

Abstract
The transduction of T cells to express chimeric T-cell antigen receptor (CAR) is an attractive strategy for adaptive immunotherapy for cancer, because the CAR can redirect the recognition specificity of T cells to tumor-associated antigens (TAAs) on the surface of target cells, thereby avoiding the limitations of HLA restriction. However, there are considerable problems with the clinical application of CAR, mostly due to its xenogeneic components, which could be immunogenic in humans. Moreover, while extensive studies on the CARs have been performed, the detailed molecular mechanisms underlying the activation of CAR-grafted T cells remain unclear. In order to eliminate potential immunogenicity and investigate the molecular basis of the CAR-mediated T-cell activation, we constructed a novel CAR (CAR57-28ζ) specific for one of the most important TAAs, epithelial cell adhesion molecule (EpCAM), using only human-derived genes. We revealed that in Jurkat T cells, lentivirally expressed CAR57-28ζ can transmit the T-cell-activating signals sufficient to induce IL-2 production upon EpCAM stimulation. An immunofluorescent analysis clearly showed that the CAR57-28ζ induces the formation of signaling clusters containing endogenous CD3ζ at the CAR/EpCAM interaction interface. These results suggest that this CAR gene may be safely and effectively applied for adaptive T-cell immunotherapy.
1. Introduction
The chimeric T-cell antigen receptor (CAR) provides a promising approach for adoptive T-cell immunotherapy for cancer [1, 2]. Commonly, CARs comprise a single chain fragment variable (scFv) of an antibody specific for a tumor-associated antigen (TAA) coupled via hinge and transmembrane regions to several cytoplasmic domains of T-cell signaling molecules, such as CD3ζ and CD28 [3, 4]. The CAR-mediated adoptive immunotherapy is thought to be suitable, especially for cancer cells that have acquired the ability to escape from immunosurveillance by HLA-downregulation and/or a lack of expression of costimulatory molecules, because the use of scFv as an antigen-binding region allows CAR-grafted T cells to directly recognize the TAAs on target tumor cells in a non-HLA-restricted manner. Therefore, the intrinsic signaling domains of CAR can transmit multiple stimulatory signals to the T cell. However, the majority of the CARs reported so far contain an scFv moiety that originated from murine-derived or “humanized” antibodies for specific recognition of TAAs, which might trigger a host immune response and have inherent risks for the production of human anti-mouse antibodies (HAMA) [5]. Of note, extensive studies on the therapeutic application of CARs have been performed, but the detailed molecular mechanisms underlying the CAR-mediated activation of the genetically modified T cells remain unclear.
We previously reported the establishment of hybridoma clones that produce a fully human monoclonal antibody specific for the epithelial cell adhesion molecule (EpCAM) [6] using the KMTM mouse, which carries human-derived immunoglobulin genes instead of the endogenous mouse genes [7]. EpCAM is a transmembrane glycoprotein expressed by both normal and malignant cells of epithelial origin, but that is overexpressed in most human carcinomas and is known to be an attractive target TAA for cancer immunotherapy [8, 9]. Here, in order to remove the undesired immunogenicity that may arise from CAR-mediated immunotherapy, we functionally cloned an anti-EpCAM scFv gene from M13-57, one of the human anti-EpCAM antibody-producing hybridomas, and a novel CAR gene specific for EpCAM was constructed using only human genes. This CAR gene (referred to hereafter as “CAR57-28ζ") was lentivirally expressed in Jurkat T cells, and its molecular characterization was carried out to investigate the molecular mechanisms by which CAR leads to T-cell activation by immunoblotting analyses, assays for cytokine production, and confocal immunofluorescence microscopy after stimulation with immobilized EpCAM. We herein reveal that EpCAM-dependent activation of CAR57-28ζ-grafted T cells and our discovery that the CAR is able to deliver activation signals via wild-type T-cell receptor (TCR-) like molecular events, including the formation of signaling clusters at the antigen-receptor interface.
2. Materials and Methods
2.1. cDNA Cloning of Anti-EpCAM scFv
Cloning of the anti-EpCAM scFv gene was performed using a phage display technique, as described previously [11]. Template mRNA was extracted from the hybridoma clone M13-57, and the cDNAs coding for variable regions of the VH and VL genes was PCR amplified using a mixture of human V-gene-specific forward and reverse primers [12] using the One-Step RT-PCR kit (Stratagene, La Jolla, CA). An scFv linker DNA that connects VH to VL was prepared by PCR amplification using modified RHuJH and RHuVκ primers and the phagemid vector pIT2 containing antiubiquitin scFv as a template [12, 13]. The VH and VL fragments were assembled into the scFv form by splice overlap extension (SOE) PCR. The PCR products were ligated into the Sfi I and Not I sites of pIT2. The resulting plasmid was transformed into Escherichia coli TG-1. Phage particles displaying anti-EpCAM scFv were prepared using the KM13 helper phage, and affinity panning was performed against an EpCAM-coated Maxisorp immunotube (Nunc, Roskilde, Denmark). Phage binders were eluted with trypsin-PBS. The eluted phages were infected into TG-1 cells, and then they were plated out on 2xTY plates containing 100 μg/mL ampicillin. The colonies were picked up, and a monoclonal phage was prepared from each isolate. The binders specific for EpCAM were selected by phage ELISA using an HRP-conjugated anti-M13 phage antibody (GE Healthcare, Buckinghamshire, UK) with TMB One Solution (Promega, Madison, WI). Plasmids were prepared from selected clones and sequenced.
2.2. Construction and Expression of Anti-EpCAM CAR Genes
The fully human anti-EpCAM CAR (CAR57-28ζ) gene was genetically constructed by exchange of the anti-CEA scFv region of the CAR45-28ζ gene, which we described previously [11], for the anti-EpCAM scFv gene that was cloned here. The CAR57-28ζ gene was temporarily cloned into a pIRES2-DsRed-Express2 vector (Clontech, Palo Alto, CA) at the Nhe I site, then a PCR fragment of the CAR with the IRES2-DsRed Express2 gene was amplified and subcloned into a lentiviral expression vector pLVSIN-EF1α (Clontech) at the Nhe I/Not I sites using an In-Fusion HD cloning kit (Clontech). The resultant vector was transfected into Lenti-X 293T cells, and the lentiviral particles were produced from the cells by using a Lenti-X HT packaging system (Clontech) according to the manufacturer's instructions. Lentiviral stocks were concentrated using Lenti-X concentrator (Clontech), and the titer was estimated with Lenti-X GoStix (Clontech). For infection, nontissue culture 35 mm plates were coated with 20 μg/mL Retronectin (Takara Bio Inc., Shiga, Japan) overnight at 4°C. The next day, the plates were washed with D-PBS containing 2% BSA and blocked with the same buffer for 1 hr at 25°C. The plates were washed and incubated with viruses for 4 hr at 37°C. Unbound viral particles were removed by washing, and Jurkat cells (2.5 × 105 cells) in RPMI 1640 medium supplemented with 10% fetal calf serum (FCS) were plated onto the plates. After 48 hr of incubation, the CAR-transduced cells were collected and inoculated into a new plate, and the cells were selected in medium containing 400 μg/mL hygromycin. The cells were analyzed for DsRed Express2 expression by a flow cytometry analysis, using a FACS Calibur analyzer (BD Biosciences, San Jose, CA).
For the immunofluorescent analysis, the pBI-ZAP-AcGFP vector [11] was transfected into CAR57-28ζ-expressing Jurkat cells with the GenePORTER 3000 Transfection Reagent (Genlantis, San Diego, CA).
2.3. IL-2 Secretion Assay
Microtiter plates were coated with an anti-CD3ε (UCHT1; 500 ng/mL; Beckman Coulter, Miami, FL) or anti-CD28 antibody (CD28.2; 500 ng/mL; Beckman Coulter) or with EpCAM (20 μg/mL). Jurkat cells (1 × 106 cells/mL) were incubated for 24 h at 37°C in coated plates. The culture supernatants were analyzed for the secretion of IL-2 using a DIAplex immunoassay kit (Gen-Probe Inc., San Diego, CA) as per the manufacturer's specifications.
2.4. Western Blotting Analyses
The Jurkat cells were stimulated with immobilized anti-CD3ε alone, anti-CD3ε plus anti-CD28, or immobilized EpCAM for 15 min at 37°C. The cells were suspended in RIPA buffer (PBS, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 2 mM EDTA) at a concentration of 1 × 107 cells/mL, then were incubated for 1 h on ice and then boiled in SDS-PAGE sample buffer with 0.1 M dithiothreitol. The samples were separated on 5–20% acrylamide gradient gels and transferred to polyvinylidene fluoride membranes. The blots were blocked with StartingBlock T20 blocking buffer (SB; Thermoscientific, Bremen, Germany), and the membranes were incubated for 1 h with phospho-specific antibodies (Anti-ACTIVE; Promega), washed five times in PBS containing 10% SB, and protein spots were detected using an HRP-conjugated secondary antibody. Immunoreactive bands were visualized using the ECL Prime Western Blotting Detection Reagent (GE Healthcare) and then were developed using the LAS3000 chemiluminescence system (Fujifilm, Tokyo, Japan).
2.5. Cell Spreading Assay
A cell spreading assay was performed as described previously, with some modifications [11, 14]. Eight-chambered coverslips (Lab-Tek II; Nunc) were treated with a 0.01% poly-l-lysine solution (Sigma, St. Louis, MO). The slides were coated overnight at 4°C with anti-CD3ε (10 μg/mL) or EpCAM (20 μg/mL). Cells (5 × 105) were seeded onto the bottoms of the chambers containing phenol red-free RPMI 1640 medium with 10% FCS and 20 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid for 3 min and fixed for 30 min at 37°C with 8% paraformaldehyde in PBS. The chambers were washed with PBS containing 10 mM glycine and blocked for 15 min in SB at room temperature. The chambers were incubated for 1 h at room temperature with a phycoerythrin-labeled antimyc antibody (Abcam, Cambridge, UK). The cells were permeabilized for 5 min with 0.1% Triton X-100, rinsed in PBS containing 10% SB, and blocked for 15 min in 100% SB. The chambers were incubated for 1 h at room temperature and then overnight at 4°C with an anti-CD3ζ antibody (Santa Cruz Biotechnologies, Santa Cruz, CA) prelabeled with Zenon Alexa Fluor 647 (Invitrogen, Carlsbad, CA). The chambers were washed with 10% SB and refixed for 15 min with 4% paraformaldehyde, then washed in PBS containing 10 mM glycine. Fluorescent images were acquired on a LSM710 confocal system (Carl Zeiss, Jena, Germany).
3. Results and Discussion
Because hybridoma cells often express more than one of the VH/VL genes due to the presence of fusion partners [15], the functional screening of VH/VL genes is an essential step for cloning the V-gene encoding a monoclonal antibody. We employed a phage display technique in order to directly clone the gene encoding the functional anti-EpCAM scFv. The cDNAs of VH and VL were amplified from reverse-transcribed mRNA of the M13-45 hybridoma with mixed primers specific for the V-gene leader and constant regions. The amplified V-gene fragments were assembled into the scFv form with a flexible linker by SOE-PCR to display the scFv protein on the phage. The scFv genes were ligated into a pIT2 phagemid vector and used to transform E. coli TG-1 for preparation of the scFv-displaying phages. Specific binders were selected by phage ELISA after two rounds of phage panning on immobilized EpCAM. Some clones clearly showed specific binding against EpCAM but negligible binding to BSA (Figure 1), thus indicating that the anti-EpCAM scFv harboring phages were successfully enriched. The DNA sequences of positive scFv clones were determined, and it was found that they had essentially the same sequence (Figure 2).
Figure 1.
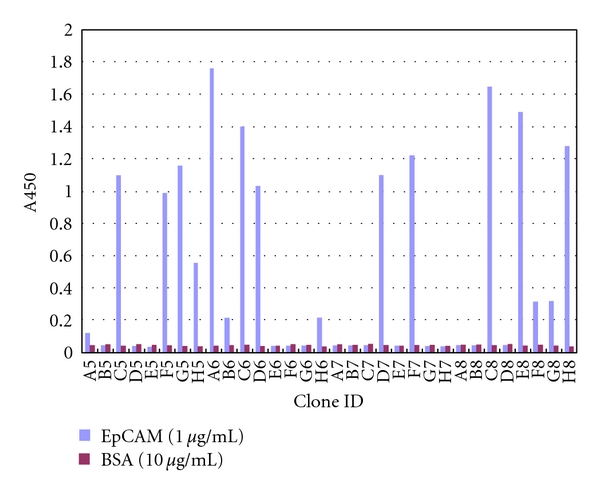
The screening of phage clones displaying anti-EpCAM scFv by monoclonal phage ELISA. The binding of phages displaying anti-EpCAM scFv to immobilized EpCAM (white bar) or BSA (gray bar) was detected by an HRP-labeled anti-M13 antibody.
Figure 2.
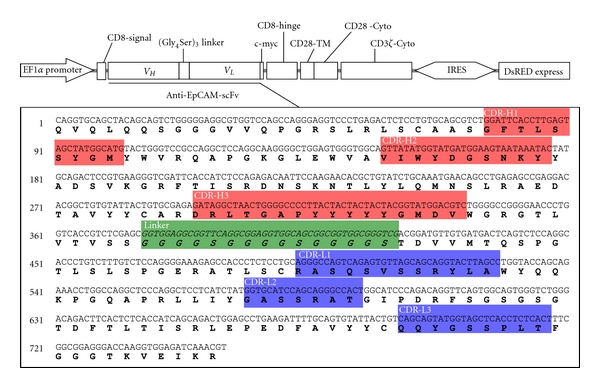
Schematic representations of CAR57-28ζ. The nucoleotide and aminoacid sequences of anti-EpCAM scFv from the M13-57 hybridoma were analyzed using the IMGT/V-QUEST and IMGT/JunctionAnalysis online tools (http://www.imgt.org/), and each complementarity-determining region (CDR) of the cloned scFv gene was determined according to Chothia's definition [10]. VH, variable region of the heavy chain; VL, variable region of the light chain; TM, transmembrane domain; Cyto, cytoplasmic domain.
Using the cloned scFv gene, a gene coding an EpCAM-specific CAR was constructed. This CAR57-28ζ gene consists of a CD8 leader sequence, scFv, the transmembrane and cytoplasmic domains of CD28, and the cytoplasmic domain of CD3ζ. All of the genes used for the construction were derived from human sources. As described in the Materials and Methods, the CAR57-28ζ, together with the gene encoding IRES-DsRed Express2, was inserted into the lentiviral expression vector pLVSIN-EF1α (Figure 2). This expression construct was then transduced into the packaging cell line, Lenti-X 293T, and lentiviral particles produced by 293T cells were used to transduce human T-cell leukemia Jurkat cells. After hygromycin selection of the transductant, the expression of the CAR57-28ζ protein was evaluated by a Western blotting analysis. The cell lysates from mock-transduced and transduced Jurkat cells were probed with an anti-CD3ζ antibody, which is capable of recognizing both the CD3ζ region of the CAR and the endogenous CD3ζ. As shown in Figure 3, a band of ca. 56 kDa consistent with the predicted size of CAR57-28ζ was detected only in lysates from CAR-transduced cells, indicating successful expression. The low fluorescence intensity in the transduced cells observed in FACS analysis might be due to the poor IRES function.
Figure 3.
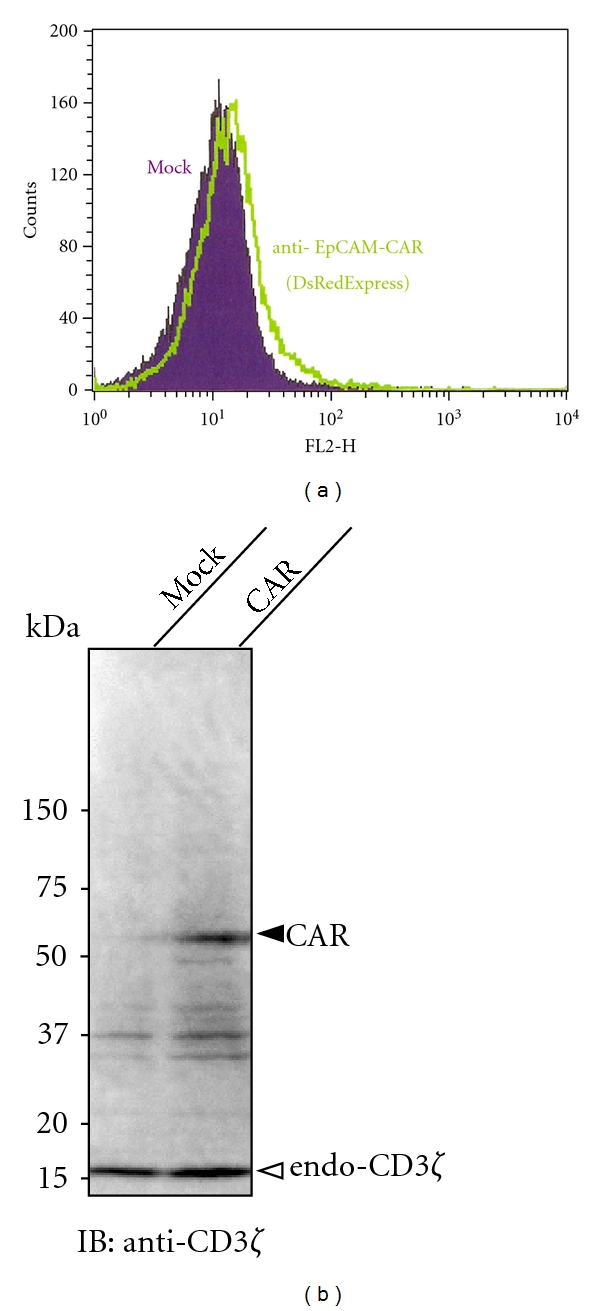
The results of the analysis of CAR57-28ζ protein expression. CAR57-28ζ-grafted Jurkat cells after hygromycin selection were analyzed by flow cytometry (a). Lysates were prepared from wild-type and CAR-expressing Jurkat cells, and immunoreactive bands were detected by a Western blotting analysis using an anti-CD3ζ antibody (b).
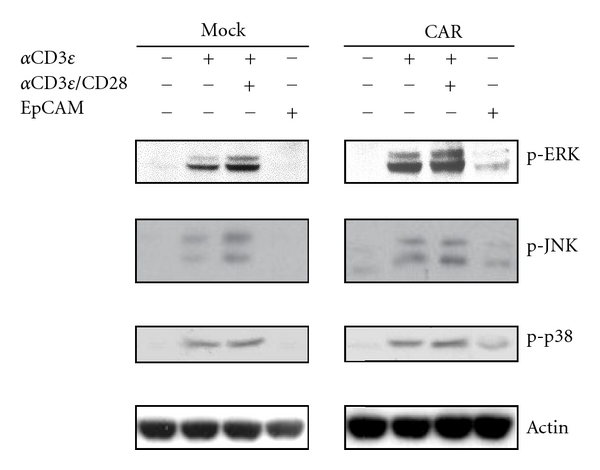
TCR activation triggers various downstream signaling events that involve the phosphorylation of cellular proteins in T cells [16, 17]. To investigate the early steps of T-cell signal transduction by the CAR, the activation status of the known signaling components downstream of the TCR was analyzed by immunoblotting with phosphospecific antibodies. When wild-type and CAR-expressing Jurkat cells were activated with stimulating antibodies (anti-CD3ε/CD28), all signaling molecules tested were phosphorylated in both cells (Figure 4). Notably, stimulation with immobilized EpCAM also induced the phosphorylation of ERK, JNK, and p38 in CAR-expressing cells, but not in wild-type cells (Figure 4). These observations clearly demonstrated that CAR57-28ζ was functionally expressed and able to transmit the T-cell activation signals in an EpCAM-dependent manner.
Figure 4.
The phosphorylation status of the signaling proteins in CAR-expressing Jurkat cells. Cells were activated for 15 min with the indicated stimulant, and the lysates were used for the DIAplex assay for IL-2 detection. Irrelevant mouse IgG was used as a negative control.
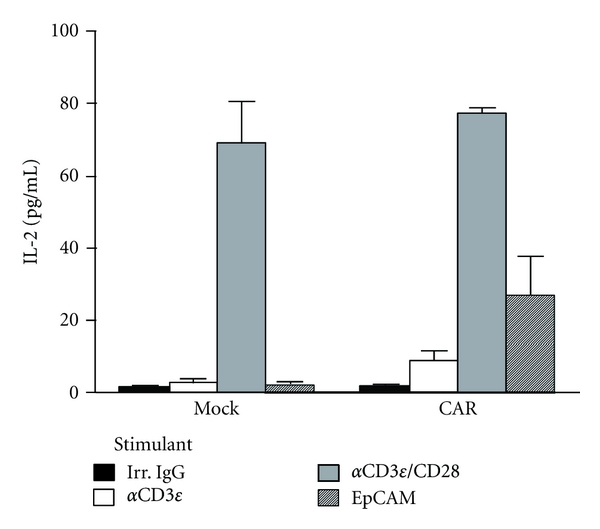
The CAR-mediated T-cell activation was further investigated by a cytokine secretion assay. No IL-2 secretion was found in the culture supernatant of the mock-transduced cells stimulated with EpCAM, nor in those treated with anti-CD3ε alone (Figure 5). In contrast, EpCAM stimulation clearly induced the production of IL-2 in CAR-expressing Jurkat cells similar to when the cells were stimulated with both anti-CD3ε/CD28 (Figure 5), thus indicating that the CD28 cytoplasmic domain in CAR57-28ζ could deliver the costimulatory signal and activate the T-cell signaling pathway(s) required for the production of IL-2 by stimulation with EpCAM.
Figure 5.
IL-2 secretion from CAR-expressing Jurkat cells. The concentration of IL-2 in the cell supernatant was measured 24 h after stimulation with an anti-CD3 antibody, anti-CD3 plus anti-CD28 antibodies, or EpCAM.
In order to gain insight into the molecular mechanisms underlying CAR-mediated T-cell activation, we performed a cell spreading assay. If T cells are dropped onto a surface coated with stimulatory molecules, this results in receptor clustering and the recruitment of signaling proteins to the points of contact with the stimulatory surface [14, 18]. In our assay system, the formation of immunoreceptor tyrosine-based activation motif (ITAM-) mediated signaling clusters was visualized by utilizing the AcGFP-tagged ZAP-70. ZAP-70 is a predominant T-cell protein tyrosine kinase containing tandem SH2 domains, which are engaged by doubly phosphorylated ITAMs of CD3ζ [17, 19]. As shown in Figure 6, when Jurkat cells expressing ZAP-AcGFP alone were introduced onto the anti-CD3ε-coated bottom sections of the chambers, the endogenous TCRs detected with anti-CD3ζ were observed in ZAP-containing signaling clusters. As expected, no such cells adhered to the coverslips when EpCAM was used as a coating protein (data not shown). On the other hand, the CAR57-28ζ-expressing cells remained on the EpCAM-coated surface even after washing, and clear colocalization of CAR57-28ζ and ZAP-70 was observed (Figure 6). These results revealed that CAR57-28ζ can induce the antigen-dependent formation of the signaling clusters at the antigen/antibody (namely, EpCAM/CAR) interaction interface. It should be noted that the immunofluorescent signals detected with the anti-CD3ζ antibody indicate the localization of endogenous TCRs, as well as that of CAR57-28ζ, implying that a cross-activation of endogenous TCRs was induced by CAR and EpCAM.
Figure 6.
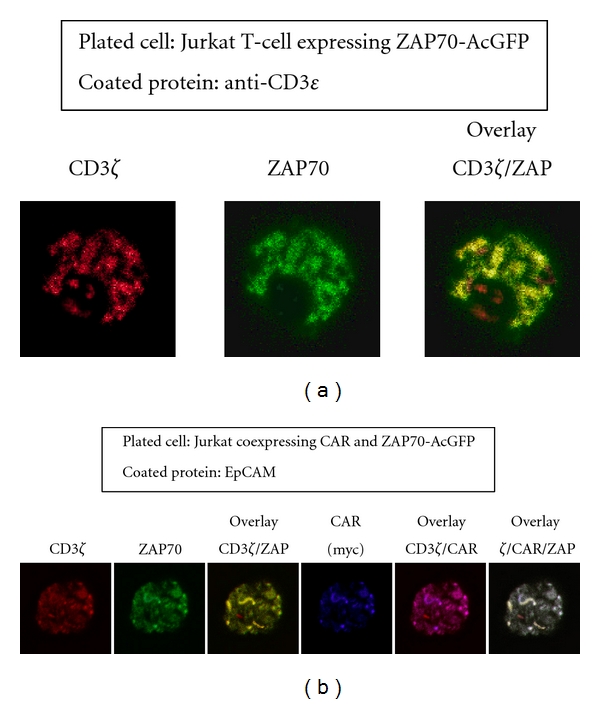
The formation of the signaling clusters in CAR-expressing cells by antigen stimulation. Jurkat cells expressing ZAP-70-AcGFP were plated on stimulatory coverslips coated with anti-CD3ε, then fixed and stained with fluorescently-labeled anti-CD3ζ. Confocal images at the interface between the attached cells and the stimulatory surface were collected (a). Jurkat cells coexpressing ZAP-70-AcGFP and CAR57-28ζ were plated onto EpCAM-coated coverslips and stained with anti-myc and Alexa-labeled anti-CD3ζ antibodies, as described in Section 2 (b).
4. Conclusion
In this study, we constructed CAR57-28ζ, a novel CAR gene specific for EpCAM, and found that the CAR gene can be functionally expressed in T cells. Because CAR57-28ζ consists of only gene products of human origin, it is likely to be less immunogenic than some alternative formats (e.g., mouse or humanized scFv) upon application for adoptive T-cell immunotherapy. We also demonstrated that the CAR-expressing T cells are activated through wild-type TCR-like molecular events mediated by the interaction of the CAR with its corresponding antigen, EpCAM. With regard to the endogenous TCRs, the lifespan and dynamics of the signaling clusters are one of the critical determinants of the activation and fate of T cells [20, 21]. Therefore, it would be important to quantitatively examine how long the CAR-containing clusters are sustained, and where the clusters migrate. To answer these questions, real-time live cell imaging of CAR-expressing cells might be helpful. Since, as compared with T-cell-stimulating antibodies, T-cell activation achieved by CAR57-28ζ was not so sufficient, further improvement of molecular design of CAR (e.g., the addition of costimulatory domain from other signaling proteins or the optimization of the signal sequence for efficient membrane sorting) is inevitable to accomplish the clinically effective CAR-mediated adaptive immunotherapy [1, 22]. Further studies are currently underway to transduce the CAR gene into human peripheral T lymphocytes and to determine the cytotoxic activity of the CAR-grafted T cells against EpCAM-positive tumor cells.
Acknowledgment
This work was supported by a Grant-in-Aid for Scientific Research (C) from the Japan Society for the Promotion of the Sciences.
References
- 1.Jena B, Dotti G, Cooper LJN. Redirecting T-cell specificity by introducing a tumor-specific chimeric antigen receptor. Blood. 2010;116(7):1035–1044. doi: 10.1182/blood-2010-01-043737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marcu-Malina V, Van Dorp S, Kuball J. Re-targeting T-cells against cancer by gene-transfer of tumor-reactive receptors. Expert Opinion on Biological Therapy. 2009;9(5):579–591. doi: 10.1517/14712590902887018. [DOI] [PubMed] [Google Scholar]
- 3.Loskog A, Giandomenico V, Rossig C, Pule M, Dotti G, Brenner MK. Addition of the CD28 signaling domain to chimeric T-cell receptors enhances chimeric T-cell resistance to T regulatory cells. Leukemia. 2006;20(10):1819–1828. doi: 10.1038/sj.leu.2404366. [DOI] [PubMed] [Google Scholar]
- 4.Kowolik CM, Topp MS, Gonzalez S, et al. CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Research. 2006;66(22):10995–11004. doi: 10.1158/0008-5472.CAN-06-0160. [DOI] [PubMed] [Google Scholar]
- 5.de Bono JS, Rha SY, Stephenson J, et al. Phase I trial of a murine antibody to MUC1 in patients with metastatic cancer: evidence for the activation of humoral and cellular antitumor immunity. Annals of Oncology. 2004;15(12):1825–1833. doi: 10.1093/annonc/mdh472. [DOI] [PubMed] [Google Scholar]
- 6.Kuroki M, Yamada H, Shibaguchi H, et al. Preparation of human IgG and IgM monoclonal antibodies for MK-1/Ep-CAM by using human immunoglobulin gene-transferred mouse and gene cloning of their variable regions. Anticancer Research. 2005;25(6 A):3733–3739. [PubMed] [Google Scholar]
- 7.Ishida I, Tomizuka K, Yoshida H, et al. Production of human monoclonal and polyclonal antibodies in TransChromo animals. Cloning and Stem Cells. 2004;4(1):91–102. doi: 10.1089/153623002753632084. [DOI] [PubMed] [Google Scholar]
- 8.Armstrong A, Eck SL. EpCAM: a new therapeutic target for an old cancer antigen. Cancer Biology and Therapy. 2003;2(4):320–326. doi: 10.4161/cbt.2.4.451. [DOI] [PubMed] [Google Scholar]
- 9.Le Naour F, Zoller M. The tumor antigen EpCAM: tetraspanins and the tight junction protein claudin-7, new partners, new functions. Frontiers in Bioscience. 2008;13:5847–5865. doi: 10.2741/3121. [DOI] [PubMed] [Google Scholar]
- 10.Al-Lazikani B, Lesk AM, Chothia C. Standard conformations for the canonical structures of immunoglobulins. Journal of Molecular Biology. 1997;273(4):927–948. doi: 10.1006/jmbi.1997.1354. [DOI] [PubMed] [Google Scholar]
- 11.Shirasu N, Shibaguci H, Kuroki M, Yamada H, Kuroki M. Construction and molecular characterization of human chimeric T-cell antigen receptors specific for carcinoembryonic antigen. Anticancer Research. 2010;30(7):2731–2738. [PubMed] [Google Scholar]
- 12.Marks JD, Bradbury A. PCR cloning of human immunoglobulin genes. Methods in Molecular Biology. 2004;248:117–134. doi: 10.1385/1-59259-666-5:117. [DOI] [PubMed] [Google Scholar]
- 13.Holt LJ, Büssow K, Walter G, Tomlinson IM. By-passing selection: direct screening for antibody-antigen interactions using protein arrays. Nucleic Acids Research. 2000;28(15):p. E72. doi: 10.1093/nar/28.15.e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bunnell SC, Barr VA, Fuller CL, Samelson LE. High-resolution multicolor imaging of dynamic signaling complexes in T cells stimulated by planar substrates. Science's STKE. 2003;2003(177):p. PL8. doi: 10.1126/stke.2003.177.pl8. [DOI] [PubMed] [Google Scholar]
- 15.Carroll WL, Mendel E, Levy S. Hybridoma fusion cell lines contain an aberrant kappa transcript. Molecular Immunology. 1988;25(10):991–995. doi: 10.1016/0161-5890(88)90005-3. [DOI] [PubMed] [Google Scholar]
- 16.Samelson LE. Signal transduction mediated by the T cell antigen receptor: the role of adapter proteins. Annual Review of Immunology. 2002;20:371–394. doi: 10.1146/annurev.immunol.20.092601.111357. [DOI] [PubMed] [Google Scholar]
- 17.Love PE, Hayes SM. ITAM-mediated signaling by the T-cell antigen receptor. Cold Spring Harbor Perspectives in Biology. 2010;2(6):p. a002485. doi: 10.1101/cshperspect.a002485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barda-Saad M, Braiman A, Titerence R, Bunnell SC, Barr VA, Samelson LE. Dynamic molecular interactions linking the T cell antigen receptor to the actin cytoskeleton. Nature Immunology. 2005;6(1):80–89. doi: 10.1038/ni1143. [DOI] [PubMed] [Google Scholar]
- 19.Au-Yeung BB, Deindl S, Hsu LY, et al. The structure, regulation, and function of ZAP-70. Immunological Reviews. 2009;228(1):41–57. doi: 10.1111/j.1600-065X.2008.00753.x. [DOI] [PubMed] [Google Scholar]
- 20.Saito T, Yokosuka T. Immunological synapse and microclusters: the site for recognition and activation of T cells. Current Opinion in Immunology. 2006;18(3):305–313. doi: 10.1016/j.coi.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 21.Bunnell SC, Singer AL, Hong DI, et al. Persistence of cooperatively stabilized signaling clusters drives T-cell activation. Molecular and Cellular Biology. 2006;26(19):7155–7166. doi: 10.1128/MCB.00507-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. The New England Journal of Medicine. 2011;365(8):725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]