line that reproducibly responds to tumor cells that express the target differentiation antigen, Melan-A/MART-1.27 As shown in Table 2, this T cell produces quantitatively significant levels of IL-2 when it is stimulated by a cell that presents the cognate HLA-A2 melanocyte peptide EAAGIGILTV.
Using IFN-beta as an internal positive control for antigen induction by the melanoma tumor cell line MU89, we were able to demonstrate that this assay is reliable and reproducible, as indicated by the Z′ score of 0.55. As shown in Figures 1 and 3, the IFN-beta treated tumor cells can be relied on to induce >2-fold increase in IL-2 production. Due to the consistent enhancement of antigen, and the resulting induction of IL-2 by our internal control, IFN-beta, we are able to exclude plates in which the expected induction by IFN-beta is not reliably detected.
The design of our assay could be adapted to evaluate other tumors and T cells. Any TCR for which the sequence, HLA specificity, and peptide are known could be evaluated by using a similar system. Other tumors and the T cells that recognize peptides presented by them could be utilized.
By screening 480 known biologically active compounds, we not only performed a proof-of-principle screening assay, but also demonstrated that most of the compounds identified as hits in the primary screen showed an excellent correlation with a series of secondary screens. Although the use of a known compound library does not allow us to identify any novel compounds, the validity of the assay should allow for future analysis of large compound libraries that include agents with unknown biological properties. Most importantly, though, this screening of a small library has rapidly identified novel bioactivities of previously known molecules, of direct interest for sustained evaluation toward therapeutic applications. The prospect for retargeting of known compounds for use in novel therapies has its own advantages,74 not the least of which is the known toxicity profiles that would allow a shorter time to clinical trial than a compound with no profile in animals or humans.
The goal of our screening assay is to identify compounds that increase tumor recognition by T cells. The assay is designed to identify hits from the primary screen by virtue of the treated tumor's ability to stimulate increased IL-2 synthesis by the responder T cells. The secondary screens are designed to identify compounds that will have the greatest therapeutic potential in a combination immunotherapy protocol. Thus, we should first confirm the hits from the primary screen over a wider dosage range to determine the optimal dosing for each compound, and then determine whether the compounds fulfill additional criteria for favorable clinical usage. Although only the repeat T-cell coculture is a truly analogous assay for efficacy of the compounds at enhancing immune recognition of the tumor cells, there are several orthogonal assays that have been identified with previously selected compounds, including the internal “gold standard,” IFN-beta. Enhanced T-cell recognition can formally be the result of numerous changes in the tumor cells that make them better stimulators of the T cells, including increased MHC antigen expression and prolonged half-life of the antigen-MHC complex on the tumor cell surface, without increasing the actual levels of antigenic protein or increased transcription of the relevant antigens.
The first secondary screen evaluates the activity of the hits on the T cells alone and allows us to eliminate false positives that are not truly increasing tumor cell recognition. The activity of PMA at inducing IL-2 by the Jurkat cells alone eliminated this compound from further evaluation. Compounds failing this test will not be further evaluated in any other secondary screens.
For those compounds that do not stimulate T cells directly, all hits should be tested in a repeat IL-2 assay using a broad dose range to determine the optimal dosing for further testing. Since the primary screen was performed at a single arbitrary dose, and many of the hits have some toxicity at high doses, or may only be marginally active at the original test dose, the retest allows us to confirm the primary hits. Testing the compounds again in the IL-2 assay allowed us to identify optimal doses for use in performing the other secondary screens. Compounds from the primary screen can be assigned priority based on the level of stimulation of tumor recognition achieved in this secondary screen. Compounds failing to repeat the primary screening result would not be followed up with other secondary screens.
It is highly desirable to be able to demonstrate that antigen increases can be achieved with a diverse panel of melanomas, and even nonmelanomas (such as the gliomas we tested), to select drugs that have the widest applicability in a diverse patient population with tumors expressing different antigens and different MHC backgrounds. By the same rationale, the ability to show enhanced expression of other melanocyte differentiation antigens is also highly desirable. Although we would not automatically exclude a candidate hit from further study because it failed to increase antibody staining of antigen in the tumor cells, the logic and feasibility of such testing is shown by the fact that the previously characterized agents IFN-beta,26 MAP-kinase inhibitors25 and topoisomerase inhibitors27 all mediate increased differentiation antigen staining. Further, in each case, this staining enhancement is observed for additional antigens beyond Melan-A/MART-1 (such as gp100). Thus, the first orthogonal secondary screen performed was the measurement of Melan-A/MART-1 and gp100 in additional melanoma cell lines and gliomas in response to the primary hit candidates. An observable increase in gp100 demonstrates that the compounds are able to positively modulate protein levels of distinct melanocyte specific antigens. If the compounds of interest increase antigen levels in multiple melanoma cell lines and two gliomas, the effects of the compounds are clearly not limited to a single melanoma cell type. Thus, Compounds able to upregulate more than one melanoma antigen and have the ability to effect multiple cell lines are given higher priority for follow-up studies.
Secondary screening with assays that are functionally orthogonal to the primary screen is useful, as hits passing such evaluations are assigned a higher probability that their observed effects are not assay artifacts, and are biologically significant. However, it can be difficult to devise truly orthogonal assays to match complex cell-based determinations at the highest levels of read-out. The primary assay we employed measures a cell-based functional result dependent on multiple signaling, processing, presentation, and activation effects. In principle, a pharmacological agent can act at numerous different levels, any of which may contribute to an observed beneficial outcome. In such circumstances, it may be expedient to use convenient orthogonal secondary assays making certain assumptions regarding an agent's mode of action. Although agents failing such a secondary screen may not necessarily lack value, agents passing both robust primary screening and secondary orthogonal assays can be confidently given higher priority in the downstream evaluation pipeline.
These considerations are directly relevant to the reporter assay where EGFP expression is driven by the Melan-A/MART-1 promoter. The orthogonality of this secondary screen in the current context derives from measurement of the modulation of Melan-A/MART-1 promoter activity by candidate compounds, independent of Melan-A/MART-1 protein expression. Increased promoter activity for Melan-A/MART-1 is not necessary for a compound to prove of value, but promoter activity is readily assessed by using our EGFP-linked MART-promoter in both MM96L+ and A375 tumor cells and provides a measure of how the compounds may induce increased T-cell recognition. Compounds passing the other secondary screens, but failing this one, may affect antigen presentation by other mechanisms such as increasing the efficiency of antigen processing, enhancing MHC Class I levels, or stabilizing the antigen/MHC complex. It is also formally possible that a compound may pass this test by a nontranscriptional mechanism, such as mRNA stabilization or direct stabilization of the EGFP reporter.
Nevertheless, compounds passing the EGFP test are assigned conditionally higher ratings, given the associated increased confidence that their biological activities are real. By this reasoning, of hits delivering approximately equal results in the IL-2 coculture assay, those which are also positive for the EGFP secondary screen will be given higher priority. However, hits with exceptional performance in the IL-2 coculture assay will be further evaluated even if they fail the EGFP secondary screen, given the alternative possible functional mechanisms just noted. Lower-level secondary screens such as the EGFP assay are useful discriminators between hits with approximately equal performance in higher-level screens, but are not to be used as absolute exclusion criteria. At the same time, a side-benefit of orthogonal secondary screens may be provision of important clues as to the utility of selected hits in a wider patient population and provide a glimpse at possible functional mechanisms. Decisions about hit-to-lead for follow-up of compounds from this screen will be made from the sum of the priorities they were given in the hierarchy of secondary screens.
The hits identified from the screen fell into five distinct categories: (1) False Positives; (2) False Negatives; (3) Strong Positives; (4) Weak Positives; and (5) Compounds that fail all secondary screens. Not surprisingly, we did uncover a known “false positive,” in PMA, which stimulates T-cell production of IL-2 on its own. Thus, this compound exemplifies a rare, but real set of compounds that are not active in tumor cells directly, but stimulate the T cells used in the assay.
A second category of “false negatives” is not directly revealed by the screening assay itself, but made apparent by virtue of our previous experience. In this regard, our library included certain agents (the MEK inhibitors U0126 and PD9805925 and doxorubicin27) that failed to produce positive assay signals, despite their validated augmentation of antigen expression on the MU89 indicators (and other melanoma cells) in independent determinations. These “false negative” effects are attributable to interference with the cell-based assay. The source of assay interference may be due either to high toxicity of the compounds toward the MU89 melanoma tumor cell line, in which case there are no surviving stimulator cells, or as a result of toxicity to the responding T cells. Toxicity toward the responder T cells could be the consequence of either direct cellular damage, or interference with the IL-2 induction; each of which would result in reduced IL-2 production even if the tumor cells had increased antigen expression. Of note, the compounds that induce antigen expression on tumor cells and allow functional T-cell activation are of particular interest for in vivo testing, as they would be most suitable for a combination immunotherapy protocol that relies on both the tumor cells expressing antigen and the responding T cells' ability to function.
Apart from the false positives and negatives, of primary interest are those compounds that are highly active not only in the screening assay, but also in a series of secondary orthogonal screens, thus indicating their future potential as therapeutics. Among the hits with strong activity are the HSP90 inhibitor 17-AAG (a semi-synthetic derivative of the natural product geldanamycin, itself a member of the broader ansamycin class of antibiotics57) and the antimetabolite aphidicolin (a tetracyclic diterpene antibiotic with inhibitory activity toward certain eukaryotic and viral DNA polymerases.75,76). Both drugs are active not only in the MU89 cells, and not only on Melan-A/MART-1 antigen expression, but also induce gp100 antigen increases in several different melanomas and two gliomas. The genes encoding both melanocyte differentiation antigens, MART-1/Melan-A and gp100 are know to be coordinately regulated at the transcriptional level, likely related to the master regulatory transcription factor MITF-M that has been shown to regulate several melanocyte differentiation genes.34–36 We have previously found both biological and small molecules impacting several melanocyte genes coordinately.25–27 Aphidicolin and 17-AAG also upregulate EGFP levels expressed from the Melan-A/MART-1 promoter, consistent with their mechanisms of activity operating at the transcriptional level, although further studies are required to confirm this hypothesis.
17-AAG is a first-in-class heat shock protein 90 (HSP90) molecular chaperone inhibitor to enter clinical trials.77 17-AAG has shown activity against human cancer in cells in both in vitro models and clinical trials. By inhibiting HSP90, 17-AAG causes the destabilization of several mutant proteins, including B-raf, c-raf and N-Ras.78,79 We have previously shown that decreasing the level of mutant B-RAF (and MAP kinase pathway signaling) increases melanocyte antigen levels.25 Through its role in regulating the conformation, stability, and function of several key oncogenic client proteins, HSP90 seems to be essential in maintaining malignant transformation and in increasing the survival, growth, and invasive potential of cancer cells.80
Aphidicolin is a tetracyclic diterpene antibiotic derived from cultures of Cephalosporium aphidicola, and is a specific reversible inhibitor of DNA polymerase α and δ. It appears that aphidicolin binds reversibly to the enzyme, not to DNA, and that the drug is selectively toxic in S phase.58 We have previously shown that topoisomerase inhibitors, which can block DNA replication similarly to aphidicolin, upregulate melanoma antigen expression.27
There were additionally several compounds that were considerably less active in the primary screen and in some of the orthogonal assays, but the fact that they are active in at least one of the orthogonal assays indicates that they can help to identify additional molecular pathways toward inducing antigen expression and tumor cell differentiation. Some of these compounds may have intrinsic therapeutic application themselves, but even more promisingly, they may represent entire new classes of drugs in which other functionally related members could prove more therapeutically efficacious. Among the drugs with moderate activity are those noted in Table 3, including the phospholipase A2 inhibitor OBAA, the Ca2+ channel blocker flunarizine and the Ca2+ active compound dantrolene, the p56lck inhibitor damnacanthol.
The fact that dantrolene and damnacanthal fail to retest in IL-2 (<2-fold stimulation) and yet have some activity (>2-fold) in the EGFP secondary screen is important in establishing the levels of performance in the secondary screens required for a hit to progress to the follow-up stage. Based on the performance of the hits from the primary screen in the secondary screens, the best hits cause increases of threefold or greater.
Finally, there are those compounds that simply do not pan out on further analysis, indicating that they are another form of “false positive.” Thus, glyburide was identified in the screening assay, but was not active in any of the secondary assays, even at widely varying test doses. Despite the demonstrable reliability and reproducibility of the assay, it is impossible to completely avoid a background level of “noise” that may deliver spurious positive signals from any library member at random intervals. These false signals are easily and rapidly excluded in favor of true positives through routine retesting in secondary screens, as we have shown.
In summary, we have successfully implemented a cell-based screening assay that can be used to identify agents which enhance antigen expression in tumor cells. The identification of two highly active compounds from a series of known biologically active compounds has allowed us to perform follow-up studies that indicate the utility of a series of drugs which could have a positive impact on immune targeting of tumor cells. The ability to insert specific TCRs also offers the possibility of implementing assays for additional tumor-associated antigens in a biological screening assay. Although our assay relied on a 96-well format, we have also implemented a 384-well assay and shown that this too could be implemented for a higher throughput assay for large-scale screening of both known and unknown compounds.
Glossary
Abbreviations
- 17-AAG
17-allylaminogeldanamycin
- DMEM
Dulbecco's modified Eagle's medium
- EGFP
enhanced green fluorescent protein
- ELISA
enzyme-linked immunosorbent assay
- FBS
fetal bovine serum
- HLA
human leukocyte antigen
- ICCB
Institute of Chemistry and Cell Biology
- IFN-beta
interferon-beta
- IL-2
interleukin 2
- MAP
mitogen-activated protein
- MART
melanoma antigen recognized by T cells
- MEK
mitogen-activated protein kinase/extracellularly regulated kinase kinase
- MHC
major histocompatibility complex
- OBAA
3-(4-octadecyl)benzoylacrylic acid
- PMA
phorbol-12-myristate-13 acetate
- RPMI
Roswell Park Memorial Institute
- TCR
T-cell receptor.
Acknowledgments
The authors would like to thank the members of the ICCB Longwood Screening Facility at Harvard Medical School; especially Caroline Shamu, Stewart Rudnicki, and Andrew Daab, for their assistance with our screen and in maintaining and providing the small molecule libraries used in this study. They would also like to thank James L. Riley (University of Pennsylvania, Philadelphia, PA) for assistance in lentiviral transduction of TCRs. This work was supported in part by grants from the NIH: R43-CA86153, R43-CA96271, R43-CA94700, R43-CA128309, and from the International Cancer Research Scholarship.
Author Disclosure Statement
T.J.H., I.S.D., L.B.R., and E.E.N. are employees of and receive compensation from CytoCure. J.T.K. and L.B.R. own shares of CytoCure. T.J.H., I.S.D., and J.T.K. are co-inventors on a patent application for the cell based assay and combination immunotherapy using compounds identified from the pilot screen.
References
- 1.Bandarchi B. Ma L. Navab R. Seth A. Rasty G. From melanocyte to metastatic malignant melanoma. Dermatol Res Pract. 2010:2010. doi: 10.1155/2010/583748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kirkwood JM. Lorigan P. Hersey P. Hauschild A. Robert C. McDermott D, et al. Phase II trial of tremelimumab (CP-675,206) in patients with advanced refractory or relapsed melanoma. Clin Cancer Res. 2010;16:1042–1048. doi: 10.1158/1078-0432.CCR-09-2033. [DOI] [PubMed] [Google Scholar]
- 3.Boon T. Cerottini JC. Van den Eynde B. van der Bruggen P. Van Pel A. Tumor antigens recognized by T lymphocytes. Annu Rev Immunol. 1994;12:337–365. doi: 10.1146/annurev.iy.12.040194.002005. [DOI] [PubMed] [Google Scholar]
- 4.Rosenberg SA. Progress in human tumour immunology and immunotherapy. Nature. 2001;411:380–384. doi: 10.1038/35077246. [DOI] [PubMed] [Google Scholar]
- 5.Mihm MC., Jr Clemente CG. Cascinelli N. Tumor infiltrating lymphocytes in lymph node melanoma metastases: a histopathologic prognostic indicator and an expression of local immune response. Lab Invest. 1996;74:43–47. [PubMed] [Google Scholar]
- 6.Uyttenhove C. Pilotte L. Theate I. Stroobant V. Colau D. Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9:1269–1274. doi: 10.1038/nm934. [DOI] [PubMed] [Google Scholar]
- 7.Willimsky G. Czeh M. Loddenkemper C. Gellermann J. Schmidt K. Wust P, et al. Immunogenicity of premalignant lesions is the primary cause of general cytotoxic T lymphocyte unresponsiveness. J Exp Med. 2008;205:1687–1700. doi: 10.1084/jem.20072016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dunn GP. Old LJ. Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21:137–148. doi: 10.1016/j.immuni.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 9.Rosenberg SA. Sherry RM. Morton KE. Scharfman WJ. Yang JC. Topalian SL, et al. Tumor progression can occur despite the induction of very high levels of self/tumor antigen-specific CD8+ T cells in patients with melanoma. J Immunol. 2005;175:6169–6176. doi: 10.4049/jimmunol.175.9.6169. [DOI] [PubMed] [Google Scholar]
- 10.Cormier JN. Panelli MC. Hackett JA. Bettinotti MP. Mixon A. Wunderlich J, et al. Natural variation of the expression of HLA and endogenous antigen modulates CTL recognition in an in vitro melanoma model. Int J Cancer. 1999;80:781–790. doi: 10.1002/(sici)1097-0215(19990301)80:5<781::aid-ijc24>3.0.co;2-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cormier J. Hijazi Y. Abati A. Fetsch P. Bettinotti M. Steinberg S, et al. Heterogeneous expression of melanoma-associated antigens and HLA-A2 in metastatic melanoma in vivo. Int J Cancer. 1998;75:517–524. doi: 10.1002/(sici)1097-0215(19980209)75:4<517::aid-ijc5>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 12.de Vries TJ. Fourkour A. Wobbes T. Verkroost G. Ruiter DJ. van Muijen GN. Heterogeneous expression of immunotherapy candidate proteins gp100, MART-1, and tyrosinase in human melanoma cell lines and in human melanocytic lesions. Cancer Res. 1997;57:3223–3229. [PubMed] [Google Scholar]
- 13.Jager E. Ringhoffer M. Altmannsberger M. Arand M. Karbach J. Jager D, et al. Immunoselection in vivo: independent loss of MHC class I and melanocyte differentiation antigen expression in metastatic melanoma. Int J Cancer. 1997;71:142–147. doi: 10.1002/(sici)1097-0215(19970410)71:2<142::aid-ijc3>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 14.Anichini A. Molla A. Mortarini R. Tragni G. Bersani I. Di Nicola M, et al. An expanded peripheral T cell population to a cytotoxic T lymphocyte (CTL)-defined, melanocyte-specific antigen in metastatic melanoma patients impacts on generation of peptide-specific CTLs but does not overcome tumor escape from immune surveillance in metastatic lesions. J Exp Med. 1999;190:651–667. doi: 10.1084/jem.190.5.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Riker A. Cormier J. Panelli M. Kammula U. Wang E. Abati A, et al. Immune selection after antigen-specific immunotherapy of melanoma. Surgery. 1999;126:112–120. [PubMed] [Google Scholar]
- 16.Thurner B. Haendle I. Roder C. Dieckmann D. Keikavoussi P. Jonuleit H, et al. Vaccination with mage-3A1 peptide-pulsed mature, monocyte-derived dendritic cells expands specific cytotoxic T cells and induces regression of some metastases in advanced stage IV melanoma. J Exp Med. 1999;190:1669–1678. doi: 10.1084/jem.190.11.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Witte MA. Jorritsma A. Kaiser A. van den Boom MD. Dokter M. Bendle GM, et al. Requirements for effective antitumor responses of TCR transduced T cells. J Immunol. 2008;181:5128–5136. doi: 10.4049/jimmunol.181.7.5128. [DOI] [PubMed] [Google Scholar]
- 18.Kurnick JT. Ramirez-Montagut T. Boyle LA. Andrews DM. Pandolfi F. Durda PJ, et al. A novel autocrine pathway of tumor escape from immune recognition: melanoma cell lines produce a soluble protein that diminishes expression of the gene encoding the melanocyte lineage melan-A/MART-1 antigen through down-modulation of its promoter. J Immunol. 2001;167:1204–1211. doi: 10.4049/jimmunol.167.3.1204. [DOI] [PubMed] [Google Scholar]
- 19.Durda PJ. Dunn IS. Rose LB. Butera D. Benson EM. Pandolfi F, et al. Induction of “antigen silencing” in melanomas by oncostatin M: down-modulation of melanocyte antigen expression. Mol Cancer Res. 2003;1:411–419. [PubMed] [Google Scholar]
- 20.Jager E. Ringhoffer M. Karbach J. Arand M. Oesch F. Knuth A. Inverse relationship of melanocyte differentiation antigen expression in melanoma tissues and CD8+ cytotoxic-T-cell responses: evidence for immunoselection of antigen-loss variants in vivo. Int J Cancer. 1996;66:470–476. doi: 10.1002/(SICI)1097-0215(19960516)66:4<470::AID-IJC10>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 21.Lozupone F. Rivoltini L. Luciani F. Venditti M. Lugini L. Cova A, et al. Adoptive transfer of an anti-MART-1(27–35)-specific CD8* T cell clone leads to immunoselection of human melanoma antigen-loss variants in SCID mice. Eur J Immunol. 2003;33:556–566. doi: 10.1002/immu.200310032. [DOI] [PubMed] [Google Scholar]
- 22.Topalian SL. Kasid A. Rosenberg SA. Immunoselection of a human melanoma resistant to specific lysis by autologous tumor-infiltrating lymphocytes. Possible mechanisms for immunotherapeutic failures. J Immunol. 1990;144:4487–4495. [PubMed] [Google Scholar]
- 23.Bitton RJ. Cancer vaccines: a critical review on clinical impact. Curr Opin Mol Ther. 2004;6:17–26. [PubMed] [Google Scholar]
- 24.Yamshchikov GV. Mullins DW. Chang CC. Ogino T. Thompson L. Presley J, et al. Sequential immune escape and shifting of T cell responses in a long-term survivor of melanoma. J Immunol. 2005;174:6863–6871. doi: 10.4049/jimmunol.174.11.6863. [DOI] [PubMed] [Google Scholar]
- 25.Kono M. Dunn IS. Durda PJ. Butera D. Rose LB. Haggerty TJ, et al. Role of the mitogen-activated protein kinase signaling pathway in the regulation of human melanocytic antigen expression. Mol Cancer Res. 2006;4:779–792. doi: 10.1158/1541-7786.MCR-06-0077. [DOI] [PubMed] [Google Scholar]
- 26.Dunn IS. Haggerty TJ. Kono M. Durda PJ. Butera D. Macdonald DB, et al. Enhancement of human melanoma antigen expression by IFN-beta. J Immunol. 2007;179:2134–2142. doi: 10.4049/jimmunol.179.4.2134. [DOI] [PubMed] [Google Scholar]
- 27.Haggerty TJ. Dunn IS. Rose LB. Newton EE. Martin S. Riley JL, et al. Topoisomerase inhibitors modulate expression of melanocytic antigens and enhance T cell recognition of tumor cells. Cancer Immunol Immunother. 2011;60:133–144. doi: 10.1007/s00262-010-0926-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kholmanskikh O. van Baren N. Brasseur F. Ottaviani S. Vanacker J. Arts N, et al. Interleukins 1alpha and 1beta secreted by some melanoma cell lines strongly reduce expression of MITF-M and melanocyte differentiation antigens. Int J Cancer. 2010;127:1625–1636. doi: 10.1002/ijc.25182. [DOI] [PubMed] [Google Scholar]
- 29.Kawakami Y. Rosenberg SA. Immunobiology of human melanoma antigens MART-1 and gp100 and their use for immuno-gene therapy. Int Rev Immunol. 1997;14:173–192. doi: 10.3109/08830189709116851. [DOI] [PubMed] [Google Scholar]
- 30.Rivoltini L. Kawakami Y. Sakaguchi K. Southwood S. Sette A. Robbins PF, et al. Induction of tumor-reactive CTL from peripheral blood and tumor-infiltrating lymphocytes of melanoma patients by in vitro stimulation with an immunodominant peptide of the human melanoma antigen MART-1. J Immunol. 1995;154:2257–2265. [PubMed] [Google Scholar]
- 31.Stevens EJ. Jacknin L. Robbins PF. Kawakami Y. el Gamil M. Rosenberg SA, et al. Generation of tumor-specific CTLs from melanoma patients by using peripheral blood stimulated with allogeneic melanoma tumor cell lines. Fine specificity and MART-1 melanoma antigen recognition. J Immunol. 1995;154:762–771. [PubMed] [Google Scholar]
- 32.Zhai Y. Yang JC. Kawakami Y. Spiess P. Wadsworth SC. Cardoza LM, et al. Antigen-specific tumor vaccines. Development and characterization of recombinant adenoviruses encoding MART1 or gp100 for cancer therapy. J Immunol. 1996;156:700–710. [PubMed] [Google Scholar]
- 33.Cheli Y. Ohanna M. Ballotti R. Bertolotto C. Fifteen-year quest for microphthalmia-associated transcription factor target genes. Pigment Cell Melanoma Res. 2010;23:27–40. doi: 10.1111/j.1755-148X.2009.00653.x. [DOI] [PubMed] [Google Scholar]
- 34.Goding C. Fisher D. Regulation of melanocyte differentiation and growth. Cell Growth Differ. 1997;8:935–940. [PubMed] [Google Scholar]
- 35.Levy C. Khaled M. Fisher DE. MITF: master regulator of melanocyte development and melanoma oncogene. Trends Mol Med. 2006;12:406–414. doi: 10.1016/j.molmed.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 36.Mitra D. Fisher DE. Transcriptional regulation in melanoma. Hematol Oncol Clin North Am. 2009;23:447–465, viii. doi: 10.1016/j.hoc.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 37.Widlund HR. Fisher DE. Microphthalamia-associated transcription factor: a critical regulator of pigment cell development and survival. Oncogene. 2003;22:3035–3041. doi: 10.1038/sj.onc.1206443. [DOI] [PubMed] [Google Scholar]
- 38.Dietrich PY. Le Gal FA. Dutoit V. Pittet MJ. Trautman L. Zippelius A, et al. Prevalent role of TCR alpha-chain in the selection of the preimmune repertoire specific for a human tumor-associated self-antigen. J Immunol. 2003;170:5103–5109. doi: 10.4049/jimmunol.170.10.5103. [DOI] [PubMed] [Google Scholar]
- 39.Jorritsma A. Gomez-Eerland R. Dokter M. van de Kasteele W. Zoet YM. Doxiadis II, et al. Selecting highly affine and well-expressed TCRs for gene therapy of melanoma. Blood. 2007;110:3564–3572. doi: 10.1182/blood-2007-02-075010. [DOI] [PubMed] [Google Scholar]
- 40.Dull T. Zufferey R. Kelly M. Mandel RJ. Nguyen M. Trono D, et al. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72:8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scholten KB. Kramer D. Kueter EW. Graf M. Schoedl T. Meijer CJ, et al. Codon modification of T cell receptors allows enhanced functional expression in transgenic human T cells. Clin Immunol. 2006;119:135–145. doi: 10.1016/j.clim.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 42.Varela-Rohena A. Molloy PE. Dunn SM. Li Y. Suhoski MM. Carroll RG, et al. Control of HIV-1 immune escape by CD8 T cells expressing enhanced T-cell receptor. Nat Med. 2008;14:1390–1395. doi: 10.1038/nm.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cohen CJ. Li YF. El-Gamil M. Robbins PF. Rosenberg SA. Morgan RA. Enhanced antitumor activity of T cells engineered to express T-cell receptors with a second disulfide bond. Cancer Res. 2007;67:3898–3903. doi: 10.1158/0008-5472.CAN-06-3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kuball J. Dossett ML. Wolfl M. Ho WY. Voss RH. Fowler C, et al. Facilitating matched pairing and expression of TCR chains introduced into human T cells. Blood. 2007;109:2331–2338. doi: 10.1182/blood-2006-05-023069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Parry RV. Rumbley CA. Vandenberghe LH. June CH. Riley JL. CD28 and inducible costimulatory protein Src homology 2 binding domains show distinct regulation of phosphatidylinositol 3-kinase, Bcl-xL, and IL-2 expression in primary human CD4 T lymphocytes. J Immunol. 2003;171:166–174. doi: 10.4049/jimmunol.171.1.166. [DOI] [PubMed] [Google Scholar]
- 46.Aarnoudse CA. Kruse M. Konopitzky R. Brouwenstijn N. Schrier PI. TCR reconstitution in Jurkat reporter cells facilitates the identification of novel tumor antigens by cDNA expression cloning. Int J Cancer. 2002;99:7–13. doi: 10.1002/ijc.10317. [DOI] [PubMed] [Google Scholar]
- 47.Ohashi PS. Mak TW. Van den Elsen P. Yanagi Y. Yoshikai Y. Calman AF, et al. Reconstitution of an active surface T3/T-cell antigen receptor by DNA transfer. Nature. 1985;316:606–609. doi: 10.1038/316606a0. [DOI] [PubMed] [Google Scholar]
- 48.Weiss A. Wiskocil RL. Stobo JD. The role of T3 surface molecules in the activation of human T cells: a two-stimulus requirement for IL 2 production reflects events occurring at a pre-translational level. J Immunol. 1984;133:123–128. [PubMed] [Google Scholar]
- 49.An WF. Tolliday NJ. Introduction: cell-based assays for high-throughput screening. Methods Mol Biol. 2009;486:1–12. doi: 10.1007/978-1-60327-545-3_1. [DOI] [PubMed] [Google Scholar]
- 50.Inglese J. Johnson RL. Simeonov A. Xia M. Zheng W. Austin CP, et al. High-throughput screening assays for the identification of chemical probes. Nat Chem Biol. 2007;3:466–479. doi: 10.1038/nchembio.2007.17. [DOI] [PubMed] [Google Scholar]
- 51.Zhang JH. Chung TD. Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 52.Kohler T. Heinisch M. Kirchner M. Peinhardt G. Hirschelmann R. Nuhn P. Phospholipase A2 inhibition by alkylbenzoylacrylic acids. Biochem Pharmacol. 1992;44:805–813. doi: 10.1016/0006-2952(92)90419-j. [DOI] [PubMed] [Google Scholar]
- 53.Miao JY. Kaji K. Hayashi H. Araki S. Inhibitors of phospholipase promote apoptosis of human endothelial cells. J Biochem. 1997;121:612–618. doi: 10.1093/oxfordjournals.jbchem.a021629. [DOI] [PubMed] [Google Scholar]
- 54.Godfraind T. Miller R. Wibo M. Calcium antagonism and calcium entry blockade. Pharmacol Rev. 1986;38:321–416. [PubMed] [Google Scholar]
- 55.Kamal A. Thao L. Sensintaffar J. Zhang L. Boehm MF. Fritz LC, et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425:407–410. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- 56.Scalzo S. Gengaro A. Boccoli G. Masciulli R. Giannella G. Salvo G, et al. Primary hypothyroidism associated with interleukin-2 and interferon alpha-2 therapy of melanoma and renal carcinoma. Eur J Cancer. 1990;26:1152–1156. doi: 10.1016/0277-5379(90)90275-x. [DOI] [PubMed] [Google Scholar]
- 57.Schulte TW. Neckers LM. The benzoquinone ansamycin 17-allylamino-17-demethoxygeldanamycin binds to HSP90 and shares important biologic activities with geldanamycin. Cancer Chemother Pharmacol. 1998;42:273–279. doi: 10.1007/s002800050817. [DOI] [PubMed] [Google Scholar]
- 58.O'Dwyer PJ. Moyer JD. Suffness M. Harrison SD., Jr Cysyk R. Hamilton TC, et al. Antitumor activity and biochemical effects of aphidicolin glycinate (NSC 303812) alone and in combination with cisplatin in vivo. Cancer Res. 1994;54:724–729. [PubMed] [Google Scholar]
- 59.Levenson V. Hamlin JL. A general protocol for evaluating the specific effects of DNA replication inhibitors. Nucleic Acids Res. 1993;21:3997–4004. doi: 10.1093/nar/21.17.3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Faltynek CR. Schroeder J. Mauvais P. Miller D. Wang S. Murphy D, et al. Damnacanthal is a highly potent, selective inhibitor of p56lck tyrosine kinase activity. Biochemistry. 1995;34:12404–12410. doi: 10.1021/bi00038a038. [DOI] [PubMed] [Google Scholar]
- 61.Ryves WJ. Evans AT. Olivier AR. Parker PJ. Evans FJ. Activation of the PKC-isotypes alpha, beta 1, gamma, delta and epsilon by phorbol esters of different biological activities. FEBS Lett. 1991;288:5–9. doi: 10.1016/0014-5793(91)80989-g. [DOI] [PubMed] [Google Scholar]
- 62.Castagna M. Takai Y. Kaibuchi K. Sano K. Kikkawa U. Nishizuka Y. Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J Biol Chem. 1982;257:7847–7851. [PubMed] [Google Scholar]
- 63.Song SK. Karl IE. Ackerman JJ. Hotchkiss RS. Increased intracellular Ca2+: a critical link in the pathophysiology of sepsis? Proc Natl Acad Sci USA. 1993;90:3933–3937. doi: 10.1073/pnas.90.9.3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Feldman JM. Glyburide: a second-generation sulfonylurea hypoglycemic agent. History, chemistry, metabolism, pharmacokinetics, clinical use and adverse effects. Pharmacotherapy. 1985;5:43–62. doi: 10.1002/j.1875-9114.1985.tb03404.x. [DOI] [PubMed] [Google Scholar]
- 65.Manger B. Hardy KJ. Weiss A. Stobo JD. Differential effect of cyclosporin A on activation signaling in human T cell lines. J Clin Invest. 1986;77:1501–1506. doi: 10.1172/JCI112464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bellelli A. Camboni C. de Luca G. Materazzi M. Mattioni M. Sezzi ML, et al. In vitro and in vivo enhancement of vincristine antitumor activity on B16 melanoma cells by calcium antagonist flunarizine. Oncology. 1987;44:17–23. doi: 10.1159/000226436. [DOI] [PubMed] [Google Scholar]
- 67.Hofmann-Wellenhof R. Fink-Puches R. Smolle J. Helige C. Tritthart HA. Kerl H. Correlation of melanoma cell motility and invasion in vitro. Melanoma Res. 1995;5:311–319. doi: 10.1097/00008390-199510000-00003. [DOI] [PubMed] [Google Scholar]
- 68.Sezzi ML. Zupi G. De Luca G. Materazzi M. Bellelli L. Effects of a calcium-antagonist (flunarizine) on the in vitro growth of B16 mouse melanoma cells. Anticancer Res. 1984;4:229–234. [PubMed] [Google Scholar]
- 69.Babchia N. Calipel A. Mouriaux F. Faussat AM. Mascarelli F. 17-AAG and 17-DMAG-induced inhibition of cell proliferation through B-Raf downregulation in WT B-Raf-expressing uveal melanoma cell lines. Invest Ophthalmol Vis Sci. 2008;49:2348–2356. doi: 10.1167/iovs.07-1305. [DOI] [PubMed] [Google Scholar]
- 70.Banerji U. Affolter A. Judson I. Marais R. Workman P. BRAF and NRAS mutations in melanoma: potential relationships to clinical response to HSP90 inhibitors. Mol Cancer Ther. 2008;7:737–739. doi: 10.1158/1535-7163.MCT-08-0145. [DOI] [PubMed] [Google Scholar]
- 71.Solit DB. Osman I. Polsky D. Panageas KS. Daud A. Goydos JS, et al. Phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with metastatic melanoma. Clin Cancer Res. 2008;14:8302–8307. doi: 10.1158/1078-0432.CCR-08-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Smith TW. Menter DG. Nicholson GL. McIntire LV. Regulation of melanoma cell adhesion stabilization to fibronectin. Melanoma Res. 1996;6:351–362. doi: 10.1097/00008390-199610000-00002. [DOI] [PubMed] [Google Scholar]
- 73.Ramirez-Montagut T. Andrews DM. Ihara A. Pervaiz S. Pandolfi F. Van Den Elsen PJ, et al. Melanoma antigen recognition by tumour-infiltrating T lymphocytes (TIL): effect of differential expression of melan-A/MART-1. Clin Exp Immunol. 2000;119:11–18. doi: 10.1046/j.1365-2249.2000.01089.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ashburn TT. Thor KB. Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discov. 2004;3:673–683. doi: 10.1038/nrd1468. [DOI] [PubMed] [Google Scholar]
- 75.Ikegami S. Taguchi T. Ohashi M. Oguro M. Nagano H. Mano Y. Aphidicolin prevents mitotic cell division by interfering with the activity of DNA polymerase-alpha. Nature. 1978;275:458–460. doi: 10.1038/275458a0. [DOI] [PubMed] [Google Scholar]
- 76.Pedrali-Noy G. Spadari S. Effect of aphidicolin on viral and human DNA polymerases. Biochem Biophys Res Commun. 1979;88:1194–1202. doi: 10.1016/0006-291x(79)91106-9. [DOI] [PubMed] [Google Scholar]
- 77.Porter JR. Fritz CC. Depew KM. Discovery and development of Hsp90 inhibitors: a promising pathway for cancer therapy. Curr Opin Chem Biol. 2010;14:412–420. doi: 10.1016/j.cbpa.2010.03.019. [DOI] [PubMed] [Google Scholar]
- 78.da Rocha Dias S. Friedlos F. Light Y. Springer C. Workman P. Marais R. Activated B-RAF is an Hsp90 client protein that is targeted by the antican-cer drug 17-allylamino-17-demethoxygeldanamycin. Cancer Res. 2005;65:10686–10691. doi: 10.1158/0008-5472.CAN-05-2632. [DOI] [PubMed] [Google Scholar]
- 79.Grbovic OM. Basso AD. Sawai A. Ye Q. Friedlander P. Solit D, et al. V600E B-Raf requires the Hsp90 chaperone for stability and is degraded in response to Hsp90 inhibitors. Proc Natl Acad Sci USA. 2006;103:57–62. doi: 10.1073/pnas.0609973103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Whitesell L. Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]