

Abstract
The ABCC6 gene encodes an organic anion transporter protein, ABCC6/MRP6. Mutations in the gene cause a rare, recessive genetic disease, pseudoxanthoma elasticum, while the loss of one ABCC6 allele is a genetic risk factor in coronary artery disease. We review here the information available on gene structure, evolution as well as the present knowledge on its transcriptional regulation. We give a detailed description of the characteristics of the protein, and analyze the relationship between the distributions of missense disease–causing mutations in the predicted three-dimensional structure of the transporter, which suggests functional importance of the domain-domain interactions. Though neither the physiological function of the protein nor its role in the pathobiology of the diseases are known, a current hypothesis that ABCC6 may be involved in the efflux of one form of Vitamin K from the liver is discussed. Finally, we analyze potential strategies how the gene can be targeted on the transcriptional level to increase protein expression in order to compensate for reduced activity. In addition, pharmacologic correction of trafficking-defect mutants or suppression of stop codon mutations as potential future therapeutic interventions are also reviewed.
Keywords: genetic disease, connective tissue, cardiovascular, transcriptional regulation, calcification, vitamin K, membrane proteins, homology model
Pseudoxanthoma elasticum
Pseudoxanthoma elasticum (PXE, OMIM 264800) is a recessive genetic disorder with a prevalence of 1: 25.000 – 100.000, affecting the elastic tissues of the body, including the skin, the arteries and the elastic Bruch’s membrane in the eye. Patients most commonly present with characteristic papules in the skin during late childhood or adolescence and subsequently develop angioid streaks of the retina. Angioid streaks are associated with subretinal neovascularisation, which can lead to hemorrhage and partial or complete loss of central vision. The diagnosis of pseudoxanthoma elasticum is suspected in individuals with characteristic skin and ocular findings and is confirmed by histological findings on biopsy of lesional skin in which fragmented calcified elastic fibers are visualized by use of special histologic stains (e.g. von Kossa staining).
The disease was first described in 1881 by D. Rigal [1] and Félix Balzer, but the term Pseudoxanthoma elasticum (PXE) was first used in 1896 by Jean-Ferdinand Darier [2]. In 1889 Robert W. Doyne was the first to describe angioid streaks, than Ester Grönblad and James Strandberg revealed the connection between PXE and angioid streaks in 1929 [3].
Cutaneous and mucosal manifestations
The first sign of PXE is usually yellowish papules on the neck and other flexor surfaces. These skin lesions vary in size from 1–5 mm and may be grouped or coalesce to form larger plaques (Figure 1/A). In most of the cases the skin loses its elasticity and becomes wrinkled and redundant. In addition to the neck, plaques may also appear on other areas, such as axillae, inguinal region, antecubital and polpliteal fossae, and periumbilical area during the progression of the disease. In some cases mucosal lesions, identical to the skin lesions, can be detected on the inside of the lower lip, vagina, and all along the digestive tract mucosal membrane [4].
Figure 1.
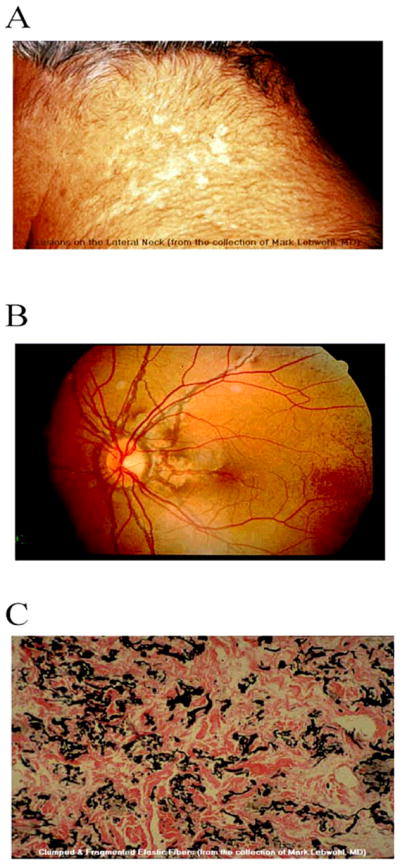
Manifestation of pseudoxanthoma elasticum. A: skin symptoms; B: angioid streaks in the eye; C: von Kossa staining of calcium deposits and fragmented elastic fibers in the skin biopsy of a PXE patient. The pictures are from the PXE International, with permission.
The classic histological findings in PXE are ultrastructural elastic tissue abnormalities in the middle and lower dermis. In PXE patients elastin becomes fragmented and degenerated (See Figure 1/C). Deposition of calcium in the abnormal elastin matrix can be visualized by von Kossa or other histological stains for calcium [5]. Similar clinical findings may also be present is some other diseases for example in beta-thalassemia [6], focal dermal elastosis [7], cutis laxa [8], calciphylaxis [9] or Paget’s disease [10]. However the ultra-structural histopathology is a hallmark of PXE. Even the skin findings in a highly related disease (PXE-like disorder with multiple coagulation factor deficiency) are in detail slightly different from the skin lesions found in PXE patients [11].
Eyes (streamlined according to Rev2 suggestions)
Symptoms eventually appear in the eyes in all cases of PXE (Figure 1/B). The affected areas are the Bruch’s membrane and the retinal-pigmented epithelium (RPE).
In PXE, calcification of dystrophic elastic fibers can be observed in the elastic layer of Bruch’s membrane, similar to what is seen in the skin. Pigment irregularities, called peau d’orange, may appear in the RPE. Calcification and thickening of the Bruch’s membrane, as well as loss of RPE pigment granules lead to the development of angioid streaks (AS) [12].
AS are dehiscences in Bruch’s membrane, forming grayish to reddish irregular lines resembling vessels, emanating from the optic disk. Due to the calcium deposition, Bruch’s membrane becomes fragile, which is thought to be the major factor resulting in the dehiscences within the membrane. Later in the course of the disease fibrovascular tissue may grow through the damaged membrane, leading to choroidal neovascularization (CNV), subretinal fibrosis, atrophy of the overlying RPE and retinal hemorrhages [13].
Hemorrhages from the fragile new vessels can lead to partial or complete vision loss in PXE patients, starting usually around the third or fourth decade of life. Central vision loss has the greatest impact on the quality of life in PXE patients, but thus far no preventive measures are available. Laser therapy can be used to stop the proliferation or the bleeding of submacular neovessels, but it may cause visual loss or central scotomas due to scarring, and a high rate of recurrence has been observed [14,15]. A promising therapy to treat ocular symptoms of PXE may be the application of antiangiogenic drugs. There have been several publications documenting efficacy of intravitreal treatment with vascular endothelial growth factor (VEGF) monoclonal antibodies in case of age-related macular degeneration resulting in CNVs [[16,17]. Similar treatment was already applied for a small group of PXE patients, showing very promising results [18].
Cardiovascular system
Calcification may affect the cardiovascular system, mostly the small and middle-sized arteries.
Cardiovascular symptoms include diminished peripheral pulses, angina pectoris, hypertension, mitral-valve prolapse and restrictive cardiomyopathy [19–23]. One of the most common cardiovascular symptoms is intermittent claudication. Bleeding, especially gastrointestinal hemorrhages may also occur probably due to calcification of the elastic fibers in the small arteries located under the mucosa [24]. Myocardial infarction or other symptoms leading to sudden death are rare, but probably the most serious complication associated with PXE is the early onset of atherosclerosis [4].
ABCC6 as a potential genetic risk factor in Coronary Artery Disease (CAD)
A strong correlation between a sequence variant of the ABCC6 gene (c.3421C>T leading to the p.R1141X non-sense mutation) and CAD has been demonstrated in a Dutch cohort [25]. However, a surprisingly high frequency of the mutant allele was observed in the control population raising doubts about the validity of this unique study on the association of ABCC6 mutation carrier status and CAD. An independent population genetic study on a large Hungarian cohort confirmed the findings of the previous report [26]. A significant association of carrier status and CAD was observed (5/361 carriers p=0.016 OR=10.5 95%CI 1.22–90.30). These findings mean that one non-functioning allele of ABCC6 increases the risk of CAD significantly.
The ABCC6 gene
In 2000 it was discovered by positional cloning that mutations in ABCC6 gene are responsible for the development of PXE [27–29]. Since the discovery of the connection between ABCC6 mutations and PXE a large number of disease-causing mutations has been identified; the most frequent ones are p.R1141X (20 – 30%) and c.EX23_29del (5 – 15). The high heterogeneity of PXE alleles in the population is comparable to that of other autosomal diseases. A locus-specific database has been established recently with the collection of the disease-causing mutations and other genetic variants and with link to other genetic databases [www.ncbi.nlm.nih.gov/lovd/home.php?select_db=ABCC6]. ABCC6 is located at 16p13.11 and codes for the ATP-binding cassette transporter protein, ABCC6/MRP6. The functional gene of 75 kb size consists of 31 exons. Two pseudogenes that are expressed at low levels and are positioned centromeric (ABCC6-ψ1) and telomeric (ABCC6- ψ2) of ABCC6 have also been mapped [30]. Both ABCC6-ψ1 and ABCC6-ψ2 share a high degree of sequence similarity (~99%) with the functional gene, but are truncated in the fourth and ninth intron, respectively. The ABCC6 locus is located in a genomic region that was subject to segmental duplications, a series of events thought to play a crucial role in the recent evolution of ABCC6 gene cluster [31]. Due to this evolutionary scenario chromosomal rearrangements, gene conversion and emergence of new genes have been observed.
There are several reports indicating that the ABCC6 locus is genetically unstable. A rare fragile site (FRA16A) has been found in close centromeric proximity to ABCC6 [32], while the breakpoint of rearranged Chromosome 16 in the acute non-lymphocytic leukemia cell line M4Eo was localized ~0.5 Mb telomeric of ABCC6 [33,34]. ABCC1, the gene located closest to ABCC6 (8 kb apart) is frequently deleted in the drug-selected M4Eo cell line, and the concomitant amplification of ABCC1 and ABCC6 is observed in the SKOV3 ovarian carcinoma cell lines after multidrug selection [35].
Transcriptional regulation
The initial characterization of the transcriptional regulation of the human ABCC6 gene identified two evolutionarily conserved regions in the 5′ sequence 10kb upstream from the translation start site [36]. Both regions harbor a CpG island (CGI), potential target of DNA methylation. Analysis of DNA methylation may give clues to the location of important regulatory regions of gene expression, as methylation is stable like an imprint. Methylated regions indicate silenced and unmethylated regions design transcriptionally active sequences [37]. Bisulfite genomic sequencing was carried out to analyze both the distal and the proximal CGI in different cell lines expressing and non-expressing ABCC6 [36]. Cell-type specific DNA methylation in the proximal CpG island was detected, which inversely correlated with the expression of the gene and suggested that this region plays an important role in the tissue-specific regulation of ABCC6.
Based on these data luciferase reporter gene assays were performed with sequential deletion promoter constructs. One silencer (between −713 and −332 bp) and one DNA methylation sensitive activator sequence (between −332 and −145 bp) were identified. These data indicated that this region confers tissue-specificity to the ABCC6 expression pattern. Further promoter mapping experiments confirmed these findings by identifying one tissue-specific regulator element (between −209 and −145 bp) and one further stronger activator sequence located between −234 and −209 bp [38].
The potential regulatory role of some transcription factors and cytokines has been suggested. The binding of the PLAG family of transcription factors and RXR has been convincingly demonstrated: they are able to transactivate the endogenous ABCC6 gene, the binding site was determined by luciferase assay and the binding to the ABCC6 promoter in the natural chromatin environment was demonstrated by chromatin immune precipitation. However, their functional role is still unclear [38,39]. The binding of NF-κB, SP1 and TGF-β has been also suggested but their functional role and their binding to the endogenous ABCC6 promoter have not been tested [40].
Signal transduction pathways leading to the modulation of ABCC6 expression have also been deciphered. Initially the activation by TGF-β and inhibition by TNF-α and IFN-γ were reported in luciferase reporter gene assays [40]. However, these effects have not yet been confirmed on the endogenous gene and the implicated signal transduction pathway was not identified. More recently, it was found that activation of the MAP kinase ERK1/2 cascade leads to the significant inhibition of the expression of ABCC6 in HepG2 and Caco-2 cell lines [41]. The detailed analysis of the molecular mechanism of this inhibition of ABCC6 expression revealed that this is a direct inhibition of transcription initiation and the response element was mapped between −166 and −154 bp relative to the transcription initiation site. This binding site is a degenerate but functional HNF4 (hepatocyte nuclear factor 4) binding site as suggested by siRNA and chromatin immune precipitation [42,43]. Luciferase assays with wild type (wt) and mutated constructs and co-expression of HNF4 with the wt construct in cells where endogenous HNF4 is not expressed also confirmed these data [41]. Interestingly this effect of HNF4 was completely prevented by the activation of ERK1/2.
These experiments also revealed the major coordinator role of HNF4 in the regulation of ABCC6. Mutated constructs with abolished HNF4 binding site have also prevented further activation of the luciferase activity by the other transcription factors [41]. This strongly suggests that HNF4 is responsible for the tissue-specific regulation of ABCC6. It is worth to note that ABCC6 and HNF4 tissue-specific expression completely overlap [44,45].
Modifying genes and PXE phenocopies
The PXE phenotype is highly variable even within a single family where patients have the same disease-causing mutations [4]. No clear genotype/phenotype correlation has been observed to date [46]. Finally, other diseases can mimic the PXE phenotype. The PXE-like syndrome is due to mutations in the gamma-glutamyl carboxylase (GGCX) gene [11], while certain genetic hemoglobinopathies (e.g. thalassemia) lead to slowly developing phenotypes similar to PXE [47]. Although the molecular mechanisms of the developing phenotype are not yet understood, in beta thalassemia mice a liver-specific down-regulation of Abcc6 gene expression was observed [48]. Christian Gotting and his co-workers identified a number of genes modifying the disease course in a German cohort. They demonstrated that the ABCC6 c.-219A>C promoter polymorphism is significantly less frequent in patients than in the control population [49]. They also showed that certain promoter polymorphism of the SPP1 (secreted phosphoprotein 1, previously called: osteopontin) gene were more frequent in PXE patients than in controls [50]. Furthermore, earlier disease onset is associated with polymorphisms of catalase, superoxide dismutase and glutathione peroxidase genes [51]. They also found that polymorphisms of the VEGF (vascular endothelial growth factor gene) are prognostic markers for ocular symptoms [52]. Similarly, it was observed that the c.2402C>G p.T801R polymorphism of the xylol-transferase II genes is associated with increased PXE severity [53].
The ABCC6/MRP6 protein
The human proteome contains 48 ABC proteins; on the basis of sequence similarity they are grouped into seven subfamilies from A to G. The ABCC-subfamily includes twelve members; most of them are active transporter while ABCC7 (CFTR) acts as a chloride channel and probably regulates the action of other ion channels (for more details, see the relevant chapter of the present issue: CFTR [ABCC7] target in Cystic Fibrosis). Two other members of the subfamily, ABCC8 and 9 are K+-channel regulators operating as intracellular ATP/ADP sensors thus reporting about the metabolic state of the cells (for more details, see the relevant chapter of the present issue: ABCC8/9 target in type 2 diabetes).
The ABCC-proteins share the general features of the ABC-kingdom: they harbor two nucleotide-binding (ABC)-domains and two transmembrane domains (TMDs), each with six membrane-spanning helices (the so called “core structure”). It is a unique feature of some ABCC-type proteins (“long MRPs” like ABCC1, 2, 3, 6, 8, 9 and 10) that two additional domains are attached to the core structure N-terminally: a transmembrane domain with five membrane spanning helices and an intracellular loop. Accordingly, the domain architecture of the long MRPs, including ABCC6/MRP6 is TMD0-L0-TMD1-ABC1-L1-TMD2-ABC2 (L0 and L1 are intracellular loop) (Figure 2 panel A). ABCC6 consists of 1503 amino acids and it is known that the protein functions as an organic anion transporter [54,55]. Indeed, in vitro studies demonstrated the transport of glutathione-conjugates like glutathione S-conjugated leukotriene C4 (LTC4), N-ethylmaleimide S-glutathione (NEM-GS) and S-(2,4-dinitrophenyl) glutathione, while the rat orthologue transports an anionic cyclopentapeptide [56]. It has also been shown by in vitro assays that some missense mutations described as causative mutations in pseudoxanthoma elasticum result in the loss of ATP-dependent transport of test substrates [54]. Compared with its sub-family members (ABCC1–5) ABCC6 is a poorly characterized transporter. The protein shows significantly lower transport rate (turnover number) in in vitro assays than the other human ABCC-type transporters, which makes its detailed biochemical/functional characterization difficult.
Figure 2.

Membrane topology and three dimensional homology models of ABCC6. Missense mutations are indicated in red. A: the membrane homology model and domain arrangements of ABCC6. B: three dimensional homology model of ABCC6 representing the outward facing conformation; C: three dimensional homology model of ABCC6 representing the nucleotide-free conformation, Insert: schematic representation of the domain swapping of ABC proteins.
It has been suggested that overexpression of ABCC6 is able to confer low levels of resistance to several commonly used natural product anticancer agents like etoposide, doxorubicin, daunorubicin and actinomycin D [55]. However, clinically relevant ABCC6-mediated drug resistance has never been found.
Homology models
No high-resolution three dimensional structure of ABCC6 is available. However, a three dimensional homology model of ABCC6 is already built and published [57], made possible by the recent publication of high resolution crystalline structures of ABC proteins [58–60]. One of the structures representing the nucleotide-saturated, outward facing conformation show that the two nucleotide-binding (ABC) domains are in close proximity to each other in the characteristic head-to-tail orientation reflecting to the previously described “nucleotide sandwich dimmer” [61]. The other shows a nucleotide-free, substrate-saturated conformation. Newly recognized structural elements are the long “rigid” extensions of the transmembrane helices, called intracellular loops (ICL). Each half of the ABC proteins has two ICLs interacting with the ABC-domains. The coupling helices contact with their “own” as well as with the “opposite” ABC-domains, hence a special type of domain swapping can be recognized in the structure (see insert on Figure 2).
We have constructed two homology models of human ABCC6 protein: one of the models is based on the Sav1866 bacterial ABC transporter structure [57] representing a nucleotide-saturated conformational state, while the other one uses the recently published mouse Abcb1 structure as template and represents the nucleotide-free (apo) conformation [Váradi et al, unpublished]. The two models are illustrated on Figure 2, Panel B and C. By performing a statistical analysis we have found a significant clustering of the missense PXE-mutations at the domain-domain interfaces: at the transmission interface that involves four intracellular loops (ICLs) and the two ABC domains as well as at the ABC - ABC interacting surfaces. In the nucleotide-saturated model the mutations affecting these regions are 2.75 and 3.53 fold more frequent than the average mutational rate along the protein sequence, respectively [57]. At the predicted ICL-ABC interfaces in the nucleotide-free model the mutational rate is 4.25-fold more frequent than the average mutational rate along the protein sequence (the ABC domains are distant in this conformation) [Váradi et al, unpublished]. The observed significant clustering means that the domain contacts are much less permissive to amino acid replacements than the rest of the protein. These results provide a “bridge” between genetic data and protein structure and can be viewed as novel proof of the importance of the studied domain-domain interactions in the ABCC6 transporter.
Animal models
Abcc6 knock out mouse models were generated and the critical role of Abcc6 in ectopic mineralization/calcification has been confirmed in the Abcc6−/− mice which recapitulates the genetic, histopathologic and ultrastructural features of PXE [62,63]. These findings suggest that the function of this transporter is conserved in the mouse. Calcification in the vibrissae capsules is the first symptom of the calcification phenotype detected at the 8 to 10 weeks of age and serves as an early biological marker of the disease [62]. A slight alteration of plasma lipid composition of the Abcc6−/− mice has also been reported [63]. The KO mouse models have been utilized for physiological and for pharmacological studies that are discussed elsewhere in this paper.
Dystrophic Cardiac Calcification (DCC) in the mouse is an autosomal recessive trait in certain laboratory strains and the Abcc6 gene locus has been recently found as a main mediator of DCC at the Dyscalc1 locus [64,65]. A splicing error in processing of Abcc6 mRNA has been identified as the causative genetic event (“splice-mutation”) of DCC. This mouse shows a more pronounced arterial calcification phenotype than the one observed in the laboratory-generated Abcc6−/− mice strains (presumably due to the different genetic background) and seems to be as good model of PXE as the latter.
The zebrafish (Danio rerio) has nearly the same ABC gene repertoire as the human and has accessible and well-characterized embryo. Two morpholinos were designed targeting two different regions of the Abcc6a gene (the Abcc6b gene was found to be inactive), and it was observed that they decrease Abcc6a expression by 54 and 81%. Both morpholinos induced a similar phenotype, cardiac edema and curled tail. Microinjecting zebrafish larvae with full-length mouse Abcc6 mRNA completely rescued the knockdown phenotype [66]. These recent results serve as basis of a novel knockdown animal model system. However, the results provided by this model may not be translated directly to human physiology, as the zebrafish gene appears to be essential for the development of the animal.
The “Vitamin K hypothesis” of PXE
ABCC6 is predominantly expressed in the liver in the basolateral compartment of the plasma membrane of the hepatocyte (and to a lesser extent in the kidney), while the symptoms are systemic affecting various organs. This apparent discrepancy led to the hypothesis that PXE is a metabolic disease suggesting that ABCC6 is involved in secretion of a metabolite from the liver into the circulation [67]. Recent experiments demonstrated that grafting of wt mouse muzzle skin onto the back of KO mice triggered mineralization, whereas grafting KO mouse muzzle skin onto wt mice was accompanied with no mineralization [68]. These transplantation experiments argue that PXE is indeed a metabolic disorder. Furthermore, in a parabiotic experiment the surgical pairing of Abcc6(−/−) mice with wild-type prevented the mineralization of the connective tissue in the knockout mice [69].
There have been a few case reports of a disease that phenotypically resembled pseudoxanthoma elasticum with respect to the mineralization of soft tissues causing cardiovascular, dermal and ocular symptoms. However, these patients suffer from a vitamin K-dependent coagulation factor deficiency which is not seen in PXE [70–72]. The disorder is extremely rare and for decades its molecular basis remained unknown. Also, no mutations in the ABCC6 gene could be detected in these individuals, suggesting that mutation of another gene could also cause PXE-like soft tissue calcification. The identity of this enigmatic gene was unraveled recently [11], and the clinical condition was classified as a novel disorder: PXE-like disease (pseudoxanthoma elasticum-like disorder with multiple coagulation factor deficiency, OMIM 610842). Six patients were found to possess compound heterozygous mutations in the gamma-glutamyl carboxylase (GGCX) gene.
The GGCX gene encodes the gamma-glutamyl carboxylase enzyme (GGCX), an ER (endoplasmic reticulum)-resident protein, responsible for post-synthetic carboxylation of Gla-domain containing proteins to which they confer Ca-binding properties [73]. During the carboxylation reaction, vitamin K (VitK) is oxidized to an epoxide form, which is then re-reduced by another enzyme, Vitamin K oxido-reductase (VKORC1), thereby completing the VitK-cycle [74]. Proteins with Gla residues bind calcium (and certain other divalent cations) and this property is required for their physiological function. The best-known members of this group of proteins are the vitamin K-dependent coagulation factors produced in and secreted from the liver. The reduced activity of the GGCX enzyme explains the coagulation deficiency in PXE-like patients. Another gamma glutamyl carboxylated protein is MGP (matrix gla protein) which is a potent inhibitor of connective tissue mineralization and its function is essentially dependent on the correct carboxylation of the protein [75–77]. Insufficient carboxylation of MGP might thus be responsible for the soft tissue calcification in PXE-like patients. MGP can be detected in mineralized tissues of individuals with classic PXE as well as in Abcc6 mutant mice [78–80]. Using antibodies specifically recognizing the non-carboxylated and the carboxylated forms of MGP, it has been shown that sites of ectopic calcification only contained the undercarboxylated form of MGP [79,80]. These data led to the hypothesis that ABCC6 could directly or indirectly influence the availability of vitamin K, or the capacity of the vitamin K cycle at peripheral tissues. The highly similar phenotypic features of the two diseases evoked hypotheses about their overlapping patho-physiology. In PXE-like disease – due to the mutations of the GGCX enzyme - the Gla-gammacarboxylation is reduced in the liver, thus resulting in blood coagulation abnormality, and also in the extrahepatic soft tissues where the control of calcification is impaired. In classical PXE gamma carboxylation is normal in the liver as there is no mutation in GGCX. According to the current hypothesis, in extrahepatic tissues gamma carboxylation is lower than normal as Vitamin K available for the carboxylation cycle may be limited in those tissues.
This notion is supported by the very recent finding that the level of circulating vitamin K1 is lower in PXE patients as compared to healthy controls [81]. It is notable that the variability between patients is high and the range of measured vitamin K1 levels in the control group overlaps with that of the PXE patient group. Collectively, these data raises the possibility that one form of Vitamin K is transported from the liver into the circulation, and this transport is mediated by ABCC6 (and is missing in PXE due to ABCC6 mutations) [82].
On the other hand, the fact that PXE is a slowly progressive disease suggests that the metabolite transported by ABCC6 may only be reduced but not completely absent in the circulation of patients. Besides being a co-factor of gamma carboxylation, VitK may have other physiological functions, including transcriptional regulation and protection of certain neuronal cells from oxidative injury [83]. These findings need to be taken into consideration when studying the connections between vitamin K status, ABCC6 and PXE. Whether there is a connection between protection against oxidative stress by VitK and the chronic oxidative stress in measured in the serum and cells of PXE patients [51,84,85] is unknown and needs further investigation.
Currently, the exact metabolic pathway of Vitamin K is not known in detail. Dietary Vitamin K (phylloquinone, VitK1) is mostly utilized in the liver to serve as a cofactor in blood clotting factor synthesis. Part of Vitamin K1 is converted by side-chain removal to Vitamin K3 (also known as menadione) probably in the enterocytes [86]. K3 can be taken up by the extrahepatic tissues and the complex aliphatic side chain is substituted to the naphtoquinone core thus generating MK4 (a menaquinone, also called Vitamin K2), which is available for the Vitamin K cycle of the extrahepatic tissues [87]. It is known that both Vitamin K1 and MK4 are metabolized to a common catabolite after beta-oxidation of the side-chains and subsequent glucoronidation and the conjugate is secreted into the urine [88]. K3 can be conjugated with glutathione. The known metabolic events and vitamin K compounds are shown in Figure 3.
Figure 3.

The major Vitamin K forms and metabolites. I: VitK1; II. MK4 (VitK2); III: VitK3 (menadione); IV: VitK3-glutathione conjugate; V: VitK aglycone; VI: aglycone glucoronide. Beta-oxidation is represented by reaction a; glucoronidation by b; glutathione conjugation by c, conversion of VitK1 to VitK3 by d, while resynthesis of the sidechain generating MK4 by e.
In principle, any of the Vitamin K metabolites or molecular forms (see Figure 3.) could be the transported substrate(s) of ABCC6, that, according to the “Vitamin K hypothesis”, control(s) indirectly - via gamma-carboxylation of Ca-binding proteins like MGP - the formation of calcium-deposits in the arterial wall and in other soft tissues. However, the key role of Vitamin K in the disease phenotypes associated with ABCC6 has not been proven. Indeed, very recent independent studies challenged the “Vitamin K theory”. In one of the papers [Jiang et al, in press] it was demonstrated that oral administration of massive amount of vitamin K2 did not alter the ectopic mineralization in Abcc6−/− mice. Similarly, intravenous administration of Vitamin K3-glutathion conjugate (K3-GSH) did not alter the degree of mineralization. Furthermore, the same authors also found that vitamin K2, K3 and K3-GSH has no effect in an in vitro calcification system, i.e. they did not trigger any mineralization inhibition.
In the other study [Brampton et al, submitted] Abcc6−/− mice were placed on a diet of either 5 or 100 mg/kg of vitamin K1 or K2 at prenatal, 3 weeks or 3 months of age. These authors also found no significant change in the levels of pathologic calcification irrespective which type of administration was used. However, measuring the plasma levels of different Vitamin K forms resulted in a very interesting observation: upon the same administration the level of Vitamin K1 in the plasma of wt mice was significantly higher than in the plasma of the Abcc6−/− animals. The same was true when Vitamin K2 was administrated orally. These results suggest that Abcc6 might be involved in vitamin K transport, absorption or metabolism in the body to some degree, which is in agreement with the observations discussed in the previous paragraphs [81].
Unequivocally, both studies provided evidence that dietary supplementation in vitamin K is not a viable approach to preventing PXE-related calcification thus suggesting that the availability of vitamin K is not a limiting factor in the pathology of PXE.
Local expression of ABCC6 has also been shown in several tissues and cell types, e.g. keratinocytes, fibroblasts, smooth muscle cells and macrophages [89–91], some of those are affected in PXE. The local effect of missing ABCC6 activity may also contribute to the progression of the disease.
ABCC6 as a drug target
Two disease conditions are associated with mutations in the ABCC6 gene: pseudoxanthoma elasticum is a recessive trait due to mutations in both ABCC6 alleles, while the loss of one functional ABCC6 allele is a genetic risk factor in coronary artery disease, CAD. As the phenotype in both cases is due to complete or partial loss of ABCC6 activity, augmentation-type gene therapy – in principle – could be an effective treatment of the disease conditions. However, even if all the safety concerns of gene therapy were solved, there are questions to be answered: is it sufficient to restore ABCC6 activity only in the liver, or it is also needed to do in other organs with lower level of expression (e.g. kidney)?
Some of the PXE-causing ABCC6 mutations may result in only partial loss of function of the protein. Induction of expression of such a low activity mutant – in theory – could potentially be therapeutic in these cases. The induction of ABCC6 expression could be achieved at distinct levels: increased transcription, increased RNA stability or increased protein stability. However, we have almost no information to date about the regulation of ABCC6 RNA and protein stability. Therefore, currently the only way to develop a hypothesis-driven therapy based on increased ABCC6 expression is to intervene at the transcriptional regulation of the gene. As discussed previously the transcriptional regulation of ABCC6 was investigated by different groups and the potential role of several transcription factors has been suggested [38–40]. However, only the role of ERK1/2-HNF4 pathway has been analyzed in a potential physiological context. As previously mentioned, HNF4 is a bona fide activator of the gene, while the activation of the ERK1/2 pathway inhibits HNF4 and thereby the expression of ABCC6 [41].
ERK1/2 regulate several physiological processes, such as cell growth and proliferation, differentiation, survival and apoptosis [92]. The pathway is activated by a number of stimuli, like growth factors and environmental stresses (e.g. oxidative stress) or through various G protein coupled receptors [93]. The pathway has a low basal activity and upon activation it is rapidly deactivated by different phosphatases the most important among them being PP2A (protein phosphatase 2A). PP2A itself is under the control of protein kinase A and other signaling cascades [94].
An option to increase the expression level of the ABCC6 gene would be the inhibition of ERK1/2 activation in hepatocytes. Due to the myriad of ERK1/2 activating factors, the low basal activity and the fast turnover of the signals, inhibitory stimuli of ERK1/2 are not easy to find. However, some of the anticancer therapies are targeting the ERK1/2 pathway and promising results have been obtained both in vivo and in vitro in inhibiting cancer progression [95,96], It is highly probable that new generations of these molecules will become available. These molecules might have beneficial effect in some patients because only a slight inhibitory effect would be necessary in the case of PXE symptoms, the toxic effect of the molecules on other tissues would be diminished. Furthermore, in some cases patients are suffering from chronic oxidative stress and develop secondary PXE (e.g. in beta thalassemia) [47,48]. These patients treated with ERK1/2 inhibitors and/or an anti-oxidative stress therapy might also receive therapeutic benefit. Similarly, the ABCC6 mutation carriers also suffer from mild oxidative stress [51,97], a condition potentially inhibiting the expression of the gene from the remaining allele. These patients could presumably benefit from the same therapy to prevent the development of CAD.
ERK1/2 has a wide variety of targets. One of them is HNF4, which is inactivated either directly or indirectly by the kinase [98,99]. HNF4 transcription factor is a major regulator of the expression of ABCC6 and also a master regulator of metabolism in hepatocytes. By influencing the global metabolic state of the liver one can induce the expression and/or the activity of HNF4. Accordingly, it has been demonstrated that in fasting states in the mouse or during high hepatic glucose levels functional HNF4 level is increased [100]. Similarly, it has been shown, that HNF4 is up-regulated by glucocorticoid hormones [101]. All these conditions might contribute to a higher ABCC6 expression level and diminishing or eliminating the PXE symptoms in some PXE and probably the beta thalassemic patients.
Missense disease-causing mutations can reduce the transport activity and/or the overall stability of the transporter, or may result in a slightly altered conformation that is not compatible with the normal trafficking of the protein to the plasma membrane. Indeed, defective protein trafficking caused by mutations underlies many human diseases and examples include several membrane-embedded ABC-proteins (like ABCC7/CFTR, ABCC2, ABCC8/SUR1 or ABCB11/BSEP). Efforts to identify pharmacologic compounds to correct the misfolding and/or misprocessing of mutant membrane proteins have already resulted in a few remarkable findings, and are considered as the molecular basis of allele-specific therapy of the given disease (see e.g. [102]). This observation raises the possibility that pharmacological compounds (acting either as “chemical chaperones” or interfering with the quality control of the protein sorting/processing mechanism) may correct the defect causing the disease in a group of patients. Substrates and modulators of ABCB1 have been demonstrated to act as chemical chaperones thus helping the appearance of fully mature protein at the cell surface in the case of processing ABCB1 mutants [103]. The first effort of promising correcting of folding of the delta508CFTR mutant was achieved by curcumin, which might act as a chemical chaperone [104]. High-throughput screening systems can be designed to test large chemical libraries for other “corrector” compounds. We have constructed a Hemagglutinin (HA) epitop-tagged version of human ABCC6: the antibody-reactive tag has been inserted into the extracellular N-terminal segment of the protein [Iliás and Váradi, unpublished]. The protein preserved transport activity and showed cell surface expression similar to the wild type, and the HA tag recognized on the surface of intact, ABCC6-overexpressing cells provided a signal for correct trafficking. However, such an approach has inherent limitations in the case of ABCC6. In PXE – in contrast to cystic fibrosis, where deltaF508CFTR mutation is present in the majority of the patients – there is no such a high frequency mutation. It is expected, that different traffick-defect mutants are arrested at different points of the trafficking pathway and may need different “intervention”.
The most frequent mutation in PXE is the R1141X nonsense mutation. It has been shown that aminoglycoside antibiotics like gentamicin can suppress premature stop codon arrest of translation by inducing the ribosome to read through (or “suppress”) the nonsense mutation via insertion of an amino acid. It was demonstrated in a muscular dystrophy mouse model that aminoglycosides could suppress stop codons not only in vitro but also in vivo [105]. However, the efficacy of the “read through” may be quite low. Enhanced production of active protein from genes with nonsense mutations can be achieved by combining treatment by inducing the promoter of the gene and the application of “read through” agents [106]. A promissing new non-aminoglycoside agent of suppressing premature termination codons is PTC124 (Ataluren), which is in clinical trial in cystic fibrosis, in Hemaphilia A and B and in Duchenne muscular dystrophy [107]. These strategies can now be tested in experiments with the aim of correcting the frequent ABCC6 R1141X stop codon mutation.
On Figure 4 we have summarized the potential targets of the therapeutical interventions discussed above.
Figure 4.
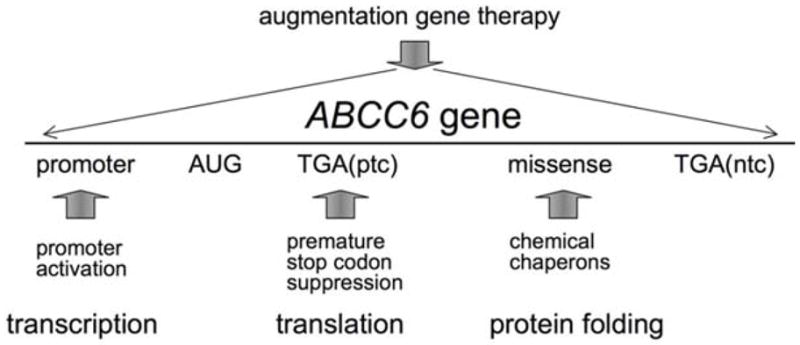
Targets of potential therapeutical interventions. Solid vertical arrows represent the target of intervention; TGA(ptc) means TGA premature termination codon (e.g. R1141X in several PXE patients); TGA(ntc) means TGA at the natural termination codon position (1504 in ABCC6 mRNA).
Acknowledgments
This work has been supported by Hungarian research grants OTKA NI 68950, NHTK-OTKA CK 80135, OTKA PD 79183, Bolyai János fellowship (to TA) and by NIH RO1 AR055225 (to AV). Images on Figure 1 were kindly provided by PXE International, while the help of László Barna (Institute of Enyzmology) in creating Figure 3 is greatly appreciated.
Abbreviations
- ABC
ATP-binding cassette
- ABCC6
ATP-binding cassette protein, family C, number 6
- AS
angioid streak
- CAD
coronary artery disease
- CGI
CpG island
- ChIP
chromatin immunoprecipitation
- CNV
choroidal neovascularization
- DCC
dystrophic cardiac calcification
- ERK1/2
extracellular signal- regulated kinase 1/2
- GGCX
γ-glutamyl carboxylase
- HNF4
hepatocyte nuclear factor 4
- K1
vitamin K1
- K2/MK4
vitamin K2/menaquinone
- K3
vitamin K3/menadione
- KO
knock out
- MAPK
mitogen-activated protein kinase
- MGP
matrix gla protein
- MRP
multidrug resistance-associated protein
- PXE
pseudoxanthoma elasticum
- RPE
retinal pigmented epithelium
- VEGF
vascular endothelial growth factor
- VKOR
vitamin K oxido-reductase
- wt
wild type
References
- 1.Rigal D. Observations pour servir á l’histoire de la cheloide diffuse xantholasmique. Ann Derm Syph. 1881;21:491–501. [Google Scholar]
- 2.Darier J. Pseudoxanthoma elasticum. Monatshefte Prakt Derm. 1896;23:609–17. [Google Scholar]
- 3.Grönblad E. Angioid streaks--pseudoxanthoma elasticum. Acta Ophthal. 1929;7:329. [Google Scholar]
- 4.Hu X, Plomp AS, van SS, Wijnholds J, de Jong PT, Bergen AA. Pseudoxanthoma elasticum: a clinical, histopathological, and molecular update. Surv Ophthalmol. 2003;48:424–38. doi: 10.1016/s0039-6257(03)00053-5. [DOI] [PubMed] [Google Scholar]
- 5.Walker ER, Frederickson RG, Mayes MD. The mineralization of elastic fibers and alterations of extracellular matrix in pseudoxanthoma elasticum. Ultrastructure, immunocytochemistry, and X-ray analysis. Arch Dermatol. 1989;125:70–6. [PubMed] [Google Scholar]
- 6.Baccarani-Contri M, Vincenzi D, Cicchetti F, Mori G, Pasquali-Ronchetti I. Immunochemical identification of abnormal constituents in the dermis of pseudoxanthoma elasticum patients. Eur J Histochem. 1994;38:111–23. [PubMed] [Google Scholar]
- 7.Limas C. Late onset focal dermal elastosis: a distinct clinicopathologic entity? Am J Dermatopathol. 1999;21:381–3. doi: 10.1097/00000372-199908000-00013. [DOI] [PubMed] [Google Scholar]
- 8.Choi GS, Kang DS, Chung JJ, Lee MG. Osteoma cutis coexisting with cutis laxa-like pseudoxanthoma elasticum. J Am Acad Dermatol. 2000;43:337–9. doi: 10.1067/mjd.2000.103188. [DOI] [PubMed] [Google Scholar]
- 9.Nikko AP, Dunningan M, Cockerell CJ. Calciphylaxis with histologic changes of pseudoxanthoma elasticum. Am J Dermatopathol. 1996;18:396–9. doi: 10.1097/00000372-199608000-00011. [DOI] [PubMed] [Google Scholar]
- 10.Gross G. Osteitis deformans (Paget)--angioid streaks (Knapp)--pseudoxanthoma elasticum (Darier)--manifestations of a common systemic disease? Arch Orthop Unfallchir. 1959;50:613–7. doi: 10.1007/BF00416348. [DOI] [PubMed] [Google Scholar]
- 11.Vanakker OM, Martin L, Gheduzzi D, Leroy BP, Loeys BL, Guerci VI, Matthys D, Terry SF, Coucke PJ, Pasquali-Ronchetti I, De Paepe A. Pseudoxanthoma elasticum-like phenotype with cutis laxa and multiple coagulation factor deficiency represents a separate genetic entity. J Invest Dermatol. 2007;127:581–7. doi: 10.1038/sj.jid.5700610. [DOI] [PubMed] [Google Scholar]
- 12.Finger RP, Charbel IP, Ladewig MS, Gotting C, Szliska C, Scholl HP, Holz FG. Pseudoxanthoma elasticum: genetics, clinical manifestations and therapeutic approaches. Surv Ophthalmol. 2009;54:272–85. doi: 10.1016/j.survophthal.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 13.Dreyer R, Green WR. The pathology of angioid streaks: a study of twenty-one cases. Trans Pa Acad Ophthalmol Otolaryngol. 1978;31:158–67. [PubMed] [Google Scholar]
- 14.Lim JI, Bressler NM, Marsh MJ, Bressler SB. Laser treatment of choroidal neovascularization in patients with angioid streaks. Am J Ophthalmol. 1993;116:414–23. doi: 10.1016/s0002-9394(14)71398-4. [DOI] [PubMed] [Google Scholar]
- 15.Pece A, Avanza P, Galli L, Brancato R. Laser photocoagulation of choroidal neovascularization in angioid streaks. Retina. 1997;17:12–6. doi: 10.1097/00006982-199701000-00003. [DOI] [PubMed] [Google Scholar]
- 16.Gragoudas ES, Adamis AP, Cunningham ET, Jr, Feinsod M, Guyer DR. Pegaptanib for neovascular age-related macular degeneration. N Engl J Med. 2004;351:2805–16. doi: 10.1056/NEJMoa042760. [DOI] [PubMed] [Google Scholar]
- 17.Rosenfeld PJ, Brown DM, Heier JS, Boyer DS, Kaiser PK, Chung CY, Kim RY. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med. 2006;355:1419–31. doi: 10.1056/NEJMoa054481. [DOI] [PubMed] [Google Scholar]
- 18.Finger RP, Charbel IP, Ladewig M, Holz FG, Scholl HP. Intravitreal bevacizumab for choroidal neovascularisation associated with pseudoxanthoma elasticum. Br J Ophthalmol. 2008;92:483–7. doi: 10.1136/bjo.2007.129916. [DOI] [PubMed] [Google Scholar]
- 19.Navarro-Lopez F, Llorian A, Ferrer-Roca O, Betriu A, Sanz G. Restrictive cardiomyopathy in pseudoxanthoma elasticum. Chest. 1980;78:113–5. doi: 10.1378/chest.78.1.113. [DOI] [PubMed] [Google Scholar]
- 20.Przybojewski JZ, Maritz F, Tiedt FA, van der Walt JJ. Pseudoxanthoma elasticum with cardiac involvement. A case report and review of the literature. S Afr Med J. 1981;59:268–75. [PubMed] [Google Scholar]
- 21.Challenor VF, Conway N, Monro JL. The surgical treatment of restrictive cardiomyopathy in pseudoxanthoma elasticum. Br Heart J. 1988;59:266–9. doi: 10.1136/hrt.59.2.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fukuda K, Uno K, Fujii T, Mukai M, Handa S. Mitral stenosis in pseudoxanthoma elasticum. Chest. 1992;101:1706–7. doi: 10.1378/chest.101.6.1706. [DOI] [PubMed] [Google Scholar]
- 23.Lebwohl M, Halperin J, Phelps RG. Brief report: occult pseudoxanthoma elasticum in patients with premature cardiovascular disease. N Engl J Med. 1993;329:1237–9. doi: 10.1056/NEJM199310213291705. [DOI] [PubMed] [Google Scholar]
- 24.Fah L. Pseudoxanthoma elasticum--a visual diagnosis. Schweiz Med Wochenschr. 1991;121:660–3. [PubMed] [Google Scholar]
- 25.Trip MD, Smulders YM, Wegman JJ, Hu X, Boer JM, ten Brink JB, Zwinderman AH, Kastelein JJ, Feskens EJ, Bergen AA. Frequent mutation in the ABCC6 gene (R1141X) is associated with a strong increase in the prevalence of coronary artery disease. Circulation. 2002;106:773–5. doi: 10.1161/01.cir.0000028420.27813.c0. [DOI] [PubMed] [Google Scholar]
- 26.Köblös G, Andrikovics H, Prohaszka Z, Tordai A, Váradi A, Arányi T. The R1141X Loss-of-Function Mutation of the ABCC6 Gene Is a Strong Genetic Risk Factor for Coronary Artery Disease. Genet Test Mol Biomarkers. 2009 doi: 10.1089/gtmb.2009.0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Le Saux O, Urban Z, Tschuch C, Csiszar K, Bacchelli B, Quaglino D, Pasquali-Ronchetti I, Pope FM, Richards A, Terry S, Bercovitch L, De PA, Boyd CD. Mutations in a gene encoding an ABC transporter cause pseudoxanthoma elasticum. Nat Genet. 2000;25:223–7. doi: 10.1038/76102. [DOI] [PubMed] [Google Scholar]
- 28.Bergen AA, Plomp AS, Schuurman EJ, Terry S, Breuning M, Dauwerse H, Swart J, Kool M, van SS, Baas F, ten Brink JB, de Jong PT. Mutations in ABCC6 cause pseudoxanthoma elasticum. Nat Genet. 2000;25:228–31. doi: 10.1038/76109. [DOI] [PubMed] [Google Scholar]
- 29.Ringpfeil F, Lebwohl MG, Christiano AM, Uitto J. Pseudoxanthoma elasticum: mutations in the MRP6 gene encoding a transmembrane ATP-binding cassette (ABC) transporter. Proc Natl Acad Sci U S A. 2000;97:6001–6. doi: 10.1073/pnas.100041297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pulkkinen L, Nakano A, Ringpfeil F, Uitto J. Identification of ABCC6 pseudogenes on human chromosome 16p: implications for mutation detection in pseudoxanthoma elasticum. Hum Genet. 2001;109:356–65. doi: 10.1007/s004390100582. [DOI] [PubMed] [Google Scholar]
- 31.Symmons O, Varadi A, Aranyi T. How segmental duplications shape our genome: recent evolution of ABCC6 and PKD1 Mendelian disease genes. Mol Biol Evol. 2008;25:2601–13. doi: 10.1093/molbev/msn202. [DOI] [PubMed] [Google Scholar]
- 32.Cai L, Struk B, Adams MD, Ji W, Haaf T, Kang HL, Dho SH, Xu X, Ringpfeil F, Nancarrow J, Zach S, Schaen L, Stumm M, Niu T, Chung J, Lunze K, Verrecchia B, Goldsmith LA, Viljoen D, Figuera LE, Fuchs W, Lebwohl M, Uitto J, Richards R, Hohl D, Ramesar R. A 500-kb region on chromosome 16p13. 1 contains the pseudoxanthoma elasticum locus: high-resolution mapping and genomic structure. J Mol Med. 2000;78:36–46. doi: 10.1007/s001090000079. [DOI] [PubMed] [Google Scholar]
- 33.Dauwerse JG, Wessels JW, Giles RH, Wiegant J, van der Reijden BA, Fugazza G, Jumelet EA, Smit E, Baas F, Raap AK. Cloning the breakpoint cluster region of the inv(16) in acute nonlymphocytic leukemia M4 Eo. Hum Mol Genet. 1993;2:1527–34. doi: 10.1093/hmg/2.10.1527. [DOI] [PubMed] [Google Scholar]
- 34.van Dongen JJ, Macintyre EA, Gabert JA, Delabesse E, Rossi V, Saglio G, Gottardi E, Rambaldi A, Dotti G, Griesinger F, Parreira A, Gameiro P, Diaz MG, Malec M, Langerak AW, San Miguel JF, Biondi A. Standardized RT-PCR analysis of fusion gene transcripts from chromosome aberrations in acute leukemia for detection of minimal residual disease. Report of the BIOMED-1 Concerted Action: investigation of minimal residual disease in acute leukemia. Leukemia. 1999;13:1901–28. doi: 10.1038/sj.leu.2401592. [DOI] [PubMed] [Google Scholar]
- 35.Buys TP, Chari R, Lee EH, Zhang M, MacAulay C, Lam S, Lam WL, Ling V. Genetic changes in the evolution of multidrug resistance for cultured human ovarian cancer cells. Genes Chromosomes Cancer. 2007;46:1069–79. doi: 10.1002/gcc.20492. [DOI] [PubMed] [Google Scholar]
- 36.Arányi T, Ratajewski M, Bardoczy V, Pulaski L, Bors A, Tordai A, Váradi A. Identification of a DNA methylation-dependent activator sequence in the pseudoxanthoma elasticum gene, ABCC6. J Biol Chem. 2005;280:18643–50. doi: 10.1074/jbc.M501139200. [DOI] [PubMed] [Google Scholar]
- 37.Arányi T, Faucheux BA, Khalfallah O, Vodjdani G, Biguet NF, Mallet J, Meloni R. The tissue-specific methylation of the human tyrosine hydroxylase gene reveals new regulatory elements in the first exon. J Neurochem. 2005;94:129–39. doi: 10.1111/j.1471-4159.2005.03173.x. [DOI] [PubMed] [Google Scholar]
- 38.Ratajewski M, Van d V, Bartosz G, Pulaski L. The human pseudoxanthoma elasticum gene ABCC6 is transcriptionally regulated by PLAG family transcription factors. Hum Genet. 2008;124:451–63. doi: 10.1007/s00439-008-0570-0. [DOI] [PubMed] [Google Scholar]
- 39.Ratajewski M, Bartosz G, Pulaski L. Expression of the human ABCC6 gene is induced by retinoids through the retinoid × receptor. Biochem Biophys Res Commun. 2006;350:1082–7. doi: 10.1016/j.bbrc.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 40.Jiang Q, Matsuzaki Y, Li K, Uitto J. Transcriptional regulation and characterization of the promoter region of the human ABCC6 gene. J Invest Dermatol. 2006;126:325–35. doi: 10.1038/sj.jid.5700065. [DOI] [PubMed] [Google Scholar]
- 41.de Boussac H, Ratajewski M, Koblos G, Sachrajda I, Tordai A, Pulaski L, Bunda S, Varadi A, Aranyi T. The ERK1/2 – HNF4a axis is a major regulator of human ABCC6 gene expression in the liver. 2010 doi: 10.1074/jbc.M110.105593. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bolotin E, Liao H, Ta TC, Yang C, Hwang-Verslues W, Evans JR, Jiang T, Sladek FM. Integrated approach for the identification of human hepatocyte nuclear factor 4alpha target genes using protein binding microarrays. Hepatology. 2010;51:642–53. doi: 10.1002/hep.23357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Odom DT, Zizlsperger N, Gordon DB, Bell GW, Rinaldi NJ, Murray HL, Volkert TL, Schreiber J, Rolfe PA, Gifford DK, Fraenkel E, Bell GI, Young RA. Control of pancreas and liver gene expression by HNF transcription factors. Science. 2004;303:1378–81. doi: 10.1126/science.1089769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sladek FM, Zhong WM, Lai E, Darnell JE., Jr Liver-enriched transcription factor HNF-4 is a novel member of the steroid hormone receptor superfamily. Genes Dev. 1990;4:2353–65. doi: 10.1101/gad.4.12b.2353. [DOI] [PubMed] [Google Scholar]
- 45.Kool M, van der LM, de HM, Baas F, Borst P. Expression of human MRP6, a homologue of the multidrug resistance protein gene MRP1, in tissues and cancer cells. Cancer Res. 1999;59:175–82. [PubMed] [Google Scholar]
- 46.Li Q, Jiang Q, Pfendner E, Váradi A, Uitto J. Pseudoxanthoma elasticum: clinical phenotypes, molecular genetics and putative pathomechanisms. Exp Dermatol. 2009;18:1–11. doi: 10.1111/j.1600-0625.2008.00795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hamlin N, Beck K, Bacchelli B, Cianciulli P, Pasquali-Ronchetti I, Le Saux O. Acquired Pseudoxanthoma elasticum-like syndrome in beta-thalassaemia patients. Br J Haematol. 2003;122:852–4. doi: 10.1046/j.1365-2141.2003.04484.x. [DOI] [PubMed] [Google Scholar]
- 48.Edinger C, Bollt O, Le Saux O. Tissue-specific Down Regulation of ABCC6 Expression in Beta-thalassemia Mice. Ethnicity & Disease. 2009;19:63–4. [Google Scholar]
- 49.Schulz V, Hendig D, Henjakovic M, Szliska C, Kleesiek K, Gotting C. Mutational analysis of the ABCC6 gene and the proximal ABCC6 gene promoter in German patients with pseudoxanthoma elasticum (PXE) Hum Mutat. 2006;27:831. doi: 10.1002/humu.9444. [DOI] [PubMed] [Google Scholar]
- 50.Hendig D, Arndt M, Szliska C, Kleesiek K, Gotting C. SPP1 promoter polymorphisms: identification of the first modifier gene for pseudoxanthoma elasticum. Clin Chem. 2007;53:829–36. doi: 10.1373/clinchem.2006.083675. [DOI] [PubMed] [Google Scholar]
- 51.Zarbock R, Hendig D, Szliska C, Kleesiek K, Gotting C. Pseudoxanthoma elasticum: genetic variations in antioxidant genes are risk factors for early disease onset. Clin Chem. 2007;53:1734–40. doi: 10.1373/clinchem.2007.088211. [DOI] [PubMed] [Google Scholar]
- 52.Zarbock R, Hendig D, Szliska C, Kleesiek K, Gotting C. Vascular endothelial growth factor gene polymorphisms as prognostic markers for ocular manifestations in pseudoxanthoma elasticum. Hum Mol Genet. 2009;18:3344–51. doi: 10.1093/hmg/ddp259. [DOI] [PubMed] [Google Scholar]
- 53.Schon S, Schulz V, Prante C, Hendig D, Szliska C, Kuhn J, Kleesiek K, Gotting C. Polymorphisms in the xylosyltransferase genes cause higher serum XT-I activity in patients with pseudoxanthoma elasticum (PXE) and are involved in a severe disease course. J Med Genet. 2006;43:745–9. doi: 10.1136/jmg.2006.040972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Iliás A, Urban Z, Seidl TL, Le SO, Sinko E, Boyd CD, Sarkadi B, Váradi A. Loss of ATP-dependent transport activity in pseudoxanthoma elasticum-associated mutants of human ABCC6 (MRP6) J Biol Chem. 2002;277:16860–7. doi: 10.1074/jbc.M110918200. [DOI] [PubMed] [Google Scholar]
- 55.Belinsky MG, Chen ZS, Shchaveleva I, Zeng H, Kruh GD. Characterization of the drug resistance and transport properties of multidrug resistance protein 6 (MRP6, ABCC6) Cancer Res. 2002;62:6172–7. [PubMed] [Google Scholar]
- 56.Madon J, Hagenbuch B, Landmann L, Meier PJ, Stieger B. Transport function and hepatocellular localization of mrp6 in rat liver. Mol Pharmacol. 2000;57:634–41. doi: 10.1124/mol.57.3.634. [DOI] [PubMed] [Google Scholar]
- 57.Fülöp K, Barna L, Symmons O, Závodszky P, Váradi A. Clustering of disease-causing mutations on the domain-domain interfaces of ABCC6. Biochem Biophys Res Commun. 2009;379:706–9. doi: 10.1016/j.bbrc.2008.12.142. [DOI] [PubMed] [Google Scholar]
- 58.Dawson RJ, Hollenstein K, Locher KP. Uptake or extrusion: crystal structures of full ABC transporters suggest a common mechanism. Mol Microbiol. 2007;65:250–7. doi: 10.1111/j.1365-2958.2007.05792.x. [DOI] [PubMed] [Google Scholar]
- 59.Dawson RJ, Locher KP. Structure of a bacterial multidrug ABC transporter. Nature. 2006;443:180–5. doi: 10.1038/nature05155. [DOI] [PubMed] [Google Scholar]
- 60.Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, Harrell PM, Trinh YT, Zhang Q, Urbatsch IL, Chang G. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323:1718–22. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Smith PC, Karpowich N, Millen L, Moody JE, Rosen J, Thomas PJ, Hunt JF. ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol Cell. 2002;10:139–49. doi: 10.1016/s1097-2765(02)00576-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Klement JF, Matsuzaki Y, Jiang QJ, Terlizzi J, Choi HY, Fujimoto N, Li K, Pulkkinen L, Birk DE, Sundberg JP, Uitto J. Targeted ablation of the abcc6 gene results in ectopic mineralization of connective tissues. Mol Cell Biol. 2005;25:8299–310. doi: 10.1128/MCB.25.18.8299-8310.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gorgels TG, Hu X, Scheffer GL, van der Wal AC, Toonstra J, de Jong PT, van Kuppevelt TH, Levelt CN, de WA, Loves WJ, Scheper RJ, Peek R, Bergen AA. Disruption of Abcc6 in the mouse: novel insight in the pathogenesis of pseudoxanthoma elasticum. Hum Mol Genet. 2005;14:1763–73. doi: 10.1093/hmg/ddi183. [DOI] [PubMed] [Google Scholar]
- 64.Meng H, Vera I, Che N, Wang X, Wang SS, Ingram-Drake L, Schadt EE, Drake TA, Lusis AJ. Identification of Abcc6 as the major causal gene for dystrophic cardiac calcification in mice through integrative genomics. Proc Natl Acad Sci U S A. 2007;104:4530–5. doi: 10.1073/pnas.0607620104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Aherrahrou Z, Doehring LC, Ehlers EM, Liptau H, Depping R, Linsel-Nitschke P, Kaczmarek PM, Erdmann J, Schunkert H. An alternative splice variant in Abcc6, the gene causing dystrophic calcification, leads to protein deficiency in C3H/He mice. J Biol Chem. 2008;283:7608–15. doi: 10.1074/jbc.M708290200. [DOI] [PubMed] [Google Scholar]
- 66.Sadowski S, Uitto J, et al. ABCC6a function in the Zebrafish Danio rerio. 2010 submitted. [Google Scholar]
- 67.Uitto J, Pulkkinen L, Ringpfeil F. Molecular genetics of pseudoxanthoma elasticum: a metabolic disorder at the environment-genome interface? Trends Mol Med. 2001;7:13–7. doi: 10.1016/s1471-4914(00)01869-4. [DOI] [PubMed] [Google Scholar]
- 68.Jiang Q, Endo M, Dibra F, Wang K, Uitto J. Pseudoxanthoma elasticum is a metabolic disease. J Invest Dermatol. 2009;129:348–54. doi: 10.1038/jid.2008.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jiang Q, Oldenburg R, Otsuru S, Grand-Pierre AE, Horwitz EM, Uitto J. Parabiotic Heterogenetic Pairing of Abcc6−/−/Rag1−/− Mice and Their Wild-Type Counterparts Halts Ectopic Mineralization in a Murine Model of Pseudoxanthoma Elasticum. Am J Pathol. 2010 doi: 10.2353/ajpath.2010.090983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Macmillan DC, Vickers HR. Pseudoxanthoma elasticum and a coagulation defect. Br J Dermatol. 1971;84:182. [PubMed] [Google Scholar]
- 71.Rongioletti F, Bertamino R, Rebora A. Generalized pseudoxanthoma elasticum with deficiency of vitamin K-dependent clotting factors. J Am Acad Dermatol. 1989;21:1150–2. doi: 10.1016/s0190-9622(89)70320-0. [DOI] [PubMed] [Google Scholar]
- 72.Le Corvaisier-Pieto C, Joly P, Thomine E, Lair G, Lauret P. Generalized pseudoxanthoma elasticum combined with vitamin K dependent clotting factors deficiency. Ann Dermatol Venereol. 1996;123:555–8. [PubMed] [Google Scholar]
- 73.Berkner KL. Vitamin K-dependent carboxylation. Vitam Horm. 2008;78:131–56. doi: 10.1016/S0083-6729(07)00007-6. [DOI] [PubMed] [Google Scholar]
- 74.Oldenburg J, Marinova M, Muller-Reible C, Watzka M. The vitamin K cycle. Vitam Horm. 2008;78:35–62. doi: 10.1016/S0083-6729(07)00003-9. [DOI] [PubMed] [Google Scholar]
- 75.Schurgers LJ, Teunissen KJ, Knapen MH, Kwaijtaal M, van DR, Appels A, Reutelingsperger CP, Cleutjens JP, Vermeer C. Novel conformation-specific antibodies against matrix gamma-carboxyglutamic acid (Gla) protein: undercarboxylated matrix Gla protein as marker for vascular calcification. Arterioscler Thromb Vasc Biol. 2005;25:1629–33. doi: 10.1161/01.ATV.0000173313.46222.43. [DOI] [PubMed] [Google Scholar]
- 76.Schurgers LJ, Spronk HM, Skepper JN, Hackeng TM, Shanahan CM, Vermeer C, Weissberg PL, Proudfoot D. Post-translational modifications regulate matrix Gla protein function: importance for inhibition of vascular smooth muscle cell calcification. J Thromb Haemost. 2007;5:2503–11. doi: 10.1111/j.1538-7836.2007.02758.x. [DOI] [PubMed] [Google Scholar]
- 77.Schurgers LJ, Cranenburg EC, Vermeer C. Matrix Gla-protein: the calcification inhibitor in need of vitamin K. Thromb Haemost. 2008;100:593–603. [PubMed] [Google Scholar]
- 78.Jiang Q, Li Q, Uitto J. Aberrant Mineralization of Connective Tissues in a Mouse Model of Pseudoxanthoma Elasticum: Systemic and Local Regulatory Factors. J Invest Dermatol. 2007 doi: 10.1038/sj.jid.5700729. [DOI] [PubMed] [Google Scholar]
- 79.Li Q, Jiang Q, Schurgers LJ, Uitto J. Pseudoxanthoma elasticum: reduced gamma-glutamyl carboxylation of matrix gla protein in a mouse model (Abcc6−/−) Biochem Biophys Res Commun. 2007;364:208–13. doi: 10.1016/j.bbrc.2007.09.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gheduzzi D, Boraldi F, Annovi G, DeVincenzi CP, Schurgers LJ, Vermeer C, Quaglino D, Ronchetti IP. Matrix Gla protein is involved in elastic fiber calcification in the dermis of pseudoxanthoma elasticum patients. Lab Invest. 2007;87:998–1008. doi: 10.1038/labinvest.3700667. [DOI] [PubMed] [Google Scholar]
- 81.Vanakker OM, Martin L, Schurgers LJ, Quaglino D, Costrop L, Vermeer C, Pasquali-Ronchetti I, Coucke PJ, De Paepe A. Low serum vitamin K in PXE results in defective carboxylation of mineralization inhibitors similar to the GGCX mutations in the PXE-like syndrome. Lab Invest. 2010 doi: 10.1038/labinvest.2010.68. in press. [DOI] [PubMed] [Google Scholar]
- 82.Borst P, van de WK, Schlingemann R. Does the absence of ABCC6 (multidrug resistance protein 6) in patients with Pseudoxanthoma elasticum prevent the liver from providing sufficient vitamin K to the periphery? Cell Cycle. 2008;7:1575–9. doi: 10.4161/cc.7.11.6005. [DOI] [PubMed] [Google Scholar]
- 83.Li J, Lin JC, Wang H, Peterson JW, Furie BC, Furie B, Booth SL, Volpe JJ, Rosenberg PA. Novel role of vitamin k in preventing oxidative injury to developing oligodendrocytes and neurons. J Neurosci. 2003;23:5816–26. doi: 10.1523/JNEUROSCI.23-13-05816.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Garcia-Fernandez MI, Gheduzzi D, Boraldi F, Paolinelli CD, Sanchez P, Valdivielso P, Morilla MJ, Quaglino D, Guerra D, Casolari S, Bercovitch L, Pasquali-Ronchetti I. Parameters of oxidative stress are present in the circulation of PXE patients. Biochim Biophys Acta. 2008;1782:474–81. doi: 10.1016/j.bbadis.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 85.Li Q, Jiang Q, Uitto J. Pseudoxanthoma elasticum: oxidative stress and antioxidant diet in a mouse model (Abcc6−/−) J Invest Dermatol. 2008;128:1160–4. doi: 10.1038/sj.jid.5701145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Thijssen HH, Vervoort LM, Schurgers LJ, Shearer MJ. Menadione is a metabolite of oral vitamin K. Br J Nutr. 2006;95:260–6. doi: 10.1079/bjn20051630. [DOI] [PubMed] [Google Scholar]
- 87.Okano T, Shimomura Y, Yamane M, Suhara Y, Kamao M, Sugiura M, Nakagawa K. Conversion of phylloquinone (Vitamin K1) into menaquinone-4 (Vitamin K2) in mice: two possible routes for menaquinone-4 accumulation in cerebra of mice. J Biol Chem. 2008;283:11270–9. doi: 10.1074/jbc.M702971200. [DOI] [PubMed] [Google Scholar]
- 88.Harrington DJ, Soper R, Edwards C, Savidge GF, Hodges SJ, Shearer MJ. Determination of the urinary aglycone metabolites of vitamin K by HPLC with redox-mode electrochemical detection. J Lipid Res. 2005;46:1053–60. doi: 10.1194/jlr.D400033-JLR200. [DOI] [PubMed] [Google Scholar]
- 89.Hendig D, Langmann T, Kocken S, Zarbock R, Szliska C, Schmitz G, Kleesiek K, Gotting C. Gene expression profiling of ABC transporters in dermal fibroblasts of pseudoxanthoma elasticum patients identifies new candidates involved in PXE pathogenesis. Lab Invest. 2008;88:1303–15. doi: 10.1038/labinvest.2008.96. [DOI] [PubMed] [Google Scholar]
- 90.Beck K, Hayashi K, Dang K, Hayashi M, Boyd CD. Analysis of ABCC6 (MRP6) in normal human tissues. Histochem Cell Biol. 2005;123:517–28. doi: 10.1007/s00418-004-0744-3. [DOI] [PubMed] [Google Scholar]
- 91.Boraldi F, Quaglino D, Croce MA, Garcia Fernandez MI, Tiozzo R, Gheduzzi D, Bacchelli B, Pasquali RI. Multidrug resistance protein-6 (MRP6) in human dermal fibroblasts. Comparison between cells from normal subjects and from Pseudoxanthoma elasticum patients. Matrix Biol. 2003;22:491–500. doi: 10.1016/j.matbio.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 92.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–2. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 93.Sugden PH, Clerk A. Regulation of the ERK subgroup of MAP kinase cascades through G protein-coupled receptors. Cell Signal. 1997;9:337–51. doi: 10.1016/s0898-6568(96)00191-x. [DOI] [PubMed] [Google Scholar]
- 94.Junttila MR, Li SP, Westermarck J. Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. FASEB J. 2008;22:954–65. doi: 10.1096/fj.06-7859rev. [DOI] [PubMed] [Google Scholar]
- 95.Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291–310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 96.Friday BB, Adjei AA. Advances in targeting the Ras/Raf/MEK/Erk mitogen-activated protein kinase cascade with MEK inhibitors for cancer therapy. Clin Cancer Res. 2008;14:342–6. doi: 10.1158/1078-0432.CCR-07-4790. [DOI] [PubMed] [Google Scholar]
- 97.Pasquali-Ronchetti I, Garcia-Fernandez MI, Boraldi F, Quaglino D, Gheduzzi D, De Vincenzi PC, Tiozzo R, Bergamini S, Ceccarelli D, Muscatello U. Oxidative stress in fibroblasts from patients with pseudoxanthoma elasticum: possible role in the pathogenesis of clinical manifestations. J Pathol. 2006;208:54–61. doi: 10.1002/path.1867. [DOI] [PubMed] [Google Scholar]
- 98.Reddy S, Yang W, Taylor DG, Shen X, Oxender D, Kust G, Leff T. Mitogen-activated protein kinase regulates transcription of the ApoCIII gene. Involvement of the orphan nuclear receptor HNF4. J Biol Chem. 1999;274:33050–6. doi: 10.1074/jbc.274.46.33050. [DOI] [PubMed] [Google Scholar]
- 99.Hatzis P, Kyrmizi I, Talianidis I. Mitogen-activated protein kinase-mediated disruption of enhancer-promoter communication inhibits hepatocyte nuclear factor 4alpha expression. Mol Cell Biol. 2006;26:7017–29. doi: 10.1128/MCB.00297-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Martinez-Jimenez CP, Kyrmizi I, Cardot P, Gonzalez FJ, Talianidis I. HNF4{alpha} coordinates a transcription factor network regulating hepatic fatty acid metabolism. Mol Cell Biol. 2009 doi: 10.1128/MCB.00927-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bailly A, Torres-Padilla ME, Tinel AP, Weiss MC. An enhancer element 6 kb upstream of the mouse HNF4alpha1 promoter is activated by glucocorticoids and liver-enriched transcription factors. Nucleic Acids Res. 2001;29:3495–505. doi: 10.1093/nar/29.17.3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hayashi H, Sugiyama Y. 4-phenylbutyrate enhances the cell surface expression and the transport capacity of wild-type and mutated bile salt export pumps. Hepatology. 2007;45:1506–16. doi: 10.1002/hep.21630. [DOI] [PubMed] [Google Scholar]
- 103.Loo TW, Clarke DM. Correction of defective protein kinesis of human P-glycoprotein mutants by substrates and modulators. J Biol Chem. 1997;272:709–12. doi: 10.1074/jbc.272.2.709. [DOI] [PubMed] [Google Scholar]
- 104.Egan ME, Pearson M, Weiner SA, Rajendran V, Rubin D, Glockner-Pagel J, Canny S, Du K, Lukacs GL, Caplan MJ. Curcumin, a major constituent of turmeric, corrects cystic fibrosis defects. Science. 2004;304:600–2. doi: 10.1126/science.1093941. [DOI] [PubMed] [Google Scholar]
- 105.Barton-Davis ER, Cordier L, Shoturma DI, Leland SE, Sweeney HL. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J Clin Invest. 1999;104:375–81. doi: 10.1172/JCI7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Xi B, Guan F, Lawrence DS. Enhanced production of functional proteins from defective genes. J Am Chem Soc. 2004;126:5660–1. doi: 10.1021/ja0318939. [DOI] [PubMed] [Google Scholar]
- 107.Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, Paushkin S, Patel M, Trotta CR, Hwang S, Wilde RG, Karp G, Takasugi J, Chen G, Jones S, Ren H, Moon YC, Corson D, Turpoff AA, Campbell JA, Conn MM, Khan A, Almstead NG, Hedrick J, Mollin A, Risher N, Weetall M, Yeh S, Branstrom AA, Colacino JM, Babiak J, Ju WD, Hirawat S, Northcutt VJ, Miller LL, Spatrick P, He F, Kawana M, Feng H, Jacobson A, Peltz SW, Sweeney HL. PTC124 targets genetic disorders caused by nonsense mutations 1. Nature. 2007;447:87–91. doi: 10.1038/nature05756. [DOI] [PubMed] [Google Scholar]