

Abstract
The ability of prions to infect some species and not others is determined by the transmission barrier. This unexplained phenomenon has led to the belief that certain species were not susceptible to transmissible spongiform encephalopathies (TSEs) and therefore represented negligible risk to human health if consumed. Using the protein misfolding cyclic amplification (PMCA) technique, we were able to overcome the species barrier in rabbits, which have been classified as TSE resistant for four decades. Rabbit brain homogenate, either unseeded or seeded in vitro with disease-related prions obtained from different species, was subjected to serial rounds of PMCA. De novo rabbit prions produced in vitro from unseeded material were tested for infectivity in rabbits, with one of three intracerebrally challenged animals succumbing to disease at 766 d and displaying all of the characteristics of a TSE, thereby demonstrating that leporids are not resistant to prion infection. Material from the brain of the clinically affected rabbit containing abnormal prion protein resulted in a 100% attack rate after its inoculation in transgenic mice overexpressing rabbit PrP. Transmissibility to rabbits (>470 d) has been confirmed in 2 of 10 rabbits after intracerebral challenge. Despite rabbits no longer being able to be classified as resistant to TSEs, an outbreak of “mad rabbit disease” is unlikely.
Keywords: in vitro replication, scrapie, transmissible spongiform encephalopathy
Transmissible spongiform encephalopathies (TSEs), or prion diseases, are infectious neurodegenerative disorders affecting humans and animals that are invariably fatal and with no currently available treatments for prevention or cure. The misfolded version (PrPSc) of the host cellular prion protein (PrPC) is the infectious agent (1), but additional fundamental aspects of TSEs, such as what dictates the generation of strains and the transmission barrier, remain elusive. The species barrier determines the ability of certain prion strains to infect some species but not others. Unsuccessful experimental challenges to overcome this species barrier and lack of documented natural TSE cases in certain families, including leporids, equids, and canids, have led to the belief that they are resistant to TSEs. Several experimental approaches have been used in an attempt to overcome the species barrier and to reduce incubation times. These include the use of transgenic mice, which overexpress PrPC, making them more susceptible to certain prion infections when compared with the original species, and intracerebral instead of oral inoculation, with the former being invariably more efficient (2). Such methodologies were the only way to investigate the species barrier even though they differ from natural transmission.
The protein misfolding cyclic amplification (PMCA) technique (3) is a powerful in vitro tool that mimics the in vivo conversion process of PrPC to PrPSc but with accelerated kinetics, amplifying minute quantities of any PrPSc present in a sample in an exponential fashion (4–7). This technique, which considerably reduces the protracted incubation periods, is potentially very important in the prion field in general (8–10), and more specifically, for the study of the transmission barrier (5, 11). Using our experience in replicating prions in vitro that can subsequently be used to infect susceptible hosts (9,11,12–16), we wanted to determine whether the lack of any documented natural cases or successful in vivo challenges was sufficient to justify rabbits being considered resistant to prion disease. We chose the rabbit as a TSE-resistant model because it is a laboratory animal, is commonly eaten by humans, is commercially reared, and is abundant in wild populations occuring worldwide and cohabiting with potential TSEs carriers, such as sheep and cattle in Europe, and deer in the Unites States and Canada.
With PMCA, we have shown, in rabbits, that a species previously classified as resistant to prion disease is both susceptible to and capable of developing clinically transmissible prion disease.
Results
In Vitro Generation of Rabbit PrPres.
Unseeded normal rabbit brain homogenate as well as normal rabbit brain homogenates seeded in vitro with different scrapie mouse adapted prion strains (ME7, RML, 22F, 79A, 139A, and 22L), sheep scrapie (SSBP/1 and CH1641), bovine spongiform encephalopathy (BSE), and chronic wasting disease (CWD) were subjected to serial automated PMCA (saPMCA) in an attempt to compare the ability of generating rabbit PrPres (RaPrPres). Bearing in mind that saPMCA is not a quantitative technique, rabbit PrP appeared to be quite susceptible to protein misfolding, as all seeds were able to generate proteinase-K (PK) resistant RaPrPres by round 7 and the unseeded homogenate produced RaPrPres by round 13, suggesting that it may be transmissible in vivo (Table 1 and Fig. 1).
Table 1.
Rounds (R1-R14) of serial automated PMCA using rabbit brain homogenate as substrate
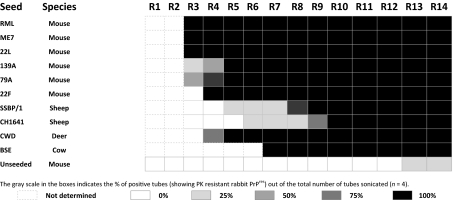 |
Fig. 1.

Biochemical analysis of different rabbit PrPres (RaPrPres) generated in vitro by serial automated PMCA (saPMCA) using a New Zealand White rabbit brain homogenate as substrate. Rabbit brain homogenates seeded with different prion strains (mouse: RML, ME7, 22L, 139A, 79A and 22F, mule deer: CWD, cattle: BSE, sheep: SSPB/1 and CH1641) or unseeded (de novo) were subjected to saPMCA. Seeded samples from round 10 and the unseeded sample from round 20 were digested with 100 μg/mL of proteinase K (PK) and analyzed by Western blot using monoclonal antibody D18. Three differential electrophoretic migration patterns are shown, depending on the seed that was used: (i) the pattern for the unglycosylated band that migrated the farthest (18–19 Kd, sheep and mouse adapted scrapie strains); (ii) the intermediate migration pattern for the same band (19–20 Kd, BSE); and (iii) the migration pattern for the unglycosylated band that migrated the least (20-21 Kd, CWD). RML and ME7 RaPrPres samples are used in all blots as reference for determining the different electrophoretic migration patterns. Control, normal rabbit brain homogenate.
On this basis, we selected saPMCA product from the unseeded normal rabbit brain homogenate, which we refer to as de novo, and the product from the homogenate seeded with the well-characterized ME7 scrapie strain, a mouse adapted prion strain that has never been transmitted successfully in previous challenges to rabbits (17), as challenge inocula in vivo. Both in vitro-derived RaPrPres products were easily amplified further in vitro (Fig. S1).
Infectivity Studies.
Ten 3-mo-old male New Zealand White rabbits (Hsdlf:NZW, Harlan, The Netherlands), of identical PrP genotype, were randomly divided into three groups and challenged intracerebrally with either de novo RaPrPres (n = 3), ME7 RaPrPres (n = 3) or original (murine) ME7 mouse brain homogenate (n = 4). Negative control material was obtained from three additional unchallenged New Zealand White rabbits. After 766 dpi, one animal from the group challenged with de novo RaPrPres developed clinical neurological signs, principally hind limb ataxia and dullness (Movies S1 and S2) and died 4 d later.
Biochemical analysis of the brain by Western blotting (WB) confirmed the presence of PrPSc (Fig. 2) showing characteristic bands with different glycosylation and electrophoretic migration patterns compared with other known PrPSc profiles (Fig. 2A). This in vivo pattern was very similar to that previously observed in the material generated in vitro (Fig. 2B). To characterize the strain generated in vivo, we performed comparative tests with two other prion strains: RML and ME7. The de novo RaPrPSc showed a greater resistance to PK treatment and guanidine denaturation (Fig. 3). Histological examination of the brain showed vacuolation of the neuropil and the cytoplasm of neurons. Immunohistochemistry (IHC) showed astrocytosis and the presence of PrPSc, and PET-blot analysis also revealed PrPres (PK resistant PrP). These findings were strikingly similar to those observed in classical cases of TSEs (Fig. 4).
Fig. 2.

Comparative electrophoretic patterns from different prion strains. (A) Brain homogenate from de novo RaPrPSc inoculated rabbit was compared biochemically with the most typical prion strains from different species. To have a clear reference and accurate comparison of the different electrophoretic migration patterns, de novo RaPrPSc was run three times (arrows). De novo RaPrPSc shows unique biochemical characteristics determined by electrophoretic migration pattern and glycosylation profile in that the unglycosylated band migrates further compared with all other strains with the exception of sporadic Creutzfeldt-Jakob disease (sCJD). However, sCJD can be easily distinguished from the RaPrPSc strain because of its glycosylation profile and the specific migration of the di-glycosylation band, which does not migrate as far. (B) In vitro generated de novo and ME7 RaPrPres (lanes 1 and 2, respectively) are biochemically compared with de novo RaPrPSc generated in vivo (lane 3). Lane 4 shows the biochemical pattern of RaPrPres generated in vitro by PMCA, using in vivo de novo RaPrPSc as seed. Although not identical, the in vivo pattern (lane 3) was very similar to that previously observed in the material generated in vitro (lanes 1, 2, and 4). All samples were digested with 100 μg/mL of proteinase K and were analyzed by Western blot using monoclonal antibody D18. Curved arrows indicate in vitro passage by PMCA. Control, normal rabbit brain homogenate.
Fig. 3.

Proteinase K and guanidine denaturation studies using an in vivo rabbit prion strain. (A) De novo RaPrPSc inoculated rabbit brain homogenate was subjected to several different PK concentrations (80–16,000 μg/mL) at 42 °C for 1 h. ME7 and RML, two well-characterized prion strains, were used as reference. Although the mouse adapted scrapie strains showed a similar PK resistance, the rabbit prion strain showed a much higher resistance to PK treatment. (B) De novo RaPrPSc inoculated rabbit brain homogenate was denatured with different guanidine (GdHCl) concentrations (1.5–4 mol/L) and then subjected to standard Proteinase K (PK) digestion. ME7 and RML prion strains were used as reference. The rabbit prion showed a greater resistance to guanidine denaturation compared with RML and ME7, which had a low or intermediate resistance respectively. Graphs represent an evaluation of Western blots by densitometric analysis from three independent experiments (mean ± SE). All samples were analyzed by Western blot using monoclonal antibody D18.
Fig. 4.

Histological and immunohistochemical studies. Sections of midbrain from a rabbit challenged with de novo RaPrPSc (A, C, E, and G) and a negative control unchallenged rabbit (B, D, F, and H). Hematoxylin and eosin–stained sections showed vacuolation, predominantly in the neuropil of the brain of the challenged rabbit (A) but none in the negative control animal (B). Immunohistochemistry using antibodies to GFAP (glial fibrilary acidic protein) and abnormal prion protein (2G11) showed respectively astrocytosis (C, brown pigment) and positive labeling of PrPSc (E, brown pigment) in the brain of the challenged rabbit. Neither astrocytosis (D) nor PrPSc (F) was found in the brain of the negative control rabbit. PET-Blot analysis using monoclonal antibody SAF84 for PrPSc was positive in the challenged rabbit (G, black pigment) but was absent in the negative control (H).
The ability of RaPrPSc to infect cells in vitro was tested using a mouse PrP knockout cell line (CF10) (18) expressing rabbit PrPC. The cells were exposed to brain homogenate derived from the clinically positive rabbit inoculated with de novo RaPrPSc, passaged five times, and analyzed for PrPSc. PrPSc, probably representing de novo RaPrPSc from the initial inoculum, was detected at passages 0 and 1 but not at passages 3, 5 (Fig. S2A), and 12. The general susceptibility of the cells to prion infection was confirmed by the ability of CF10 cells to produce PrPSc over multiple passages following exposure to 22L mouse scrapie infectivity (Fig. S2B). Thus, the inability of de novo RaPrPSc to trigger PrPSc formation suggested that it was unable to persistently infect the cells.
After 818 d post inoculation (dpi), one animal challenged with ME7 RaPrPres died without clinical signs 1 day after being moved to a new cage. Histological and biochemical analysis of the brain from this animal failed to show any lesions typical of TSE or PrPsc. However, PMCA amplification of this sample using normal rabbit brain homogenate as a substrate revealed glycosylation bands and electrophoretic migration patterns characteristic of a TSE (Fig. S3). PMCA data generated using serially diluted de novo RaPrPSc as a control showed the levels from the ME7 RaPrPSc challenged rabbit to be 103 to 104 times less than the de novo RaPrPSc case (Fig. S3). This low quantity could explain the failure of IHC, WB or Pet-Blot to detect PrPSc. Two additional rabbits had to be culled because of intercurrent diseases, one rabbit from the ME7 group and one from the ME7 RaPrPSc group, at 1,078 and 1,085 dpi, respectively; both were negative for PrPSc expression by WB and IHC.
At the time of writing (∼1,380 dpi), all of the remaining rabbits were clinically healthy and devoid of any signs of TSE infection.
Transmission Studies.
Three 8-wk-old male transgenic mice overexpressing rabbit PrP (raPrPTg) were challenged intracerebrally with a 1% (wt/vol) brain homogenate prepared from the clinically affected rabbit containing abnormal prion protein. Five raPrPTg mice were inoculated with 1% (wt/vol) healthy brain homogenate as negative controls. The three prion-inoculated raPrPTg mice started showing clinical prion disease at a mean (± SEM) of 266 ± 7 dpi and were killed at 253, 262, and 283 dpi. Biochemical analysis of their brains by WB confirmed the presence of PrPSc with similar characteristic bands seen in the de novo RaPrPSc sample (Fig. 5A). The raPrPTg mice used as negative controls remain healthy after 350 dpi, with the exception of one animal dying at 281 dpi, but showing no PrPSc by WB.
Fig. 5.

Electrophoretic patterns of rabbit and rabbit PrP over-expressing transgenic (raPrPTg) mouse adapted de novo RaTgPrPSc strains. (A) Brain homogenate from de novo RaPrPSc-inoculated rabbit (lanes 1 and 5) was compared biochemically with three brain homogenates from de novo RaPrPSc-inoculated raPrPTg mice (coded as 01, 02, and 03). Both types of samples [rabbit (first passage) and raPrPTg (second passage)–derived prion strains] were indistinguishable according to the electrophoretic migration pattern and glycosylation profile, suggesting that de novo RaPrPSc strain was stable after its transmission in a rabbit PrP transgenic model. (B) Brain homogenate from de novo RaPrPSc-inoculated rabbit (first passage, lane 1) was compared biochemically with two brain homogenates from de novo RaPrPSc-inoculated rabbits (second passage, lanes 2–5). Two different areas [frontal cortex (FC) and midbrain (MD)] from two animals (477 dpi, lanes 2–3 ; 540 dpi, lanes 4–5) were analyzed. Both types of samples [rabbit (first passage) and rabbit (second passage)–derived prion strains] were indistinguishable according to the electrophoretic migration pattern and glycosylation profile, suggesting that de novo RaPrPSc strain was stable after its transmission in rabbits. Samples were digested with 100 μg/mL of proteinase K and were analyzed by Western blot using monoclonal antibodies L42 (Left) and SAF84 (Right). Control, normal rabbit brain homogenate.
Transmissibility in rabbit has been studied through a second passage experiment performed in New Zealand White rabbits (Hsdlf:NZW, Harlan, The Netherlands). Ten 3-mo-old male NZW rabbits were randomly divided into two groups and challenged intracerebrally with the 10−1 (wt/vol; n = 5) or 10−3 (wt/vol; n = 5) brain homogenate prepared from the clinically affected rabbit containing abnormal prion protein. After 477 dpi, one animal belonging to the 10−3 group died without apparent clinical signs. Biochemical analysis of the brain by WB confirmed the presence of PrPSc (Fig. 5B), which was indistinguishable from that previously observed in the material generated in the first passage (Fig. 2B). A second rabbit, this time from the 10−1 group, showed pododermatitis and loss of weight and had to be culled at 540 dpi for ethical reasons. Similarly, WB confirmed the presence of PrPSc in all of the brain areas examined (Fig. 5B). The rest of the rabbits remained clinically normal at ∼560 dpi.
Discussion
Slightly more than 25 y ago, cattle were considered free of prion diseases. No one would have predicted the BSE epidemic with the considerable human and animal health repercussions, and the political and economic impacts that it had in Europe during the 1990s. At that time, the scientific knowledge of prions was too limited to determine its role in the development of spontaneous cases of BSE and their subsequent impact on human health. By 2011, however, several new prion strains, naturally occurring (19, 20) or artificially generated (10), have been described, indicating that their number has increased and that a species should be considered resistant to disease only after careful consideration.
The degree of resistance and susceptibility to prion disease(s) differs within species. Animals can be both extremely susceptible to the majority of prion diseases or strains yet remain resistant to others on first passage (21). Therefore, it would be unwise to assume that a species generally resistant to TSEs would represent a minor risk to human health, as new TSEs and strains are continually being detected.
It is notoriously difficult to predict how a new TSE or strain will behave in different species, so great caution must be exercised when determining the transmissibility of prions between species. To evaluate the potential risk of transmission, every tool in the prion toolkit is essential, including artificial methods, such as the use of transgenic animals, or secondary in vivo transmission, which can exaggerate the possibility of infection (2, 22). Even these extreme measures, which probably do not reflect the normal mechanisms of infection, have to be considered to avoid future epidemics similar to that observed with the new variant Creutzfeldt-Jakob disease (vCJD). For this reason, we selected PMCA with its associated advantages, including the ability to overcome the transmission barrier (8), as the preferred tool to evaluate the absolute susceptibility of rabbits to TSEs.
Using only normal rabbit brain homogenate as a substrate for PMCA, PrPC was efficiently converted to rabbit PrPres when seeded with strains originating from four different species (Table 1), and at least three biochemically distinguishable rabbit PrPres strains were obtained (Fig. 1). This suggests that rabbit PrPC is more readily misfolded than originally thought. However, a prion disease is more than a simple misfolding process, and other factors are probably required for successful disease progression in vivo.
To address this point, we inoculated three groups of rabbits, two groups with ME7 based inocula (murine and leporine) and a further group with the de novo rabbit PMCA derived PrPres. Selection of these particular strains was carried out on the basis that ME7 was stable and well characterized and the de novo strain was an unknown entity. The number of strains tested and rabbits challenged were limited because of the necessity of housing these animals in containment level 3 facilities for protracted periods of time that maximized the possibility of transmission and development of disease.
Although rabbits were inoculated with prions derived from the same species, a 25-mo period was necessary to observe the standard prion disease. The three animals that died without clinical signs of TSE were all negative by IHC and WB, although the first of these deaths was in a rabbit challenged with ME7 RaPrPSc, which had PrPSc levels in the brain detectable only by PMCA. This result indicates that even though the animal died of a non–TSE-related disease, it might have been at an early preclinical stage of prion disease. However, we cannot rule out the possibility that the limited PrPSc level in the brain sample was due to residual inoculum, as shown previously in PrP knock-out mice (23). To date, the rabbits inoculated with mouse ME7 remain healthy after more than 43 mo, which is in agreement with the experiments performed approximately 35 y ago by Barlow and Rennie (17).
In summary, after 3 y postchallenge with three different rabbit-derived inocula, we have obtained one positive clinical case, one possible preclinical case, two intercurrent deaths, and six animals that have remained healthy. Although the incubation periods do not directly correlate with the degree of susceptibility, these data might indicate that rabbits are poorly susceptible to prion infection. Although the rabbits used in this study were not inbred, they all had identical full-length PrP sequences and, to date, no difference has been detected in the ORF PrP sequence in any other published rabbit PrP sequence placed in GenBank. To further investigate this, two types of second passage experiment were performed; three raPrPTg mice and 10 rabbits were all intracerebrally inoculated using brain homogenate from the clinically affected rabbit. In contrast to 100% of the de novo RaPrPSc-inoculated transgenic mice having succumbed to a standard clinical prion disease and thereby demonstrating a high rate of transmissibility in vivo, two of 10 rabbits developed a TSE (477 and 540 dpi, respectively) to date. A plausible explanation for the evident differences between these two transmission studies would be the high level of rabbit PrPC expression (4- to 6-fold) in the murine model. In addition, it is well known that even if overexpression does not increase susceptibility, it can significantly reduce the incubation time of disease (2). However, the two positive TSE cases in the second rabbit passage, even though 8 rabbits remained clinically normal at 560 dpi, have led us to conclude that rabbits can no longer be considered a prion-resistant species. The long incubation times, even after a second passage, might be due to the presence of some unknown, and probably rare, susceptibility factor in rabbits, which may also be present, for example, in equids and canids.
To critically evaluate this risk, several experiments are currently underway to characterize this new prion disease in rabbits and other species to examine its ability to cross the species barrier. In addition, supplementary experiments have been initiated in rabbits and also in transgenic mice that overexpress rabbit PrPC, to evaluate their susceptibilities to other important prion diseases including CWD and BSE. There are several factors that any potential new TSE epidemic would require: (i) the new prion should be efficiently transmitted through the homologous species; (ii) animals should be edible by humans and should be slaughtered at an age at which the disease has developed, thereby increasing the chance that prions have replicated (especially for those prions that require long incubation times); and (iii) the meat and bone meal should be recycled and fed to new members of the same species. In the light of these data and taking into account the previous three factors, it is unlikely there will be an outbreak of “mad rabbit disease,” and consumers of rabbit meat face much less of a risk than consumers of cattle or sheep products.
Methods
Inocula Preparation for in Vitro Prion Replication Studies.
Brain homogenates (10−1 in PBS) were prepared using scrapie mouse adapted prion strains (ME7, RML, 22F, 79A, 139A, and 22L), scrapie sheep prion strains (SSBPB/1 and CH1641), and deer CWD and cattle BSE prion strains from the brains of clinically positive animals.
Generation of RaPrPres by saPMCA.
The in vitro prion replication, including the PrPres detection of amplified samples, was performed following the basic conditions described previously (5). Briefly, 5-μL aliquots of 10% (wt/vol) brain homogenate from animals infected by each strain were diluted into 50 μL of 10% normal rabbit brain homogenate and loaded onto 0.2-mL PCR tubes. Tubes were positioned on an adaptor placed on the plate holder of a microsonicator (Misonix Model 4000, Farmingdale, NY) and programmed to perform cycles of 30-min incubation at 38 °C followed by a 20-s pulse of sonication set at potency of 80. Samples were incubated without shaking in the water bath of the sonicator. Serial rounds of PMCA consisted of 48 h of standard PMCA followed by serial in vitro 1:10 passages in fresh rabbit brain substrate. The detailed protocol for PMCA, including reagents, solutions, and troubleshooting, has been published elsewhere (4).
Biochemical Characterization of in Vitro- and in Vivo-Generated Prion Strains.
The standard procedure to digest PrPSc was performed following the basic conditions described previously (5). To study the profile of PK sensitivity for the in vivo-generated RaPrPres, samples were incubated for 60 min at 42 °C with different concentrations of PK ranging from 0 to 16,000 μg/mL. Digestion was stopped by adding electrophoresis loading buffer, and the samples were analyzed by Western blotting.
Stability Studies with Denaturating Agents.
The RaPrPSc sample produced in vivo was treated with different concentrations (1.5–4 mol/L) of guanidine hydrochloride (GdHCl) for 2 h before being centrifuged at 25,000 × g for 1 h at 4 °C. ME7 and RML prion strains were used as reference. The pellets were resuspended and treated with PK (50–100 μg/mL). PrPSc signal was detected using Western blotting.
Preparation of ME7 and RaPrPSc Inocula.
Brain homogenate, diluted 10−1 in sterile 0.9% saline solution, was prepared from the brains of mice challenged intracerebrally with ME7 that had developed clinical signs of TSE. ME7 and de novo RaPrPSc samples generated in vitro during round 20 of saPMCA were diluted 10−1 in sterile PBS.
Rabbit DNA Sequencing.
Chromosomal DNA was extracted from rabbit tissue using DNeasy blood and tissue kit (Qiagen) for use as a template for PCR using HotStarTaq DNA polymerase (Qiagen) with the forward primer RaPrP/F (5′-ATGGCGCACCTCGGCTAC-3′) and the reverse primer RaPrP/R (5′-TCATCCCACGATCAGGAAG-3′). Primers were designed according to the published sequence of the Oryctolagus cuniculus prnp gene (GenBank accession no. U28334). PCR was performed in a Hybaid MBS 0.2G Thermal Cycler and included 5 min of initial denaturation at 95 °C followed by 35 cycles of 95 °C for 20 s, 56 °C for 45 s, and 72 °C for 1 min. The amplified product from each reaction was purified with a kit (Qiagen) and sequenced in both directions by Eurofins MWG Operon using the same primers as above. All sequences were compared at the nucleotide level and translated to provide the corresponding amino acid sequence. DNA and amino acid sequences were compared using SeqMan software (DNASTAR).
Inoculation.
Ten 3-mo-old New Zealand White male rabbits (Harlan) were subjected to gaseous anesthetic (isoflurane; Abbott Laboratories). The left side of the scalp was shaved and prepared as for aseptic surgery, and 0.1 mL of prewarmed (37 °C) inoculum was injected into the left cerebral hemisphere using a 23-gauge hypodermic needle with a collar applied to restrict penetration to ∼7 mm.
Transgenic Mice.
A raPrPTg mouse line was generated and characterized as previously described (24). These mice express ∼4- to 6-fold the normal rabbit PrP protein under the control of the murine PrP promoter in a murine PrP0/0 background.
Cell Culture Studies.
Mouse neuronal cells in which PrP expression had been ablated (CF10 cells) (18) were transduced with a retrovirus encoding rabbit PrPC, mouse PrPC containing the epitope to the mouse monoclonal antibody 3F4 (Mo3F4PrPc), or vector alone (pSFF) using previously described techniques (18). For mouse scrapie infection, cells were exposed to 200 μL of a 1% brain homogenate derived from a 22L-infected, transgenic mouse displaying clinical disease. These transgenic mice express mouse PrPC without the glycophosphotidyl inositol membrane anchor. After 4 h at 37 °C, an additional 1 mL of medium was added, and the culture was incubated for an additional 3 d at 37 °C.
When the cells were confluent, the medium was removed and the cells in one well were trypsinized and replated into a single well of a six-well plate (passage 1). Thereafter, cells were trypsinized and passaged in six-well plates at a 1:10 dilution. The cells from the duplicate well were trypsinized, lysed in 500 μL of lysis buffer (0.5% Triton X-100, 0.5% sodium deoxycholate, 150 mmol/L NaCl, 50 mmol/L Tris-HCl, 5 mmol/L EDTA), centrifuged at 16,000 × g for 5 min, and the resultant lysate supernatant stored at −20 °C for PrPSc analysis (passage 0). For detection of PrPSc at different cell passes, 500 μL of cell lysate, derived as described above, was digested with 20 μg/mL of proteinase K (PK; Roche) for 1 h at 37 °C. Following the addition of 1 mmol/L Pefabloc (Roche), lysates were centrifuged at 200,000 × g for 1 h at 4 °C. The resultant pellet was sonicated into 15 μL of sample buffer (5% SDS, 3 mmol/L EDTA, 2% β-mercaptoethanol, 5% glycerol, 0.02% bromophenol blue, 63 mmol/L Tris⋅HCl), and boiled for 5 min, and the entire sample was loaded onto 16% Tris⋅glycine precast gels (Invitrogen). Following transfer to PVDF membrane (Millipore), rabbit PrP was detected with the mouse monoclonal antibody L42 (1:3,000 dilution, R-Biopharm AG) and Mo3F4 PrP with the mouse monoclonal antibody 3F4 (1:25,000 dilution, mouse ascitic fluid). Blots were developed using the enhanced chemiluminescence system according to the instructions of the manufacturer (GE Healthcare).
Transmission Studies Using Rabbit PrP Transgenic Mice.
Groups of three and five mice (8 wk old) were housed following the guidelines of the Code for Methods and Welfare Considerations in Behavioral Research on Animals (Directive 86/609EC). Mice were inoculated in the right parietal lobe using a disposable 25-gauge hypodermic syringe. A 20-μL quantity of 1% de novo raPrPSc inoculated brain or healthy brain homogenates was delivered to each animal. The neurological status of the inoculated mice was assessed twice weekly. The presence of three signs of neurological dysfunction of 10 established diagnostic criteria was needed to score a mouse as positive for prion disease. When progression of the disease was evident, the animals were killed for ethical reasons, and their brains were harvested for subsequent biochemical and histological analysis.
Histology.
Brain was collected postmortem from rabbits and sliced sagittally down the middle, with one half stored frozen (−80 °C) for use in biochemical studies and the preparation of inocula for future experiments, and the other half fixed in 10% buffered formalin. After 1 wk in fixative selected areas of brain (frontal cortex, midbrain, cerebellum, and obex) were processed using standard protocols (dehydrated through graded alcohols, embedded in paraffin wax, sectioned in 4-μm sections, mounted on glass microscope slides) and stained with hematoxylin and eosin.
Immunohistochemistry.
Additional tissue sections were cut and mounted onto Superfrost Plus microscope slides (Menzel) and dried overnight at 37 °C. The method for labeling of glial fibrillary acidic protein (GFAP) and PrPSc was identical, except for the primary antibody used and the omission of formic acid treatment for GFAP. Sections were rehydrated through graded alcohols to water and were then immersed in 98% formic acid for 10 min at room temperature (RT). Antigen retrieval consisted of autoclaving the sections in 0.01 mol/L citrate buffer, pH6, at 121 °C for 10 min. Endogenous peroxidase activity was blocked by placing sections in 3% hydrogen peroxide in methanol (vol/vol) for 20 min at RT. Subsequently slides were placed in the Sequenza slide rack system (ThermoFisher Scientific) for the rest of the method and 0.3 mol/L PBS containing 0.05% Tween 20 was used for all dilutions of antibodies and washes. Nonspecific binding was inhibited by applying 25% normal goat serum (Vector Laboratories) for 30 min at RT. Sections were incubated overnight at 4 °C with either mouse anti-prion protein antibody 2G11 (SPiBio, Montigny, Le Bretonneux, France) at 0.5 μg/mL or mouse anti-GFAP antibody [6F2] (clone ab86144; Abcam) prediluted by the manufacturer. Visualization was performed via goat anti-mouse IgG antibody with a peroxidase labeled polymer (EnVision+ System-HRP; DakoCytomation) that was applied for 30 min at RT followed by DAB chromogen (Dako) for 8 min before being washed in water, counterstained with hematoxylin, blued up with Scot's tap water substitute, dehydrated, cleared, and mounted.
Pet-Blot.
Further tissue sections (4-μm) were cut and mounted onto 0.45-μm nitrocellulose membrane (Sigma, Dorset, United Kingdom) and dried for 48 hours at 60 °C. The nitrocellulose membrane–mounted section was de-paraffinized with xylene and then rehydrated using isopropanol gradients. All washes were carried out using TBS with Tween 20 (TBST) buffer pH 7.6 (0.1 mol/L Tris⋅HCl, 0.1 mol/L di-NaCl, 0.05% Tween 20) unless stated. Sections were subjected to 50 μg/mL PK at 55 °C overnight. PK digestion was halted using 1 mmol/L PEFA block (Pierce Biotechnology) for 15 min at RT. Further denaturation of the protein was carried out using 4 mol/L guanidine thiocyanate for 15 min. Sections were then washed three times before addition of 2% casein for 60 min to block nonspecific binding. Subsequent to this, sections were incubated with antibody SAF 84 (SPi-Bio, at 2 μg/mL in blocking solution) for 90 min. After washing, sections were incubated with rabbit anti-mouse IgG:alkaline phosphotase antibody (Pierce Biotechnology), diluted 1:200, for 90 min. Sections were washed twice for 5 min in TMN solution (0.1 mol/L Tris/HCl, 0.1 mol/L di-NaCl, 0.005 mol/L di-MgCl2 at pH9) before incubation with the chromagen nitro-blue tetrazolium chloride/5-bromo-4-chloro-3′-indolyphosphate p-toluidine salt (NBT/BCIP; Perbio Science) for 1–5 min, until labeling of the positive control sections was clear). Sections were then washed with an EDTA buffer (0.1 mol/L Tris⋅HCl, 0.1 mol/L di-NaCl, 0.02 mol/L EDTA) twice for 2 min and air dried at RT.
Supplementary Material
Acknowledgments
The authors acknowledge the IKERBASQUE Foundation; support for vivarium and maintenance from CIC bioGUNE; Drs. Jean E. Jewel and Tomás Mayoral for the CWD and BSE brain tissue samples; Drs. Silvia Siso (AHVLA Lasswade), Ashwin Woodhoo (CIC bioGUNE), and Mike Fontaine (MRI) for useful discussion and advice; the Roslin Institute for use of equipment; and Moredun Bioservices for care and maintenance of animals. This work was financially supported by national grants from Spain (AGL2009-11553-C02-01 and AGL2008-05296-C02); Basque government Grant PI2010-18; National Institutes of Health (NIH) Grant 1R01NS060790-01A2; Intramural Research Program of the NIH, National Institute of Allergy and Infectious Diseases 1-Z01-AI000752-12; and Etortek Research Programs 2011/2013, Scripps and the Moredun Research Institute.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1120076109/-/DCSupplemental.
References
- 1.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 2.Sigurdson CJ, et al. Strain fidelity of chronic wasting disease upon murine adaptation. J Virol. 2006;80:12303–12311. doi: 10.1128/JVI.01120-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Saborio GP, Permanne B, Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature. 2001;411:810–813. doi: 10.1038/35081095. [DOI] [PubMed] [Google Scholar]
- 4.Saá P, Castilla J, Soto C. Cyclic amplification of protein misfolding and aggregation. Methods Mol Biol. 2005;299:53–65. doi: 10.1385/1-59259-874-9:053. [DOI] [PubMed] [Google Scholar]
- 5.Castilla J, Saá P, Hetz C, Soto C. In vitro generation of infectious scrapie prions. Cell. 2005;121:195–206. doi: 10.1016/j.cell.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 6.Saá P, Castilla J, Soto C. Ultra-efficient replication of infectious prions by automated protein misfolding cyclic amplification. J Biol Chem. 2006;281:35245–35252. doi: 10.1074/jbc.M603964200. [DOI] [PubMed] [Google Scholar]
- 7.Castilla J, et al. Protein misfolding cyclic amplification for diagnosis and prion propagation studies. Methods Enzymol. 2006;412:3–21. doi: 10.1016/S0076-6879(06)12001-7. [DOI] [PubMed] [Google Scholar]
- 8.Fernández-Borges N, de Castro J, Castilla J. In vitro studies of the transmission barrier. Prion. 2009;3:220–223. doi: 10.4161/pri.3.4.10500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Green KM, et al. Accelerated high fidelity prion amplification within and across prion species barriers. PLoS Pathog. 2008;4:e1000139. doi: 10.1371/journal.ppat.1000139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soto C, et al. Pre-symptomatic detection of prions by cyclic amplification of protein misfolding. FEBS Lett. 2005;579:638–642. doi: 10.1016/j.febslet.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 11.Castilla J, et al. Cell-free propagation of prion strains. EMBO J. 2008;27:2557–2566. doi: 10.1038/emboj.2008.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim JI, et al. Mammalian prions generated from bacterially expressed prion protein in the absence of any mammalian cofactors. J Biol Chem. 2010;285:14083–14087. doi: 10.1074/jbc.C110.113464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang F, Wang X, Yuan CG, Ma J. Generating a prion with bacterially expressed recombinant prion protein. Science. 2010;327:1132–1135. doi: 10.1126/science.1183748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weber P, et al. Generation of genuine prion infectivity by serial PMCA. Vet Microbiol. 2007;123:346–357. doi: 10.1016/j.vetmic.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 15.Deleault NR, Harris BT, Rees JR, Supattapone S. Formation of native prions from minimal components in vitro. Proc Natl Acad Sci USA. 2007;104:9741–9746. doi: 10.1073/pnas.0702662104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Castilla J, et al. Crossing the species barrier by PrP(Sc) replication in vitro generates unique infectious prions. Cell. 2008;134:757–768. doi: 10.1016/j.cell.2008.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barlow RM, Rennie JC. The fate of ME7 scrapie infection in rats, guinea-pigs and rabbits. Res Vet Sci. 1976;21:110–111. [PubMed] [Google Scholar]
- 18.Greil CS, et al. Acute cellular uptake of abnormal prion protein is cell type and scrapie-strain independent. Virology. 2008;379:284–293. doi: 10.1016/j.virol.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Casalone C, et al. Identification of a second bovine amyloidotic spongiform encephalopathy: Molecular similarities with sporadic Creutzfeldt-Jakob disease. Proc Natl Acad Sci USA. 2004;101:3065–3070. doi: 10.1073/pnas.0305777101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Benestad SL, et al. Cases of scrapie with unusual features in Norway and designation of a new type, Nor98. Vet Rec. 2003;153:202–208. doi: 10.1136/vr.153.7.202. [DOI] [PubMed] [Google Scholar]
- 21.Bencsik A, et al. Scrapie strain transmission studies in ovine PrP transgenic mice reveal dissimilar susceptibility. Histochem Cell Biol. 2007;127:531–539. doi: 10.1007/s00418-007-0276-8. [DOI] [PubMed] [Google Scholar]
- 22.Hill AF, et al. Species-barrier-independent prion replication in apparently resistant species. Proc Natl Acad Sci USA. 2000;97:10248–10253. doi: 10.1073/pnas.97.18.10248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sailer A, Büeler H, Fischer M, Aguzzi A, Weissmann C. No propagation of prions in mice devoid of PrP. Cell. 1994;77:967–968. doi: 10.1016/0092-8674(94)90436-7. [DOI] [PubMed] [Google Scholar]
- 24.Castilla J, et al. Subclinical bovine spongiform encephalopathy infection in transgenic mice expressing porcine prion protein. J Neurosci. 2004;24:5063–5069. doi: 10.1523/JNEUROSCI.5400-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.