

Abstract
There has been intense interest in the development of selective bioorthogonal reactions or “click” chemistry that can proceed in live animals. Until now however, most reactions still require vast surpluses of reactants because of steep temporal and spatial concentration gradients. Using computational modeling and design of pharmacokinetically optimized reactants, we have developed a predictable method for efficient in vivo click reactions. Specifically, we show that polymer modified tetrazines (PMT) are a key enabler for in vivo bioorthogonal chemistry based on the very fast and catalyst-free [4 + 2] tetrazine/trans-cyclooctene cycloaddition. Using fluorescent PMT for cellular resolution and 18F labeled PMT for whole animal imaging, we show that cancer cell epitopes can be easily reacted in vivo. This generic strategy should help guide the design of future chemistries and find widespread use for different in vivo bioorthogonal applications, particularly in the biomedical sciences.
Keywords: in vivo chemistry, pharmacokinetics, PET imaging, intravital microscopy, pretargeting
The ability to perform selective chemistries in living systems such as single cells, 3D cultures, invertebrates, or mammals would have far reaching applications in tracking biomolecules, designing new therapeutic approaches, and in visualizing medically relevant biomarkers. To date, only a few practical bioorthogonal reactions have been reported, the most popular being the Staudinger ligation and the [3 + 2] cycloaddition “click” reaction between azides and alkynes (1, 2). The latter click reaction involves copper(I) catalyzed coupling of an azide and terminal alkyne to generate a stable triazole (2). Until recently, the necessity of the copper catalyst precluded use of this reaction in biological systems due to toxicity concerns (3, 4). Bertozzi and others elegantly solved this problem by developing several new ring strained dienophile derivatives that do not require catalysts (5–9). However, many of these derivatives have low water solubility, require complex multistep synthesis, and possess suboptimal kinetics. Our search for alternative rapid, selective, and chemically accessible coupling reactions without need for a catalyst led us and others to investigate the [4 + 2] inverse Diels-Alder cycloaddition (10–13). We realized that this set of chemistries is more uniquely suited to biological applications and may indeed represent a universal platform technology (Fig. 1 A and B). Specifically we and others have shown that the cycloaddition between tetrazine (Tz) and trans-cyclooctene (TCO) can proceed orders of magnitude faster than previously studied azide and alkyne click reactions and importantly does not require the action of a catalyst. This reaction has also been adapted for single cell imaging using newer fluorogenic Tz probes (14).
Fig. 1.

In Vivo Bioorthogonal Reactions. (A) Tetrazine cycloaddition with trans-cyclooctene forming a dihydropyrazine. (B) Schematic of PMT used in this study. The scaffold consists of dextran that has been aminated to allow attachment of tetrazine reactive groups as well as imaging agents such as near-infrared fluorophores and radioisotopes. (C) In vivo multistep delivery of imaging agent. A slow clearing targeting agent is administered first (green) and is given 24 h for localization and background clearance. Next, a lower molecular weight secondary agent (red) is delivered that rapidly reacts and is cleared from the background tissue much faster than the primary agent. (D) Kinetic parameters of consideration for in vivo clicking. The secondary tetrazine agent reacts with transcyclooctene antibodies at a given rate (kreaction). This rate is in competition with other rates including the clearance of the secondary agent from the body (kclearance) and internalization of the antibody (kendocytocis).
Although initial work focused on in vitro labeling there has been a surge of recent work applying this reaction for various in vivo applications (15–17). Despite the above progress, our results with initial TCO/Tz reactions in live animals were disappointing unless we used excessive amounts of reactants. This brute force approach would ultimately be cost prohibitive and likely not be biologically viable. We therefore systematically eliminated different reasons for the observed discrepancies (stability, bioconversion of reactants, target delivery, etc.) and identified simple pharmacokinetic principles as the primary reason why the reaction did not proceed at target sites as originally anticipated. We then performed detailed studies on how the reaction rate and pharmacokinetics (PK) can affect the efficiency of an in vivo chemical reaction. Using reactants with modified PK profiles, such as polymer modified tetrazines [(PMT); Fig. 1C] and a newly developed computational model to predict reaction efficiency, we now observe highly efficient in vivo bioorthogonal reaction. Using cancer cell epitopes as a model target (Fig. 1D), we show the specificity and sensitivity of the method in a mouse model of colon cancer. Specifically, we observe in vivo labeling with cellular resolution using near infrared fluorophores. Furthermore, we demonstrate use of bioorthogonal reactions with rapidly decaying positron emission tomography (PET) isotopes for imaging cancer cell epitopes in solid tumors. We believe our work and findings will guide further development of in vivo chemistries for a number of different biomedical applications.
Results
We first compared different reactants in mice to better understand achievable in vivo click reaction efficiencies as a function of PK profiles (Table 1). These studies were performed as serial blood draws, single time point biodistribution studies, or longitudinal imaging studies at different resolutions. In a first set of experiments, mice were injected with 30 micrograms of anti-CD45 monoclonal antibodies labeled with TCO or norbornene dienophiles. The goal was to provide a reactive target in the plasma to avoid the additional complexities of delivering agents to extravascular sites. The dienophile labeling was controlled so on average each antibody possessed approximately three reactive groups. After waiting 3 h, an excess of tetrazine chaser was administered. Because the pharmacokinetics of the chaser would likely affect the efficiency of labeling, we administered either a small molecular tetrazine fluorophore (MW 1.3 kDa) or polymer modified tetrazines (PMT) using two differently sized dextrans: 10 kDa (PMT10) and 40 kDa (PMT40). Dextrans were chosen as polymer scaffold due to their low cost, high stability, and numerous previous in vivo applications (18–20). These amine modified scaffolds were modified on average with approximately one fluorochrome and either two (PMT10) or eleven (PMT40) tetrazines as measured using absorption spectroscopy. Such high coupling efficiencies to amino dextran may be the result of the low molecular weight and lipophilic nature of tetrazine affinity probes. Measurement of reaction kinetics demonstrated that conjugation combined with multivalent presentation did not significantly impede overall probe reactivity compared to small molecule tetrazines (see SI Text). The clearance kinetics of these chaser imaging agents were measured separately through serial blood draws (Fig. 2A). The tetrazine fluorophore is a low molecular weight and highly charged probe, and thus it is not surprising that the PK showed rapid tissue redistribution and clearance (21). In contrast, the PMT’s persisted in the blood and exhibited biexponential clearances with the larger molecular weight dextran PMT40 circulating longer.
Table 1.
Reported Bioorthogonal Rate Constants and Pharmacokinetic Parameters
| Reaction type | Rate constant (/M·s) | Reference |
| azide-DIFO | 0.076 (CH3CN) | (7) |
| phen-tetrazine-norbornene | 1.6 (H2O, 20 °C) | (11) |
| pyr-tetrazine-TCO | 2,000 (9∶1 MeOH/H2O, 25 °C) | (12) |
| phen-tetrazine-TCO | 6,000 (H2O, 37 °C) | (10) |
| pyr-tetrazine-TCO | 13,090 (H2O, 37 °C) | (15) |
| pyr-tetrazine-strained-TCO | 22,000 (MeOH, 25 °C) | (13) |
Fig. 2.

Optimization of In Vivo Chemistry with Polymer Modified Tetrazines (PMT). (A) Clearance kinetics of tetrazine-VT680 (red), PMT10 (blue) and PMT40 (green). (B) Efficiency of in vivo labelling of trans-cyclooctene modified leukocytes using various tetrazine cycloadditions. (C) Heat map showing predicted efficiency of reaction given a reaction rate and secondary clearance rate. The 1.3 kDa point represents the small molecule tetrazine-VT680. The highest efficiencies were observed for the fast tetrazine/trans-cyclooctene reaction using slow clearing PMT10 or PMT40. Use of either a faster clearing tetrazine or a much slower reaction greatly diminishes the efficiency of reaction as seen in plot in (B).
Fig. 2B shows the labeling of dienophile modified white blood cells, measured by flow cytometry, 3 h after administration of a fluorescent tetrazine chaser. The labeling efficiency is strongly dependent on the kinetics of the reaction (e.g., trans-cyclooctene≫norbornene) and the clearance kinetics of the secondary agent. TCO/Tz reaction efficiencies were very high for both the fluorescent PMT10 and PMT40 (680 nm). However, tetrazine-VT680 TCO reactions showed a much lower labeling efficiency, presumably due to its much faster clearance rate. Likewise, targeting PMT40 to a slow reacting norbornene antibody led to nearly negligible reaction. In order to gain greater quantitative insight, computational modeling of the in vivo reaction was performed (see Materials and Methods). A plot of predicted labeling efficiency vs. both the kinetics of cycloaddition reaction and the area under the concentration curve (AUC) is displayed in Fig. 2C. The fraction of primary agent reacted is an exponential function of the reaction rate and AUC of the secondary agent. Small molecule tetrazines, while possessing rapid reaction rates, do not possess the requisite AUC for very efficient labeling, explaining our experimental results.
Based on the results from the initial in vivo experiments, we decided to use PMT10 to demonstrate the utility of our method for imaging cancer cell epitopes in a more challenging but clinically relevant solid tumor model. PMT10 showed good clearance after several hours yet persisted long enough to allow highly efficient labeling using the TCO/Tz reaction. As a disease model, we targeted the A33 glycoprotein which is overexpressed in > 95% of all human colorectal cancers including the human colorectal cancer LS174T cell line (22). In addition, this antigen is associated with tight junction proteins, giving it a surface persistence with a half life greater than 2 d (23).
Initially we reacted near-infrared fluorescent PMT imaging agents to obtain cellular resolution of the in vivo bioorthogonal reaction. In addition, fluorescent agents avoid the cost and handling complexities of radionuclides, and are increasingly being used for in vivo applications (24). To facilitate the in vivo imaging, we used dorsal window chambers for intravital microscopy. Window chamber mice bearing LS174T tumors were injected first with anti-A33 monoclonal antibodies bearing TCO (n = 3) dienophiles (25). After waiting 24 h for antibody targeting and clearance, the mice were subsequently injected with 30 μg of fluorescent PMT10 (750 nm). After waiting 3 h to allow for the reaction and clearance to occur, the tumors were visualized using confocal microscopy. The images revealed beds of LS174T cells whose surfaces were brightly stained with the fluorescent antibody as well as the tetrazine dextran probe (Fig. 3). Merging of the two channels demonstrated excellent colocalization between the location of the dienophile antibody and the secondary tetrazine dextran, indicative of a specific bioorthogonal reaction. Control experiments involving mice which received an antibody lacking a dienophile also showed significant antibody staining of LS174T cell surfaces but lacked subsequent tagging by the tetrazine dextran. These in vivo images and controls looked remarkably similar to corresponding in vitro experiments. These initial studies demonstrated that use of a higher molecular weight PMT10 agent, previously validated in the blood, is also able to label specific cell surface biomarkers in the tumor microenvironment.
Fig. 3.

In Vitro and In Vivo Microscopy of Fluorescent PMT10 Targeting a Cancer Cell Epitope. Confocal imaging demonstrating targeting of PMT10 (750 nm) to trans-cyclooctene (TCO) antibodies (680 nm) on the surface of LS174T cells both in vitro and in vivo. (A) LS174T cells in culture labeled with TCO monoclonal antibodies followed by PMT10 (750 nm) labeling. (B) Same as previous except monoclonal antibodies lack TCO. (C) Cells in a LS174T xenograft in vivo labeled with TCO monoclonal antibodies followed by PMT10 (750 nm) labeling. (D) Same as previous except monoclonal antibodies lack TCO. See Materials and Methods for experimental details.
To demonstrate the utility of our method for clinically relevant PET imaging we performed longitudinal studies using a radiolabeled 18F-PMT10 (26). Mice were implanted subcutaneously with LS174T xenografts, and once tumors had grown to approximately 1 cm in diameter, these mice were injected with 30 μg of TCO-antibody against A33 (also labeled with VT680 fluorophore). After allowing 24 h for accumulation and clearance of the TCO-modified antibody, 18F-PMT10 was injected (30 μg; 150 μCi). Cycloaddition of 4 mCi of TCO-18F to 60 μg of dextran proceeded cleanly, leading to an 89.2% decay corrected radiochemical yield of 18F-PMT10. This construct was used for all PET/Computed Tomography (CT) and subsequent autoradiography experiments. PET/CT scans were initiated 3 h after injection of 18F-PMT10. The images revealed significant renal clearance of PMT10 as well as widespread tissue distribution. Fig. 4A shows one representative axial slice from the PET/CT reconstruction. Compared to control mice which were administered antibodies lacking TCO, there was significant accumulation of signal in the vascularized region of the tumor mediated by the in vivo TCO/Tz reaction. To investigate further, mice were euthanized 3 h after injection, the tumors were excised, and 1 mm slices were imaged to determine the extent of cycloaddition. Near infrared fluorescence was used to determine antibody/TCO localization and autoradiography was used to reveal the 18F-PMT10. Fig. 4B shows two representative tumor slices. The localization patterns of the chemical probes were distinct and often showed greatest activity in the periphery of the tumor, likely reflecting the necrotic nature of the tumor core. Comparison of the pattern to the localization of the fluorescent antibody demonstrated good colocalization, indicating that the Tz probe was primarily localized to the site of TCO binding. Tumors from mice which were administered the control antibody lacking reactive TCO showed much lower uptake of Tz and a lack of correlation to antibody/TCO fluorescence signal.
Fig. 4.

PET and Autoradiography using 18F Tetrazine Agents. (A) PET/CT fusion of LS174T tumor xenograft labeled using either trans-cyclooctene (TCO) monoclonal antibodies (MAb TCO) or control unlabeled antibodies (MAb) followed by 18F-PMT10. Arrows indicate location of the tumor xenograft. Bladder has been omitted for clarity. (B) Imaging using autoradiography (left side) and fluorescence reflectance (right side) of 1 mm LS174T tumor slices after targeting with fluorescent TCO monoclonal antibody and 18F-PMT10. (C) PET/CT fusion of mouse bearing A431 and LS174T tumors after targeting with anti-A33 TCO monoclonal antibodies followed by 18F-PMT10. Arrows indicate location of tumors and the liver has been omitted for clarity. (D) Autoradiography of representative 1 mm LS174T or A431 tumor slices after multistep targeting.
We also tested for biomarker specificity with mice simultaneously implanted with LS174T tumors and A431 tumors which lack expression of the A33 glycoprotein epitope (27). Mice were administered anti-A33 TCO antibody and 18F-PMT10 as previously described and imaged using PET/CT. Fig. 4C shows one representative coronal slice from the PET/CT reconstruction. LS174T tumors can be clearly visualized whereas the control A431 tumor shows much lower Tz uptake. Autoradiography of excised tumor slices further demonstrate the greater uptake of Tz in biomarker positive LS174T tumors vs. the control A431 tumors (Fig. 4D).
Discussion
This work demonstrates that bioorthogonal TCO/Tz reactions can efficiently occur in live animals when reaction partner pharmacokinetics are optimized. Based on modeling of reaction kinetics and PK clearance rates, we designed and tested specific polymer-tetrazine adducts bearing “imaging” reporters (near infrared fluorochromes and 18F) to visualize reactions in live mice both at the cellular and whole animal level. The current generation of polymer-tetrazine conjugates included dextran derivatives (PMT10 and PMT40) because dextrans have a long history of in vivo use and the PK are well understood (18–20). Based on the kinetics of the TCO component, we would envision that the PMT could be based on a number of different polymers for further optimization (e.g., PEGs, PL, PLGA, nanoparticles, etc.). We expect that the methodology used for developing these probes and modulating the reaction rate and clearance kinetics should be applicable to other bioorthogonal reactions, imaging agents, and therapeutics.
The TCO/Tz reaction is an ideal bioorthogonal chemical system for in vivo use given its fast reactions kinetics (> 6,000 M-1 sec-1)(10). Despite these impressive kinetics for benzylamine Tz/TCO, PK modifications are required for in vivo use. These improved PK properties can theoretically be achieved by small molecule tetrazine modifications, such as those that would alter pharmacokinetics without affecting reaction rates such as side groups with protein binding abilities. In this respect we have recently looked at reaction rates of a number of different TZ analogs. However, although binding to proteins would decrease blood clearance, it was unclear if the protein bound Tz would be viable for chemical reaction with TCO targets. Instead, to improve PK of the reactants we decided to conjugate Tz to dextran creating PMT of different chain lengths but with identical reactivities to decouple and independently tune the clearance and reaction rates.
We developed a simple mathematical model that takes TCO/Tz reaction rates and pharmacokinetics of reaction partners into account (Fig. 2) to better predict the ideal design and dosages of reaction partners for in vivo use. The model predicts that the secondary agent must remain at a high enough concentration for a sufficient time to undergo the reaction in vivo. These experimental and computational results allow approximation of labeling efficiency in vivo given a chemical reaction rate and secondary agent clearance rate when the secondary agent is administered in excess. These conditions are commonly encountered when labeling surface exposed molecules and is directly comparable to the experiments targeting cell surface antigens in the blood. For other targets, such as solid tumors, the concentration profile of the primary and secondary agents are more complex given the blood flow, extravasation, diffusion, binding, and clearance rates found in tumors (28), but the basic principles still apply. We believe these insights are not only important for our study but may offer helpful guidance to future studies where one is interested in performing in vivo reactions.
We envision a number of practical applications of the developed in vivo click approach based on TCO/PMT and other small molecule probes. For instance, the short positron decay half-life (1.8 h) of 18F requires affinity ligands that show significant target accumulation and rapid background clearance in hours. These results are simply not possible by directly labeling high AUC affinity agents such as monoclonal antibodies. Multistep techniques with bioorthognal chemistries could overcome this problem. Additionally, in vivo assembly could be used to improve therapeutic drug delivery by delivering components as small molecules to facilitate transport (e.g., across the blood brain barrier, blood vessel endothelium, intestinal wall) followed by specific covalent assembly. The technique may also provide information on the concentration of drugs or metabolic markers labeled with chemical tags in different tissue compartments. Finally, as we have demonstrated in this work, in vivo chemistry shows great promise for imaging biomarkers and targeting agents with rapidly decaying isotopes.
Materials and Methods
Measurement of Clearance Kinetics of Tetrazine Agents.
Fluorescent tetrazine agents (tetrazine-VT680, PMT10, PMT40) were injected via tail vein into mice. Serial retroorbital 30 μL blood draws were taken starting at 1 min after injection and periodically up to 2 h. The blood was diluted in equal volume 5 mM EDTA in phosphate buffered saline, and the fluorescence intensity was measured using a plate reader. The concentration of the fluorescent agent in the blood was determined by comparison with a standard curve using blood mixtures with known concentrations of fluorescent agent.
Modeling of Reaction Efficiency.
The efficiency of tetrazine imaging agent reaction with trans-cyclooctene modified antibodies on the surface of white blood cells was modeled using simple mass action kinetics. Assuming a mouse plasma volume of approximately 2 mL, the initial antibody bolus dose should result in an antibody plasma concentration of 100 nM. After 24 h, the plasma concentration will drop due to clearance (28) and potentially from uptake by target white blood cells (29). After clearance, the tetrazine probe is delivered at an initial concentration of 2 μM, well in excess of the antibody plasma concentration. Given the large excess of tetrazine probe, the loss in tetrazine due to reaction is considered negligible. The beta phase of antibody clearance has a 7 d half life, so, after 24 h, antibody clearance is assumed to be negligible over 3 h. Because the reaction occurs in the plasma, no tissue transport parameters were required.
The mass balance for antibody concentration is therefore:
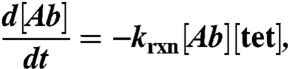 |
where [Ab] is the trans-cyclooctene modified antibody concentration, krxn is the second order reaction rate constant, and [tet] is the concentration of tetrazine probe.
The highly dispersive nature of the mouse vascular system is likely to rapidly result in homogenous distribution of reactive agents in the plasma, so a biexponential decay model is used for plasma concentration:
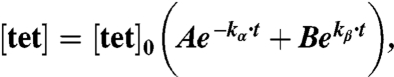 |
where [tet]0 is the initial plasma concentration of tetrazine, A and B are the fractions of alpha vs. beta phase clearance, t is the time after injection of the tetrazine agent, and kα and kβ are the alpha and beta phase clearance rate constants, respectively. It is important to capture both phases in measurements, particularly for imaging agents that are given as a single bolus dose. While small molecular weight agents may have beta (i.e., clearance) phases of several minutes or longer, this often is preceded by a rapid drop in concentration, which can be greater than an order of magnitude, with a half life of tens of seconds (21).
Because the tetrazine concentration is assumed independent from the reaction:
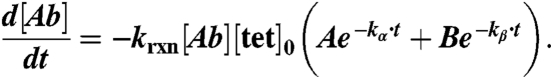 |
Integrating:
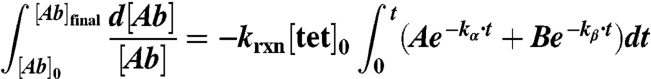 |
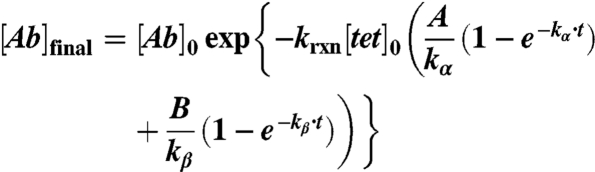 |
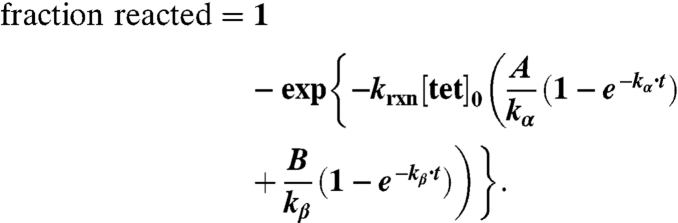 |
The integral of the tetrazine concentration-time curve is commonly defined as the area under the curve or AUC:
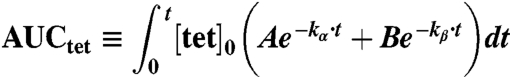 |
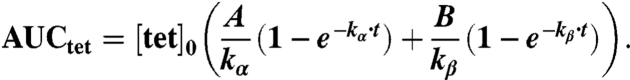 |
Substituting into the equation for fraction of TCO reacted:
This equation is graphed as a heat map in Fig. 2C.
Measurement of Reaction Efficiency on Circulating White Blood Cells.
Dienophile modified anti-CD45 antibody was injected into mice (n = 3) via tail vein. After 1 h, a 30 μL retroorbital blood sample was taken from each mouse and exposed to an excess of tetrazine agent to determine the saturated labeling density. The tetrazine agent was then administered via tail vein into the mice, and after 3 h, a second retroorbital blood sample was taken. The blood samples were exposed to 1 mL BD pharmlyse solution for 15 min at room temperature, centrifuged, and the pellet resuspended in order to remove lysed red blood cells. The white blood cells were run on a flow cytometer to measure the average fluorescent intensity. The in vivo intensity measurements were then divided by the in vitro saturated intensity in order to determine the labeling efficiency.
Fluorescence Imaging of Tetrazine Cycloadditions In Vitro and In Vivo.
LS174T cells were trypsinized, plated on cover glass dishes, and allowed to attach overnight. Cells were then exposed to 100 nM of TCO modified anti-A33 modified with a VT680 probe. After washing, the cells were incubated with 100 nM of fluorescent PMT10 (750 nm). The reaction was allowed to proceed for 45 min after which the cells were washed and imaged using scanning confocal microscopy. Control cells were treated identically except that they received an antibody lacking TCO modification.
Window chambered nude mice (n = 3) were prepared as previously described. To create the LS174T xenograft, approximately 1 million LS174T cells were subcutaneously injected and allowed to develop into a tumor of approximately 2 mm in diameter. 30 μg of TCO modified anti-A33 modified with a VT680 probe was injected by tail vein. After 24 h, a secondary injection of 30 μg of fluorescent PMT10 (750 nm) was injected via tail vein. After 3 h, the tumors were imaged using scanning confocal microscopy. Control mice (n = 3) were treated identically except that they received an antibody lacking TCO modification.
PET Imaging of Tetrazine Cycloadditions.
Approximately 106 LS174T or 1.5 × 106 A431 cells were injected subcutaneously in either the hind limb or upper dorsal region in nude mice (n = 3). The difference in cell number was used to ensure similar sized tumors at the time of imaging due to different growth rates. Tumors were allowed to grow for 2 w after which mice were injected via tail vein with 30 μg of TCO modified anti-A33 modified with a VT680 label. After allowing 24 h for targeting and clearance of the antibody, the mice received 30 μg of 18F-PMT (30 μg, 150 μCi) via a tail vein injection. All PET-CT images were acquired on a Siemens Inveon PET-CT. Each PET acquisition was approximately 60 min in duration. PET was reconstructed from 600 million coincidental, 511 keV photon counts on a series of LSO (lutetium oxyorthosilicate) scintillating crystal rings. Counts were rebinned in 3D by registering photons spanning no more than three consecutive rings, then reconstructed into sinograms by utilizing a high resolution Fourier Rebin algorithm. A reconstruction of sinograms yielded a 3D mapping of positron signal using a 2D filtered back-projection algorithm, with a Ramp filter at a Nyquist cut-off of 0.5. Image pixel size was anisotropic, with dimensions of 0.796 mm in the z direction and 0.861 mm in the x and y directions, for a total of 128 × 128 × 159 pixels.
CT was reconstructed from 360 projections of X-rays with a cone beam angle of 9.3 ° over 360 ° perpendicular to the animal bed. 80 keV X-rays were transmitted from a 500 μA anode source, 347 mm from the center of rotation and recorded on a CCD detector, containing 2,048 transaxial and 3,072 axial pixels. Projections were calibrated using 70 dark and 70 light images, interpolated bilinearly, processed through a Shepp-Logan filter, then reconstructed using a filtered back projection algorithm. Isotropic CT pixel size was 110.6 μm , with a total of 512 × 512 × 768 pixels. Scaling to Hounsfield Units, calibration was done using a 8.0 cm cylindrical phantom containing water prior to CT acquisition. During CT acquisition, iodine contrast was infused into the tail vein at a rate of 35 μL/ min to enhance intravascular contrast. Projections were acquired at end expiration using a BioVet gating system (M2M Imaging) and CT acquisition time was approximately 10 min. Reconstruction of datasets, PET-CT fusion, and image analysis were done using IRW software (Siemens). 3D visualizations were produced with the DICOM viewer OsiriX (The OsiriX foundation).
Imaging Antibody and Dextran Uptake in Tumor Slices.
Xenograft bearing mice were euthanized following PET-CT, tumors were excised and cut into 1 mm thick slices using a heart slicer (Zivic Laboratories). Tumor slices were then exposed to a phosphor screen (GE Healthcare) for 24 h. The next day, the screen was scanned using a phosphor imager (Typhoon 9410; GE) yielding autoradiographic imaging of 18F TCO localization in tumors. In the exact positioning used for autoradiography exposure, samples then underwent fluorescence reflective imaging (OV-110; Olympus) with a 0.56X objective and 280 ms exposure for localization of VT680 conjugated A33 antibody.
Supplementary Material
Acknowledgments.
The authors gratefully acknowledge S.A. Hilderbrand, J.B. Haun, and R. Mazitschek for many helpful discussions and suggestions. This work was funded in part by National Institutes of Health (NIH) grants P50CA86355, R01EB010011, K01EB010078, and T32CA079443.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1113466109/-/DCSupplemental.
References
- 1.Prescher JA, Dube DH, Bertozzi CR. Chemical remodelling of cell surfaces in living animals. Nature. 2004;430:873–877. doi: 10.1038/nature02791. [DOI] [PubMed] [Google Scholar]
- 2.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective "ligation" of azides and terminal alkynes. Angew Chem Int Ed Engl. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 3.Hong V, Steinmetz NF, Manchester M, Finn MG. Labeling live cells by copper-catalyzed alkyne-azide click chemistry. Bioconjug Chem. 2010;21:1912–1916. doi: 10.1021/bc100272z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jiang H, et al. Imaging glycans in zebrafish embryos by metabolic labeling and bioorthogonal click chemistry. Journal of visualized experiments. 2011;JoVE (52) doi: 10.3791/2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Agard NJ, Prescher JA, Bertozzi CR. A strain-promoted [3 + 2] azide-alkyne cycloaddition for covalent modification of biomolecules in living systems. J Am Chem Soc. 2004;126:15046–15047. doi: 10.1021/ja044996f. [DOI] [PubMed] [Google Scholar]
- 6.Agard NJ, et al. A comparative study of bioorthogonal reactions with azides. ACS Chemical Biology. 2006;1:644–648. doi: 10.1021/cb6003228. [DOI] [PubMed] [Google Scholar]
- 7.Baskin JM, et al. Copper-free click chemistry for dynamic in vivo imaging. Proc Natl Acad Sci USA. 2007;104:16793–16797. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ning XH, Guo J, Wolfert MA, Boons GJ. Visualizing metabolically labeled glycoconjugates of living cells by copper-free and fast huisgen cycloadditions. Angew Chem Int Ed. 2008;47:2253–2255. doi: 10.1002/anie.200705456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Codelli JA, Baskin JM, Agard NJ, Bertozzi CR. Second-generation difluorinated cyclooctynes for copper-free click chemistry. J Am Chem Soc. 2008;130:11486–11493. doi: 10.1021/ja803086r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Devaraj NK, Upadhyay R, Haun JB, Hilderbrand SA, Weissleder R. Fast and sensitive pretargeted labeling of cancer cells through a tetrazine/trans-cyclooctene cycloaddition. Angew Chem Int Ed Engl. 2009;48:7013–7016. doi: 10.1002/anie.200903233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Devaraj NK, Weissleder R, Hilderbrand SA. Tetrazine-based cycloadditions: application to pretargeted live cell imaging. Bioconjug Chem. 2008;19:2297–2299. doi: 10.1021/bc8004446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blackman ML, Royzen M, Fox JM. Tetrazine ligation: fast bioconjugation based on inverse-electron-demand Diels-Alder reactivity. J Am Chem Soc. 2008;130:13518–13519. doi: 10.1021/ja8053805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taylor MT, Blackman ML, Dmitrenko O, Fox JM. Design and synthesis of highly reactive dienophiles for the tetrazine-trans-cyclooctene ligation. J Am Chem Soc. 2011;133:9646–9649. doi: 10.1021/ja201844c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Devaraj NK, Hilderbrand S, Upadhyay R, Mazitschek R, Weissleder R. Bioorthogonal turn-on probes for imaging small molecules inside living cells. Angew Chem Int Ed Engl. 2010;49:2869–2872. doi: 10.1002/anie.200906120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rossin R, et al. In vivo chemistry for pretargeted tumor imaging in live mice. Angew Chem Int Ed Engl. 2010;49:3375–3378. doi: 10.1002/anie.200906294. [DOI] [PubMed] [Google Scholar]
- 16.Selvaraj R, et al. Tetrazine-trans-cyclooctene ligation for the rapid construction of integrin alpha(v)beta(3) targeted PET tracer based on a cyclic RGD peptide. Bioorg Med Chem Lett. 2011;21:5011–5014. doi: 10.1016/j.bmcl.2011.04.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reiner T, Keliher EJ, Earley S, Marinelli B, Weissleder R. Synthesis and in vivo imaging of a 18F-labeled PARP1 inhibitor using a chemically orthogonal scavenger-assisted high-performance method. Angew Chem Int Ed Engl. 2011;50:1922–1925. doi: 10.1002/anie.201006579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wingardh K, Strand SE. Evaluation in vitro and in vivo of two labelling techniques of different 99 mTc-dextrans for lymphoscintigraphy. Eur J Nucl Med. 1989;15:146–151. doi: 10.1007/BF00254628. [DOI] [PubMed] [Google Scholar]
- 19.Bisht S, Maitra A. Dextran-doxorubicin/chitosan nanoparticles for solid tumor therapy. Wiley Interdisciplinary Reviews-Nanomedicine and Nanobiotechnology. 2009;1:415–425. doi: 10.1002/wnan.43. [DOI] [PubMed] [Google Scholar]
- 20.Niemi TT, Miyashita R, Yamakage M. Colloid solutions: a clinical update. J Anesth. 2010;24:913–925. doi: 10.1007/s00540-010-1034-y. [DOI] [PubMed] [Google Scholar]
- 21.Orcutt KD, Nasr KA, Whitehead DG, Frangioni JV, Wittrup KD. Biodistribution and clearance of small molecule hapten chelates for pretargeted radioimmunotherapy. Mol Imaging Biol. 2011;13:215–221. doi: 10.1007/s11307-010-0353-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heath JK, et al. The human A33 antigen is a transmembrane glycoprotein and a novel member of the immunoglobulin superfamily. Proc Natl Acad Sci USA. 1997;94:469–474. doi: 10.1073/pnas.94.2.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ackerman ME, et al. A33 antigen displays persistent surface expression. Cancer Immunol Immunother. 2008;57:1017–1027. doi: 10.1007/s00262-007-0433-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weissleder R, Pittet MJ. Imaging in the era of molecular oncology. Nature. 2008;452:580–589. doi: 10.1038/nature06917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lehr HA, Leunig M, Menger MD, Nolte D, Messmer K. Dorsal skinfold chamber technique for intravital microscopy in nude mice. Am J Pathol. 1993;143:1055–1062. [PMC free article] [PubMed] [Google Scholar]
- 26.Keliher EJ, Reiner T, Turetsky A, Hilderbrand SA, Weissleder R. High-yielding, two-step 18F labeling strategy for 18F-PARP1 inhibitors. ChemMedChem. 2011;6:424–427. doi: 10.1002/cmdc.201000426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee FT, et al. Enhanced efficacy of radioimmunotherapy with 90Y-CHX-A”'-DTPA-hu3S193 by inhibition of epidermal growth factor receptor (EGFR) signaling with EGFR tyrosine kinase inhibitor AG1478. Clin Cancer Res. 2005;11:7080s–7086s. doi: 10.1158/1078-0432.CCR-1004-0019. [DOI] [PubMed] [Google Scholar]
- 28.Thurber GM, Zajic SC, Wittrup KD. Theoretic criteria for antibody penetration into solid tumors and micrometastases. J Nucl Med. 2007;48:995–999. doi: 10.2967/jnumed.106.037069. [DOI] [PubMed] [Google Scholar]
- 29.Mager DE. Target-mediated drug disposition and dynamics. Biochem Pharmacol. 2006;72:1–10. doi: 10.1016/j.bcp.2005.12.041. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.