

Abstract
17β-Estradiol (E2) is an important hormone signal that regulates multiple tissues and functions in the body. This review focuses on the neuroprotective actions of E2 in the brain against cerebral ischemia and the potential underlying mechanisms. A particular focus of the review will be on the role of E2 to attenuate NADPH oxidase activation, superoxide and reactive oxygen species (ROS) generation and reduce oxidative stress in the ischemic brain as a potentially key neuroprotective mechanism. Evidence of a potential novel role of extranuclear estrogen receptors in mediating E2 signaling and neuroprotective actions will also be discussed. An additional subject covered by the review is the growing evidence that periods of long term estrogen deprivation (LTED), such as occur after menopause or surgical menopause, may lead to loss or attenuation of E2 signaling and neuroprotective actions in the brain, and to enhanced sensitivity of the hippocampus to ischemic stress damage. These findings have important implications with respect to the “critical period hypothesis”, which proposes that estrogen replacement must be initiated at peri-menopause in humans to exert its beneficial cardiovascular and neural effects. Insights gained from these various studies will be valuable in guiding future directions of the field.
Keywords: Stroke, hippocampus, cerebral cortex, menopause, ovariectomy
Introduction - Estradiol and Sex Differences in Stroke Risk and Outcome
17β-Estradiol (E2) is a steroid hormone that is released into the blood where it can exert trophic or regulatory effects on many different target tissues such as the breast, ovary, uterus, bone and brain (1). The major source of circulating E2 in the female is the ovary, although other tissues such as adipose and brain have some capacity for E2 synthesis due to expression of the E2 synthesizing enzyme, aromatase (2-4). E2 levels in the blood fluctuate throughout the cycle in females, with peak circulating levels observed at midcycle in humans, and late diestrus II to proestrus in rodents (1, 5). Interestingly, stroke infarct size has been shown to have an inverse correlation with serum E2 levels, with smaller infarct size noted upon proestrus in rats, when E2 levels are highest (6-7). Administration of an estrogen receptor antagonist, ICI182,780 to intact female rats has also been shown to result in an increase in infarct size following focal cerebral ischemia (FCI), suggesting a role for endogenous E2 and estrogen receptors in mediating neuroprotection against cerebral ischemia (8). Sex differences in stroke have been reported in humans, with studies focusing primarily on incidence, age of first stroke, and stroke outcome (9-13). The studies suggest that women are “protected” against stroke relative to men – at least until the years of menopause, when E2 levels fall due to follicular depletion and stroke incidence increases in women (9, 11-13). Intriguingly, stroke outcome in postmenopausal women has been shown to be worse as compared to males, with postmenopausal women having a significantly higher disability and fatality rate as compared to men (9-10, 12-13).
While the ovary is a significant source of circulating E2 in women, there is significant evidence that E2 can be produced in extragonadal tissues as well. Of interest to this review, the enzyme for production of E2 from androgens, aromatase, has been shown to be expressed in several brain regions, including the hypothalamus, cortex, and hippocampus in male and female rats (2, 14), humans (4, 15) and monkeys (16). The roles and importance of brain-derived E2 are currently not fully understood. In vitro studies using aromatase inhibitors have suggested that brain-derived E2 has a role in regulating connectivity/plasticity of neurons (17-18). In addition, in vivo studies using aromatase knockout (KO) mice have shown that infarct volume is significantly increased in the aromatase KO animals following FCI as compared to wild-type mice (19-20). Intriguingly, infarct size was reported to be smaller in ovariectomized wild-type mice than in the aromatase KO mice, suggesting that brain-derived E2 production may have a role in neuroprotection (19). Aromatase expression has also been reported to increase in the peri-infarct region at 24h after FCI in the rat, with at least part of this increased expression occurring in astrocytes (21). Our laboratory has also observed that E2 increased aromatase expression in the hippocampal CA1 region at 48h after global cerebral ischemia (GCI) (Brann et al, unpublished observation). Collectively, the studies suggest that endogenous E2 production from gonadal and extragonadal sources have a neuroprotective role in the brain against cerebral ischemia.
Estrogen Receptor-alpha (ER-α) Mediates E2 Neuroprotection Against Cerebral Ischemia
Estradiol is thought to exert the majority of its biological actions in the body via interaction with two primary estrogen receptors: estrogen receptor-alpha (ER-α) and estrogen receptor-beta (ER-β). The two receptors exhibit significant homology in their structures, but display differential function, localization and pattern of expression in the brain (22-23). Both receptors are composed of seven domains, bind E2 with high affinity, and they both dimerize and utilize the classical estrogen response elements in a similar fashion. However, several differences do exist between ER-α and ER-β, as it has been shown that they contain different ligand binding domains, and each receptor is encoded by a different gene. The receptors also signal differently at the AF-1 site in the presence of E2, where E2 activates transcription at ER-α while it inhibits transcription at ER-β, respectively (24). ER-α and ER-β are primarily localized in the nucleus of cells, but extranuclear localization has also been demonstrated in the cytoplasm and membrane of cells and neurons, as will be discussed later in a subsequent section (25-29). Thus, both receptors have been implicated to mediate genomic signaling as well as non-genomic signaling in cells (30-32). Another difference between ER-α and ER-β is that they differ in their tissue distribution - with ER-α being expressed in breast, ovary, uterus, and brain (33-35), while ER-β is expressed in bone, heart, lungs, kidney, endothelial cells and brain (33, 36-37). In the brain, localization studies have demonstrated that ER-α is localized most densely in the hypothalamus, hippocampus, and preoptic area, with moderate to light density in the cerebral cortex (34-35). Conversely, ER-β localization has been documented predominantly in the cortex, throughout the hippocampus, in the olfactory bulb, septum, preoptic area, nucleus of striata terminalis, amygdala, paraventricular hypothalamus, thalamus, ventral tegmental area, substantia nigra and cerebellum (33, 38-39).
With respect to which receptor is thought to mediate E2 neuroprotection against cerebral ischemia, the majority of the literature suggests that ER-α has the primary and critical mediator role for E2-induced neuroprotection. In support of this contention, E2 neuroprotection against FCI has been shown to be lost in ER-α KO mice, but preserved in ER-β KO mice (40-41). In addition, antisense knockdown studies confirmed a critical role for ER-α, but not ER-β in mediating E2 neuroprotection in the hippocampal CA1 region in rats following GCI (42). Furthermore, administration of a selective ER-α agonist, propyl pyrazole triol (PPT) has also been shown to exert neuroprotection in the hippocampal CA1 region following GCI, and rescue the ischemia-induced deficit in long-term potentiation (43-44). E2 may achieve its neuroprotective effects through a multitude of effects upon a variety of cell types in the brain, including neurons, astrocytes, microglia and endothelial cells (1). However, emerging evidence suggests that a direct effect of E2 upon neurons mediated via neuronal ER-α is critical for mediating the neuroprotective effect of E2 against FCI, as E2 neuroprotection has been shown to be lost in neuron-specific ER-α knockout mice, but not in microglia-specific ER-α knockout mice (45). The study did not assess E2 neuroprotective ability in astrocyte- or endothelial-specific ER-α KO mice, so no definitive conclusion can be inferred about the role of these non-neuronal cell types in E2 neuroprotection against cerebral ischemia. There is a significant literature suggesting E2 can act on astrocytes to influence release of neuroprotective factors such as growth factors, as reviewed previously by our lab and others (46-48). In addition, E2 and the ER-α selective agonist, PPT, have been shown to directly enhance endothelial cell viability in vitro of immortalized mouse brain endothelial cells following an ischemic insult, suggesting E2 could act directly on endothelial cells and exert protection of the vasculature following ischemia (49).
While the majority of the literature appears to support a critical role for ER-α in mediating E2 neuroprotective effects against cerebral ischemia, there are studies suggesting that ER-β may have a neuroprotective role in certain situations. For instance, administration of a selective ER-β agonist, WAY 200070-3, has been shown to exert neuroprotection in the rat hippocampal CA1 region following GCI (44), and another study found that the ER-β agonist, DPN, reduced global cerebral ischemia damage in the mouse hippocampal CA1 region by 55% (50). In addition, the plant phytoestrogen, genistein, has also been shown to exert neuroprotection in the hippocampus against global cerebral ischemia, and this effect was blocked by treatment with an ER-β specific antagonist (51). These studies suggest that exogenous activation of ER-β can exert neuroprotection against cerebral ischemia. However, evidence of a role for ER-β in mediating endogenous E2 neuroprotection against cerebral ischemia is currently lacking, as E2 is fully capable of exerting neuroprotection against cerebral ischemia in ER-β knockout mice (40-41). Nevertheless, there is evidence that ER-β may have a role in basal neuronal survival, as it has been reported that there is substantial neuronal loss in the brains of ER-β knockout mice at 2 years of age as compared to wild type mice (52).
In addition, a novel, putative third ER, G-Protein-Coupled ER (GPR30, also known as GPER1), has recently been described (53). GPR30 is a seven transmembrane domain G-protein- coupled receptor known to be primarily localized in the plasma membrane and endoplasmic reticulum (53-54) of neurons in the brain and is expressed in several brain regions, including the islands of calleja, striatum, hypothalamus, area postrema, nucleus of the solitary tract, and hippocampus (54). Evidence supporting the role of GPR30 in neuroprotection was obtained from studies using a purported selective agonist for GPR30, G-1 (55-56). The studies showed that G-1 pretreatment significantly attenuated glutamate-induced neuronal cell death in hippocampal cell cultures (55). G-1 has also been recently shown to exert neuroprotection against FCI in female mice (57). While these studies are intriguing, they rely on exogenous agonist studies and do not demonstrate conclusively a role for GPR30 in mediating endogenous E2 neuroprotective actions. More definitive conclusions on the role of GPR30 in mediating E2 neuroprotection must await the results from studies using GPR30 KO mice, as well as selective GPR30 antagonist and knockdown approaches.
Finally, there is also evidence that non-feminizing estrogen analogues that lack affinity for estrogen receptors can also exert neuroprotection in cerebral ischemia (58-61). As reviewed recently by Yi et al (61), eight different nonfeminizing estrogens have been shown to be neuroprotective against cerebral ischemia. These findings are very intriguing, as nonfeminizing estrogens, since they lack ER affinity, would be predicted to lack negative side effects common to E2, such as stimulation of the breast, uterus, an enhancement of blood clotting. Further work has shown that estrogen analogues with large bulky groups at the 2 and/or 4 carbon of the phenolic A ring eliminate ER binding but enhance neuroprotective potency in cell culture screening models (61). It is not known whether the nonfeminizing estrogens bind to GPR30 to mediate their effects. Further studies are needed to address this interesting question. Further studies are also needed to elucidate the mechanism of action underlying the neuroprotective effects of nonfeminizing estrogens and establish whether they might have efficacy for postmenopausal hormone therapy.
Estrogen Regulation of Reactive Oxygen Species and Oxidative Stress
ROS, particularly superoxide, have been implicated to play a key role in neuronal cell death following cerebral ischemia (62-66). The superoxide anion radical (O2-) is the product of a one electron reduction of oxygen and it is the precursor of most ROS, including the highly toxic and damaging hydroxyl ion and peroxynitrite (67-68). While ROS are suggested to mediate physiological processes at low concentrations, when they are over-produced in pathological situations they can be highly injurious to adjacent structures in cells and neurons, including lipid membranes, DNA, and proteins (63). It is well known that following the onset of either permanent or transient FCI, ROS increase significantly in the cerebral cortex and other brain regions (1, 62-66). Along these lines, it has been shown that there is a marked steady elevation of ROS in the penumbra (infarct border) of the parietal cortex during a 3h measurement period post ischemia in permanent cerebral ischemia (64). Likewise, studies using a marker of O2- production, hydroethidine (HEt), have yielded a similar pattern of increased O2- production in the cortex of male mice and ovariectomized female rats within 1-3h hrs of permanent cerebral ischemia (1, 65-66). In addition, as shown in Figure 1a, work by our laboratory has shown that O2- production increases rapidly in the hippocampal CA1 region following GCI in both male and female rats, with an elevation occurring as early as 30 min after reperfusion and peak levels observed at 3h after reperfusion (42, 69). As also shown in Figure 1a, E2 treatment strongly attenuated the elevation of O2- levels in the hippocampal CA1 region following cerebral ischemia, which correlated with its neuroprotective effect (42). Further studies showed that the E2 attenuation of O2- levels was associated with a dramatic attenuation of oxidative stress damage in the hippocampal CA1 region at 24h after cerebral ischemia, as determined by measurement of oxidative damage markers for lipid peroxidation (4-HNE) and DNA damage (8-OHdG) (Figure 1b-c) (42). A similar E2 suppression of O2- production was demonstrated in the cerebral cortex following FCI (1). In the next section, we will discuss how E2 may regulate ROS generation in cerebral ischemia with a particular focus on an emerging key enzyme for O2- production, Nicotinamide Adenine Dinucleotide Phosphate (NADPH) oxidase.
Figure 1. E2 attenuates superoxide production and oxidative damage in hippocampal CA1 after global cerebral ischemia.
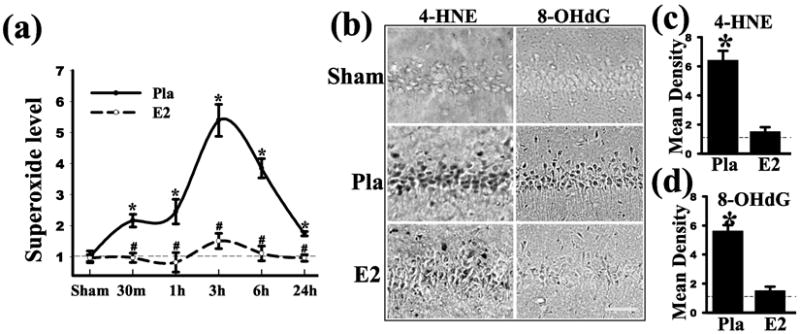
Adult ovariectomized rats were treated with E2 for 1 week prior to 10-min GCI and killed at various times after reperfusion. The E2 minipumps produced serum levels of 10-15pg/ml. (a) Superoxide production in the hippocampal CA1 region from sham, placebo (Pla) and E2 treated rats was measured using a luminol-based photoemissions assay (b-d) Effect of E2 on oxidative damage markers for lipid peroxidation (4-HNE) and DNA damage (8-OHdG) 1 day after ischemia. Note that E2 strongly decreased 4-HNE and 8-OHdG staining. Values are means ±SE of 4-5 rats in each group and expressed as fold changed vs. sham+Pla group. *P <0.05 vs. sham; #P <0.05 vs. Pla at the same time point. Reproduced with permission from QG Zhang et. al.: Journal of Neuroscience 29:13823-13836, 2009 (42).
E2 Attenuates NADPH Oxidase Activation Following Global Cerebral Ischemia
In vitro studies have suggested that there may be three distinct mechanisms for generating ROS in hippocampal and cortical neurons during hypoxia/reoxygenation (70). The studies provided evidence that the mitochondria generates the initial ROS burst during hypoxia, followed by xanthine oxidase (XO) during the delayed phase, and ending with NADPH oxidase-generated ROS production in reperfusion. It is well known that E2 can have beneficial effects upon mitochondria to preserve mitochondrial function. These effects include regulation/preservation of ATP generation, ROS production, mitochondrial apoptotic factors, and antioxidant mechanisms. E2 effects upon mitochondria have been extensively reviewed previously, and the reader is referred to these excellent reviews for additional information (71-72). New emerging evidence suggests that the membrane, via NADPH oxidase, may play an additional critical role in ROS generation in neurons following cerebral ischemia. The NADPH oxidase enzyme is composed of key subunits from the NOX family, whose primary job is to transport electrons across biological membranes to reduce molecular oxygen to O2- (73-76). The NOX family is composed of five isoforms (NOX1-NOX5). Despite their similar structure and enzymatic function, NOX family isoforms differ in their mechanism of activation. NOX1 activity requires the subunits p22phox, NOXO1 and NOXA1, and is Ras-Related C3 Botulinum Toxin Substrate 1 (Rac1)-dependent, while NOX 3 requires similar subunits for its activation, but is Rac1-independent. NOX4 and NOX5 isoforms do not appear to require many subunits for their activation, as they are thought to be constitutively active and Rac1-independent (73). The activation of NOX2, the most studied and best characterized NOX isoform and a major focus of our studies, involves interaction with the subunits p22phox, p67phox, p40phox and p47phox subunits. In addition, the GTPase, Rac1 has been shown to be critical for NOX2 activation (69, 73, 75). NOX2 and p22phox are found primarily on the membrane, in resting cells, existing in close association and stabilizing one another. Upon cell activation/stress, there is an exchange of GDP for GTP on Rac1, a Rho GTPase, leading to its activation and translocation to the membrane. Simultaneously, phosphorylation of cytosolic p47phox allows for its binding with other membrane subunits (p67phox and p40phox), leading to conformational changes to allow interaction with p22phox on the membrane. This activates the NOX2 enzyme complex, which transports electrons from cytoplasmic NADPH to oxygen and generates O2- (73).
Localization of the NOX family isoforms has been studied extensively in many tissues throughout the body. In 2001, Lambeth and his group documented strong NOX2 mRNA expression and faint RT-PCR bands of NOX4 and NOX5 in the brain (77). Moreover, further studies by our group and others revealed NOX2 (42, 69, 78) and NOX4 (79) expression in the hippocampus, as well as NOX2 localization in the cerebral cortex (78). Of the different NOX enzyme isoforms, the greatest evidence to date implicates a critical role for NOX2 in ROS generation following cerebral ischemia and the resultant oxidative stress damage. In support of this contention, infarct volume was shown to be significantly reduced in NOX2 knockout mice as compared to their wild-type litter mates (80-81). Furthermore administration of the NADPH oxidase inhibitor, apocynin was shown to reduce infarct size after FCI (82) and significantly reduced neurological deficit score in mice, thus achieving an improved behavioral cognitive outcome (80-82). The ability of apocynin to reduce infarct volume, neurological impairment and mortality was lost when it was administered in NOX2 KO mice, which strongly suggests that its beneficial neuroprotective effects are due specifically to inhibition of NOX2 NADPH oxidase (81). Apocynin neuroprotection against cerebral ischemia was associated with reduced levels of apoptotic factors and markers, such as Bax, Bcl-2 and TUNEL staining (83), suggesting that NADPH oxidase activation plays a key role in the induction of apoptosis following cerebral ischemia. Additional work by our laboratory showed that administration of a specific competitive NOX2 inhibitor, gp91ds-tat, significantly attenuated elevation of NADPH oxidase activity and O2- levels in the hippocampal CA1 region following GCI, and was strongly neuroprotective (42). This suggests that NOX2 NADPH oxidase plays a significant role in the elevation of O2- and resultant neuronal damage in the hippocampus following cerebral ischemia. Further work by our laboratory and others demonstrated that NOX2 is predominantly localized in neurons in the hippocampus following cerebral ischemia (42), but also appears in microglia at later time-points after cerebral ischemia (84). In situ O2- determination using the hydroethidine method also revealed O2- elevation in neurons, with some occurring in microglia/macrophages, and little in endothelial cells in the cortex and hippocampus at early time-points after cerebral ischemia (42, 85). There is also some evidence that NOX2-derived O2- from circulating lymphocytes that infiltrate the infract area may also contribute to O2- elevation at the infarct site (86).
As shown in Fig. 2, work by our laboratory showed that NADPH oxidase activity increases rapidly in the hippocampal CA1 region following GCI in ovariectomized female rats, with peak levels observed at 3h after reperfusion (42). Note that the pattern of NADPH oxidase activation following cerebral ischemia is similar to that we observed for O2- elevation. As also shown in Fig. 2, E2 treatment strongly attenuated the elevation of NADPH oxidase activity in the hippocampal CA1 region following cerebral ischemia, which correlated with its suppression of O2- levels and its neuroprotective effect (42). As shown in Fig. 3, the ability of E2 to exert neuroprotection and attenuate the elevation of NADPH oxidase activity and O2- in the hippocampal CA1 region after global cerebral ischemia was lost in animals in which ER-α was knocked by antisense oligonucleotides, but was preserved in ER-β antisense knockdown animals (Fig. 3a-d) (42). This suggests that the neuroprotective and antioxidant effects of E2 in global cerebral ischemia are primarily mediated by ER-α. We further showed that E2 inhibited activation of the GTPase, Rac1, in an Akt-dependent manner following cerebral ischemia, which is critical for NOX2 NADPH oxidase activation (42). Additional work showed that administration of a Rac1 inhibitor markedly attenuated NADPH oxidase and superoxide generation in the hippocampal CA1 region following cerebral ischemia and was neuroprotective and preserved cognitive function (69).
Figure 2. E2 attenuates NADPH oxidase activity in hippocampal CA1 after global cerebral ischemia.
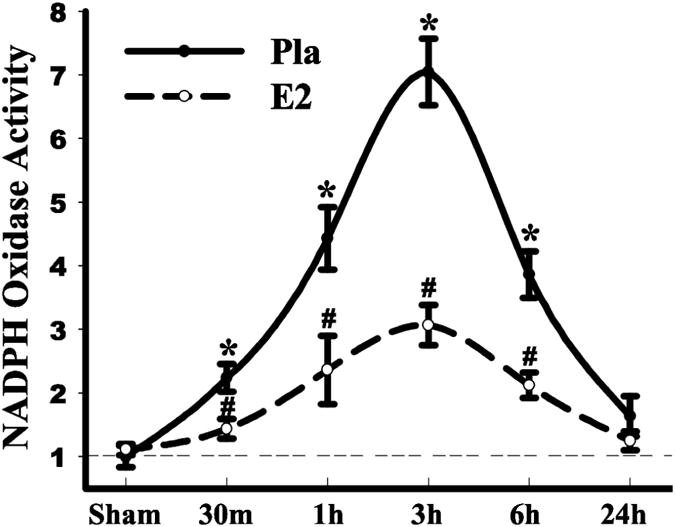
Adult ovariectomized rats were treated with E2 for 1 week prior to 10-min GCI and killed at various times after reperfusion. The E2 minipumps produced serum levels of 10-15pg/ml. NADPH oxidase activity in the hippocampal CA1 region from sham, placebo (Pla) and E2 treated rats was measured using a lucigenin-based photoemissions assay. Values are means ±SE of 4-5 rats in each group and expressed as fold changed vs. sham+Pla group. *P <0.05 vs. sham; #P <0.05 vs. Pla at the same time point. Reproduced with permission from QG Zhang et. al.: Journal of Neuroscience 29:13823-13836, 2009 (42).
Figure 3. Evidence that ER-α mediates E2 antioxidant and neuroprotective effects in the hippocampal CA1 region following cerebral ischemia.
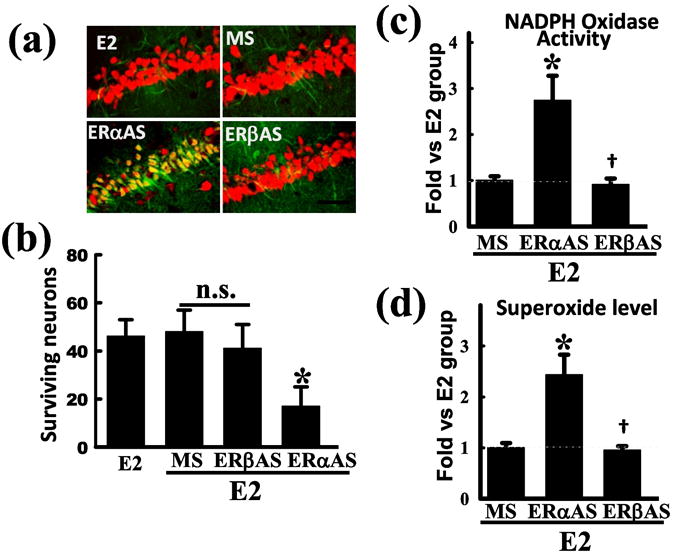
Ovariectomized rats were treated with E2 for 1 week prior to 10-min GCI. The E2 minipumps produced serum levels of 10-15pg/ml. (a) Missense (MS) oligodeoxynucleotides, ER-α or ER-β antisense (AS) oligodeoxynucleotides (10 nmol) were injected bilaterally icv every 24h for 4d prior to GCI reperfusion. Hippocampal CA1 sections were collected at 7 days after reperfusion and assessed for neuroprotection by immunohistochemistry for NeuN (neuronal marker - red) and staining for FluoroJadeB (neuronal degeneration marker – green). E2 neuroprotection was imaged and visualized using confocal microscopy. Note that E2 neuroprotection was abolished only in the ERα AS treated animals. Values are means ± SE from 6-7 animals. (b) Quantification of surviving neurons by counting NeuN positive and FluoroJade B negative cells. *P <0.05 vs. E2+MS group. Scale bar, 50 μm; 40×. (c-d) NADPH oxidase activation (c) and superoxide production (d) was assessed at 3h reperfusion using a lucigenin and luminol-based photoemission assay, respectively. Note that E2 attenuation of NADPH oxidase activity and superoxide elevation was abolished in ER-α AS treated animals but not ER-β treated rats *P <0.05 vs. E2+MS, †P >0.05 vs.E2+ERα-AS. Reproduced with permission from QG Zhang et. al.: Journal of Neuroscience 29:13823-13836, 2009 (42).
Estrogen Extranuclear Receptor Signaling and E2 Neuroprotection
It has been predominantly thought that E2 neuroprotection in the brain is mediated principally by the “classical” nuclear ER-mediated genomic signaling pathway, which involves E2 interaction with nuclear ER and regulation of transcription of various genes that mediate neuroprotection. For instance, E2 has been shown to increase the expression of the anti-apoptotic gene, bcl-2, in the ischemic penumbra following FCI and GCI (87). E2 also increases bcl-2 in vitro in rat hippocampal neurons and human NT2 neurons (88-89), while it inhibits expression of pro-apoptotic BAD (bcl-2-antagonist of cell death) (87-90). Additionally, E2 enhances expression of the anti-apoptotic pro-survival factor, survivin in the hippocampus CA1 following GCI, which facilitates neuronal survival (91). E2 has also been shown to enhance expression of brain derived neurotrophic factor (BDNF) in the brain, which has been implicated as a neuroprotective factor and to be important for synaptic plasticity, learning, and memory (92-93).
In addition to genomic signaling, there is increasing evidence that rapid non-genomic signaling via membrane localized extranuclear ER may also play a role in mediating E2 neuroprotective effects in the brain (30, 94-95). Along these lines, several laboratories have shown that the rapid activation of extracellular signal-regulated kinases 1,2 (ERKs) by E2 is critical for its neuroprotective effects, as administration of a MEK inhibitor blocks E2 neuroprotection in neurons in vitro (94-96). Furthermore, E2-induced ERK activation in the CA1 region after GCI, which is critical for its neuroprotective effects as treatment with a MEK inhibitor blocked E2-induced ERK activation and E2 neuroprotection in the hippocampus (97). Likewise, a role for the pro-survival serine kinase Akt in E2 neuroprotection has been implicated, as E2 rapidly up-regulates Akt activation in cortical neurons in vitro (98), and in the hippocampus CA1 in vivo following GCI (99), while treatment with a PI3K inhibitor attenuates the neuroprotective effects of E2 both in vitro and in vivo (98-99). In addition, we recently demonstrated that E2 attenuates the rapid activation of the pro-apoptotic signaling kinase, JNK in the hippocampal CA1 region after GCI (91). As a whole, these findings suggest that E2-induced rapid non-genomic signaling may play a critical role in E2 neuroprotection.
However, since the above studies principally used E2, which can activate both extranuclear and nuclear estrogen receptors, it has been difficult to distinguish the importance and contribution of extranuclear receptor-mediated signaling in E2 neuroprotective effects. To address this issue, we employed two E2 conjugates, E2-BSA conjugate (100-102) and the newer E2 dendrimer conjugate (EDC) (103), which due to their size and charge cannot enter the cell nucleus. EDC and E2-BSA retain their ability to induce rapid extranuclear-mediated nongenomic signaling, but lack significant nuclear ER-mediated genomic signaling ability due to their inability to enter the cell nucleus and interact with nuclear ER (102-103). Using FITC-labeled EDC and E2-BSA we demonstrated that following intracerebroventrically (icv) injection in the lateral ventricle, the compounds are heavily localized in the hippocampal CA1 region and display a membrane/cytoplasmic localization without any appearance of nuclear localization (104). The results of the study further revealed that EDC and E2-BSA administered icv rapidly activates ERK, Akt and CREB signaling pathways in the hippocampus, enhances levels of the CREB transcriptional target, BDNF, strongly protects the hippocampal CA1 region against neuronal cell death, and significantly improves hippocampal-dependent cognitive function in the Morris water maze following GCI (104). The effects required estrogen receptor mediation, as they were blocked by administration of the estrogen receptor antagonist, ICI182,780. In addition, further studies showed that EDC attenuated Rac1 and NADPH oxidase activation and elevation of O2- in the hippocampal CA1 region after cerebral ischemia, and that its effects involved activation of the pro-survival kinase, Akt (42). The results of these studies thus provides important new evidence supporting an important role for extranuclear estrogen receptor activation in estrogen-induced neuroprotection and improved functional cognitive outcome following GCI, and suggests that ERK-Akt-CREB-BDNF signaling is an important component mediating extranuclear estrogen receptor beneficial neural effects. It should be mentioned that in addition to the proposed neuroprotective role of ERK1/2 activation in cerebral ischemia, there is also evidence for a pro-death role of ERK activation. For instance, administration of MEK inhibitors has been shown to significantly reduce ischemic damage to the brain following GCI or FCI (105-107), which suggests a neurodegenerative role for ERK activation after cerebral ischemia. It has been postulated that enhanced ERK1/2 activation may send a neuroprotective signal that involves the eventual down-regulation of its own activation, thereby preventing a prolonged elevation of ERK. However, in our studies in vivo in the GCI model, we found that ERK activation in the vehicle-treated rat is biphasic, with an early elevation at 10 min and 30 min after reperfusion, a fall to control levels at 3h and 6h after reperfusion, followed by a secondary elevation at 24h after reperfusion (104). Interestingly, acute EDC treatment significantly elevated ERK activation at 10 min, 30 min, 3h and 6h post-reperfusion as compared to the vehicle-treated group, but did not enhance the secondary elevation that occurred at 24h after reperfusion. Hence, in our studies acute estrogen analogue treatment enhanced and prolonged ERK activation in vivo in the hippocampal CA1 region following GCI. Thus, our studies did not show an estrogen-induced reduction of ERK activation that would fit the proposed model of ERK activation leading to its own inactivation. However, our study only examined up to 24h after GCI, and thus studies at more prolonged timepoints after GCI may be needed to determine if there is a subsequent down-regulation of ERK at later timepoints. The seemingly contradictory “good role” versus “bad role” of ERK activation in cerebral ischemia could depend on many factors, including 1) cell type of induction (neuron, glia, or endothelial cell), 2) pattern/duration of induction (acute, biphasic, chronic), and 3) subcellular localization of ERK (nucleus versus cytoplasm). For an elegant discussion and treatment of this complex subject, the reader is referred to an excellent review by Sawe et. al. (108) on the dual role of ERK activation in cerebral ischemia.
Currently, it is unclear which extranuclear estrogen receptor mediates the rapid effects of E2 or E2 conjugates in neurons. Previous work has shown that ER-α and ER-β can exist as dimers in the plasma membrane of cells (32, 109), and that COS-7 cells engineered to express ER-α and ER-β display localization of ∼2-5% of ER-α and ER-β protein to the plasma membrane (102). These studies suggest that classical ERs can be targeted to the plasma membrane. Key mechanisms for targeting ER-α and ER-β to the plasma membrane include palmitoylation of ER-α and ER-β and interaction of ERs with the scaffold protein, caveolin-1 (110-111). While these studies were conducted in non-neuronal cells, numerous studies have confirmed the presence of both ER-α and ER-β at the plasma membrane of neurons in various brain regions including the hippocampus, and at other extranuclear sites, such as in dendrites and spines (25, 28, 112-116). Furthermore, membrane localization of ER-α and ER-β has been demonstrated in glia cells in different brain regions (113, 115, 117-118), and glia cells have also been implicated to potentially participate in mediating estrogen neuroprotection via the release of growth factors and neuroactive steroids (48, 119-120).
Finally, there is evidence that estrogen extranuclear receptor-induced non-genomic signaling can crosstalk to the nucleus to effect genomic signaling. Along these lines, Katzenellenbogen and coworkers (121) have demonstrated that EDC can regulate gene expression in cells in vitro and that the effect does not involve interaction with or activation of nuclear ER genomic signaling. Rather, EDC effected changes in gene expression via its activation of rapid ERK and Src kinase signaling, which can regulate phosphorylation of transcription factors, histones and other factors and thereby modulate gene transcription. The study further showed that EDC was incapable of recruiting nuclear ER-α to estrogen responsive regions of genes, whereas ER-α recruitment by E2 was very effective. Thus, EDC non-genomic signaling can induce genomic signaling that is independent of nuclear ER. Intriguingly, previous work by Pfaff and coworkers has also demonstrated that non-genomic signaling by E2 in the hypothalamus can actually potentiate E2 genomic actions to induce lordorsis behavior (122-123), suggesting that rapid effects of E2 may also modulate genomic effects of E2. Interestingly, our own findings revealed that EDC and E2-BSA enhanced phosphorylation of the transcription factor, CREB in a rapid fashion following reperfusion, and that this effect is ERK- and Akt-dependent. Among the best known CREB transcriptional targets is the growth factor, BDNF, and intriguingly our study also demonstrated it to be elevated by EDC. This finding raises the possibility that EDC activation of extranuclear estrogen receptors may involve a non-genomic to genomic signaling cascade via kinase-induced activation of the transcription factor, CREB. As a whole the studies, suggest that both extranuclear and nuclear receptor signaling mediates E2 neuroprotective actions and that there may be crosstalk between the two signaling pathways.
Long-Term E2 Deprivation Alters the Sensitivity of the Brain to E2
Basic science and clinical observation studies have provided evidence of a beneficial effect of E2 upon cardiovascular disease, neuroprotection and neurodegenerative diseases such as stroke and Alzheimer's disease (1, 124-128). However, the Women's Health Initiative (WHI) surprisingly failed to observe a protective effect of hormone replacement therapy upon the cardiovascular and neural system, and in fact reported a small, but significant increase in risk for stroke and dementia (129-131). The average age of subjects in the WHI study was 63 years of age, far past the onset of menopause. It has been suggested that there may be a “critical period” for beneficial protective effect of E2 upon the brain, and that estrogen may need to be administered at peri-menopause or earlier to observe a beneficial effect upon the cardiovascular and neural system (132-134).
In support of a “critical period” hypothesis for E2 beneficial effects in the brain, a significant body of work has emerged which has shown in animal and human studies that long-term E2 deprivation (LTED) (long-term ovariectomy) leads to a loss of many E2 effects in the brain, such as neuroprotection, synaptic plasticity and cognitive function, and enhances risk of neurological diseases and mortality. As shown in Table 1, LTED has also been shown to lead to a loss of E2 ability to enhance LTP, spine density, attention processes, working memory, and exert vascular protective actions in rodents (135-138). In addition, surgical menopause (long-term ovariectomy) in humans has been shown to increase risk for cognitive decline, dementia, Parkinson's disease, depression, and increased mortality for neurological and mental diseases (139-142) (Table 1). Intriguingly, E2 replacement has been shown to reverse these effects in surgical menopausal subjects, indicating it is the loss of E2 that leads to these increased risks and negative outcomes (124, 143). Recent work by our group and other has shown that E2 neuroprotection in animal models of FCI and GCI is lost following LTED (42, 144). Along these lines, Fig. 4a shows that E2 treatment administrated after a 10 week period of E2 deprivation (ovariectomy) was no longer able to exert neuroprotection against GCI. Interestingly, the uterus was still responsive to E2, as evidenced by a robust uterotrophic response to E2 in the LTED animals (Fig. 4b). Thus there was a tissue-dependent loss of sensitivity to E2 in the LTED animals. We thus examined whether the loss of E2 sensitivity in the hippocampal CA1 region could be due to an alteration in estrogen receptor levels. As shown in Fig. 4c-d, Western blot analysis revealed a dramatic attenuation of ER-α, but not ER-β protein levels in the hippocampal CA1 region of LTED animals (10W) as compared to animals who received immediate E2 replacement after ovariectomy (Imm). Note that the reduction in ER-α protein levels occurred in all groups, including sham controls, suggesting that LTED leads to lower ER-α levels regardless of treatment and that E2 and ischemia cannot reverse the suppression of ER-α protein levels (42). This decrease in ER-α and E2 sensitivity was tissue-specific, as ER-α did not decrease in the uterus following LTED (Fig. 4 e-f). It should be noted that LTED has been shown to lead to a significant decrease of ER-α in the vasculature as well, which was correlated with a loss of E2 vascular protective actions (145-146). Additional work by our group has shown that the hippocampal CA3 region, which is resistant and not normally damaged following global cerebral ischemia, becomes heavily damaged in LTED rats following global cerebral ischemia (42). There is also a dramatic induction of Alzheimer's Disease (AD)-related proteins such as beta-amyloid, amyloid precursor protein, and phospho-tau in the hippocampal CA3 region of LTED rats following GCI (147). It is speculated that the hypersensitivity of the hippocampal CA3 region to ischemic stress damage and AD-related protein induction observed in our study could help explain the increased risk for cognitive decline and dementia observed in women following surgical menopause. Finally, a new 10 year reevaluation of a component of the WHI study has provided important support for the critical period hypothesis (148). The study examined 11,000 women ages 50-79 that had hysterectomies and were treated with either placebo or estrogen alone. The WHI study was stopped in 2004 due to increased stroke risk and the women stopped taking estrogen at that time. The 10 year follow-up study found significant beneficial cardiovascular effects of estrogen in women in their 50s, neutral effects for those in their 60s, and increasingly negative effects in women in their 70s. Women who were treated with estrogen in their 50s had a 41% lower coronary disease risk, 46% lower heart attack risk, significantly decreased invasive breast cancer risk, and a significant decrease in overall mortality. In contrast, estrogen treatment begun in women in their 70s had increased risk of cardiovascular disease, colorectal cancer and mortality. The study shows that age has an important effect on outcome of estrogen replacement therapy in humans, and that estrogen replacement in women in their 50s exerts many beneficial effects that are lost if E2 treatment is delayed to later in life (e.g. age 70 or greater). These findings are consistent with the “critical period” hypothesis that estrogen replacement, to be beneficial, must be given prior to a long-term period of estrogen deprivation such as occurs after the menopause. It should be mentioned that there are several other large clinical trials ongoing on estrogen replacement therapy benefits in humans, and it will be interesting to see the outcomes of these studies.
Table 1. Neural and Cardiovascular Effects of Long Term Ovariectomy.
| Group | Species | Tissue | Effect |
|---|---|---|---|
| Rocca et al 2007 (141) | Human | Brain | ↑ risk cognitive impairment and dementia |
| Rocca et al 2008 (142) | Human | Brain | ↑ risk Parkinson's Disease |
| Rocca et al 2008 (140) | Human | Brain | ↑ risk depression & anxiety |
| Rocca et al 2009 (139) | Human | Brain | ↑ mortality for neurological and mental diseases |
| Suzuki et al 2007 (144) | Rat | Cortex | Loss of E2 neuroprotective effect |
| Zhang et al 2009 (42) | Rat | Hippocampus | Loss of E2 neuroprotection, ↓ ERα, ↑ ischemic damage to hippocampal CA3 region |
| Daniel et al 2006 (137) | Rat | Cortex & Hippocampus | Loss of E2 enhancement of working memory |
| Bohacek & Daniel 2010 (138) | Rat | Cortex & Hippocampus | Loss of E2 enhancement of attention processes |
| Smith et al 2010 (136) | Rat | Hippocampus | Loss of E2 enhancement of spine density and LTP |
| Wu et al 2011 (135) | Rat | Hippocampus | ↓ intrinsic excitability and loss of E2 sensitivity |
| Pinna et al 2008 (146) | Rat | Aorta | ↓ ERα and loss of E2 protective vascular actions |
| Jesmin et al 2003 (145) | Rat | Cerebral vessels | ↓ ERα & ERβ, and ↓ cerebral capillary density |
Figure 4. Attenuation of hippocampal CA1 region ER-α levels and loss of E2 neuroprotective ability against GCI following long-term E2 deprivation (LTED.
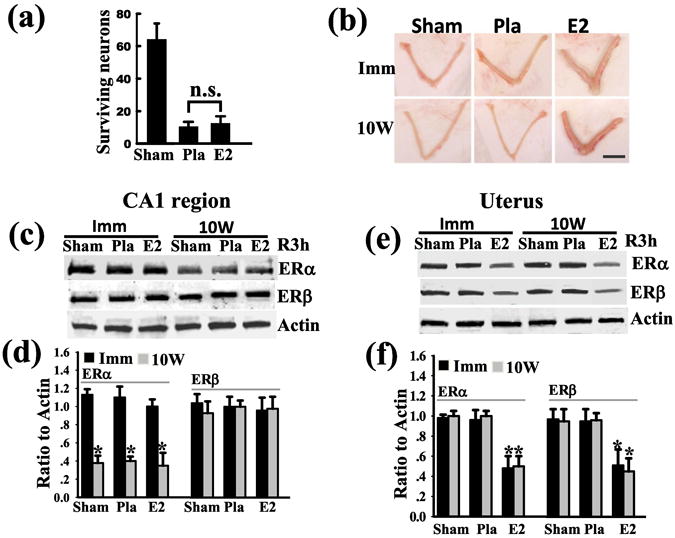
(a) Adult female rats were ovariectomized and 10 weeks later treated with Placebo (Pla) or E2 for 1 week and then subjected to 10 min GCI. Sham animals were included as controls and were subjected to the surgeries but no cerebral ischemia. The animals were killed at 7 days after reperfusion and the number of surviving neurons (NeuN positive and FluoroJadeB negative) in the hippocampal CA1 region was counted. Note that E2 does not protect against GCI in the LTED animals. N.S. = No significant difference. (b) Rats were ovariectomized and treated either immediately (Imm) or 10 weeks later (10W) with Placebo (Pla) or E2. One week after Pla or E2 treatment, the animals underwent 10-min GCI, and 7 days after reperfusion the animals were killed and uterus examined for uterotrophic effect of E2. Note that E2 exerted a robust uterotrophic effect in both Imm and 10W (LTED). Scale bar, 1cm; 1×. (C) Western blot analysis for ER-α and ER-β protein levels in the hippocampal CA1 region of Immediate (Imm) versus 10W (LTED) animals at 3h reperfusion show a profound reduction of ER-α, but not ER-β, levels in the hippocampal CA1 region of 10W (LTED) animals as compared to the Imm animals. (d) Semi-quantitative analysis of data from Western blot analysis in Panel c. Data is expressed as Means ± SE (n=4-6 rats per group) and as a ratio to actin.*P <0.05 vs. Imm group. (e) In contrast, Western blot analysis of uterine samples reveal that 10W (LTED) animals have the same pattern and levels of ER-α and ER-β levels as Imm animals (e.g. no decrease of either ER-α or ER-β levels by LTED). Note that E2 exerts a significant down-regulation of both ER-α and ER-β in the uterus in both Imm and 10W (LTED) rats, further indicating that the uterus maintains sensitivity to E2 following LTED. (f) Semi-quantitative analysis of data from Western blot analysis in Panel e. Data is expressed as Mean ± SE per group (n= 4-6) and expressed as ratio to actin *P < 0.05 vs. Pla group. Reproduced with permission from QG Zhang et. al.: Journal of Neuroscience 29:13823-13836, 2009 (42).
Conclusions
Based on the literature summarized in this review, there is abundant evidence that E2 has a significant neuroprotective effect against cerebral ischemia. Diagram 1 provides a summary pathway for the mechanisms of E2 neuroprotection discussed in this review. As shown in Diagram 1, E2 neuroprotection is suggested to be mediated by both extranuclear and nuclear estrogen receptor-signaling pathways. Based on knockout and knockdown studies, as well as selective agonist studies, the predominant view is that E2 neuroprotection against cerebral ischemia is mediated by ER-α. Exogenous agonist studies suggest that activation of GPR30 and ER-β exogenously may also exert neuroprotection against cerebral ischemia, but studies showing these receptors mediate endogenous E2 neuroprotection against cerebral ischemia are lacking. As further shown in Diagram 1, E2 activation of nuclear ER leads to genomic signaling in which the expression of pro-survival and anti-apoptotic genes are up-regulated and pro-death/apoptotic genes are down-regulated. In contrast, E2 activation of extranuclear ER is proposed to modulate activation of kinases which can post-translationally modify the activity of other key cellular proteins to exert neuroprotection. For instance, our studies showed that extranuclear signaling by E2 can activate the pro-survival kinase, Akt which phosphorylates Rac1 and inhibits its activation. The inhibition of Rac1 activation is proposed to lead to a profound inhibition of NADPH oxidase activation, and a resultant attenuation of cerebral ischemia induced O2- elevation, and oxidative stress damage, as well as decreased mitochondrial damage and apoptosis. Although not shown, there is also abundant evidence that E2 can act directly on mitochondria as well to preserve ATP production, decrease ROS generation and inhibit apoptotic signaling. Finally, the extranuclear non-genomic signaling pathway may crosstalk to the genomic signaling pathway, as E2 activation of kinases can lead to their translocation to the nucleus, where they can regulate gene expression by post-translationally modifying the transcription factors and thus changing their activity. It should be pointed out that this diagram is obviously not “all inclusive” of the many possible signaling roles and actions of E2. Nevertheless, it highlights some important signaling pathways that have been recently elaborated and thought to play a key role in E2 neuroprotection in cerebral ischemia. Finally, LTED can lead to a loss of E2 neuroprotection and other key neural effects in the brain. For the hippocampus, the loss of E2 neuroprotective effect following LTED was shown to be correlated with a significant decrease of ER-α levels in the hippocampal CA1 region. LTED was also shown to lead to hypersensitivity of the hippocampal CA3 region to ischemic stress. As a whole, the findings of decreased sensitivity of certain brain regions to E2 provide support for the “critical period” hypothesis that estrogen replacement therapy may need to be administered at peri-menopause to observe many of its beneficial neural effects. In support of this contention, new results from the WHI study 10-year evaluation on estrogen alone replacement in women with prior hysterectomy provides supports for the “critical period” hypothesis by demonstrating that beneficial effects of estrogen alone on cardiovascular disease, heart attack, invasive breast cancer, and mortality were observed when administered to subjects in their 50s, but not observed when administered to subjects in their 70s (148). Finally, the studies by our group and others on LTED may also provide insights into why surgical menopausal patients have increased risks for cognitive decline, dementia, and increased mortality for neurological diseases.
Diagram 1.
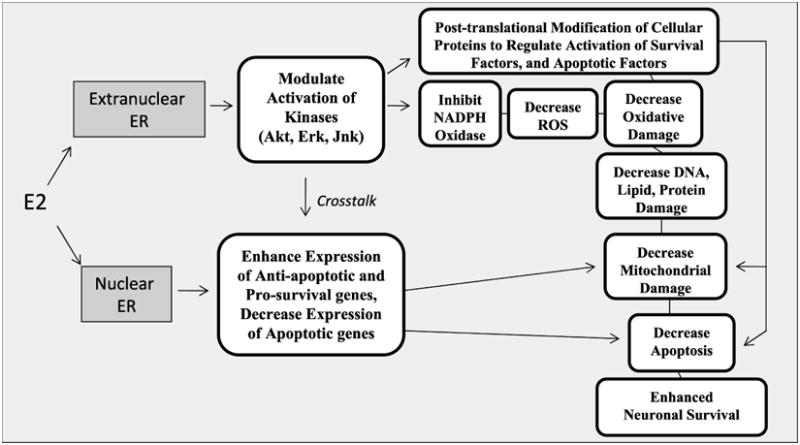
Summary diagram depicting the neuroprotective mechanisms of E2 via nuclear and extranuclear signaling pathways. See text for discussion.
Acknowledgments
This study was supported by a research grant to DWB from the NINDS (NS050730), National Institutes of Health, United States of America, and a Scientist Development research grant to QZ from the American Heart Association.
Abreviations
- AD
Alzheimer's Disease
- BDNF
Brain Derived Neurotrophic Factor
- E2
17β-Estradiol
- EDC
Estrogen Dendrimer Conjugate
- ER
Estrogen Receptor
- ER-α
Estrogen Receptor Alpha
- ER-β
Estrogen Receptor Beta
- ERK
Extracellular Signal-Regulated Kinase
- FCI
Focal Cerebral Ischemia
- GCI
Global Cerebral Ischemia
- GPR-30
G-Protein-Coupled-Receptor
- ICV
Intracerebroventricular
- KO
Knockout
- LTED
Long Term Estrogen Deprivation
- NADPH Oxidase
Nicotinamide Adenine Dinucleotide Phosphate Oxidase
- O2-
Superoxide
- PPT
Propyl Pyrazole Triol
- Rac1
Ras-Related C3 Botulinum Toxin Substrate 1
- ROS
Reactive Oxygen Species
- WHI
Women Health Initiative
References
- 1.Brann DW, Dhandapani K, Wakade C, Mahesh VB, Khan MM. Neurotrophic and neuroprotective actions of estrogen: basic mechanisms and clinical implications. Steroids. 2007;72(5):381–405. doi: 10.1016/j.steroids.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hojo Y, Hattori TA, Enami T, Furukawa A, Suzuki K, Ishii HT, Mukai H, Morrison JH, Janssen WG, Kominami S, Harada N, Kimoto T, Kawato S. Adult male rat hippocampus synthesizes estradiol from pregnenolone by cytochromes P45017alpha and P450 aromatase localized in neurons. Proc Natl Acad Sci U S A. 2004;101(3):865–70. doi: 10.1073/pnas.2630225100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balthazart J, Ball GF. New insights into the regulation and function of brain estrogen synthase (aromatase) Trends Neurosci. 1998;21(6):243–9. doi: 10.1016/s0166-2236(97)01221-6. [DOI] [PubMed] [Google Scholar]
- 4.Sasano H, Takashashi K, Satoh F, Nagura H, Harada N. Aromatase in the human central nervous system. Clin Endocrinol (Oxf) 1998;48(3):325–9. doi: 10.1046/j.1365-2265.1998.00390.x. [DOI] [PubMed] [Google Scholar]
- 5.Brann DW, Mahesh VB. The aging reproductive neuroendocrine axis. Steroids. 2005;70(4):273–83. doi: 10.1016/j.steroids.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 6.Liao S, Chen W, Kuo J, Chen C. Association of serum estrogen level and ischemic neuroprotection in female rats. Neurosci Lett. 2001;297(3):159–62. doi: 10.1016/s0304-3940(00)01704-3. [DOI] [PubMed] [Google Scholar]
- 7.Carswell HV, Dominiczak AF, Macrae IM. Estrogen status affects sensitivity to focal cerebral ischemia in stroke-prone spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2000;278(1):H290–4. doi: 10.1152/ajpheart.2000.278.1.H290. [DOI] [PubMed] [Google Scholar]
- 8.Sawada M, Alkayed NJ, Goto S, Crain BJ, Traystman RJ, Shaivitz A, Nelson RJ, Hurn PD. Estrogen receptor antagonist ICI182,780 exacerbates ischemic injury in female mouse. J Cereb Blood Flow Metab. 2000;20(1):112–8. doi: 10.1097/00004647-200001000-00015. [DOI] [PubMed] [Google Scholar]
- 9.Niewada M, Kobayashi A, Sandercock PA, Kaminski B, Czlonkowska A. Influence of gender on baseline features and clinical outcomes among 17,370 patients with confirmed ischaemic stroke in the international stroke trial. Neuroepidemiology. 2005;24(3):123–8. doi: 10.1159/000082999. [DOI] [PubMed] [Google Scholar]
- 10.Roquer J, Campello AR, Gomis M. Sex differences in first-ever acute stroke. Stroke. 2003;34(7):1581–5. doi: 10.1161/01.STR.0000078562.82918.F6. [DOI] [PubMed] [Google Scholar]
- 11.Murphy SJ, McCullough LD, Smith JM. Stroke in the female: role of biological sex and estrogen. ILAR J. 2004;45(2):147–59. doi: 10.1093/ilar.45.2.147. [DOI] [PubMed] [Google Scholar]
- 12.Di Carlo A, Lamassa M, Baldereschi M, Pracucci G, Basile AM, Wolfe CD, Giroud M, Rudd A, Ghetti A, Inzitari D. Sex differences in the clinical presentation, resource use, and 3-month outcome of acute stroke in Europe: data from a multicenter multinational hospital-based registry. Stroke. 2003;34(5):1114–9. doi: 10.1161/01.STR.0000068410.07397.D7. [DOI] [PubMed] [Google Scholar]
- 13.Hochner-Celnikier D, Manor O, Garbi B, Chajek-Shaul T. Gender gap in cerebrovascular accidents: comparison of the extent, severity, and risk factors in men and women aged 45-65. Int J Fertil Womens Med. 2005;50(3):122–8. [PubMed] [Google Scholar]
- 14.Mukai H, Kimoto T, Hojo Y, Kawato S, Murakami G, Higo S, Hatanaka Y, Ogiue-Ikeda M. Modulation of synaptic plasticity by brain estrogen in the hippocampus. Biochim Biophys Acta. 2010;1800(10):1030–44. doi: 10.1016/j.bbagen.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 15.Yague JG, Azcoitia I, DeFelipe J, Garcia-Segura LM, Munoz A. Aromatase expression in the normal and epileptic human hippocampus. Brain Res. 2010;1315:41–52. doi: 10.1016/j.brainres.2009.09.111. [DOI] [PubMed] [Google Scholar]
- 16.Yague JG, Wang AC, Janssen WG, Hof PR, Garcia-Segura LM, Azcoitia I, Morrison JH. Aromatase distribution in the monkey temporal neocortex and hippocampus. Brain Res. 2008;1209:115–27. doi: 10.1016/j.brainres.2008.02.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hojo Y, Murakami G, Mukai H, Higo S, Hatanaka Y, Ogiue-Ikeda M, Ishii H, Kimoto T, Kawato S. Estrogen synthesis in the brain--role in synaptic plasticity and memory. Mol Cell Endocrinol. 2008;290(1-2):31–43. doi: 10.1016/j.mce.2008.04.017. [DOI] [PubMed] [Google Scholar]
- 18.Kretz O, Fester L, Wehrenberg U, Zhou L, Brauckmann S, Zhao S, Prange-Kiel J, Naumann T, Jarry H, Frotscher M, Rune GM. Hippocampal synapses depend on hippocampal estrogen synthesis. J Neurosci. 2004;24(26):5913–21. doi: 10.1523/JNEUROSCI.5186-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McCullough LD, Blizzard K, Simpson ER, Oz OK, Hurn PD. Aromatase cytochrome P450 and extragonadal estrogen play a role in ischemic neuroprotection. J Neurosci. 2003;23(25):8701–5. doi: 10.1523/JNEUROSCI.23-25-08701.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roselli CE, Liu M, Hurn PD. Brain aromatization: classic roles and new perspectives. Semin Reprod Med. 2009;27(3):207–17. doi: 10.1055/s-0029-1216274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carswell HV, Dominiczak AF, Garcia-Segura LM, Harada N, Hutchison JB, Macrae IM. Brain aromatase expression after experimental stroke: topography and time course. J Steroid Biochem Mol Biol. 2005;96(1):89–91. doi: 10.1016/j.jsbmb.2005.02.016. [DOI] [PubMed] [Google Scholar]
- 22.Deroo BJ, Korach KS. Estrogen receptors and human disease. The Journal of clinical investigation. 2006;116(3):561–70. doi: 10.1172/JCI27987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McEwen BS. Invited review: Estrogens effects on the brain: multiple sites and molecular mechanisms. J Appl Physiol. 2001;91(6):2785–801. doi: 10.1152/jappl.2001.91.6.2785. [DOI] [PubMed] [Google Scholar]
- 24.Levin ER. Rapid signaling by steroid receptors. Am J Physiol Regul Integr Comp Physiol. 2008;295(5):R1425–30. doi: 10.1152/ajpregu.90605.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kalita K, Szymczak S, Kaczmarek L. Non-nuclear estrogen receptor beta and alpha in the hippocampus of male and female rats. Hippocampus. 2005;15(3):404–12. doi: 10.1002/hipo.20066. [DOI] [PubMed] [Google Scholar]
- 26.Clarke CH, Norfleet AM, Clarke MS, Watson CS, Cunningham KA, Thomas ML. Perimembrane localization of the estrogen receptor alpha protein in neuronal processes of cultured hippocampal neurons. Neuroendocrinology. 2000;71(1):34–42. doi: 10.1159/000054518. [DOI] [PubMed] [Google Scholar]
- 27.Milner TA, Lubbers LS, Alves SE, McEwen BS. Nuclear and extranuclear estrogen binding sites in the rat forebrain and autonomic medullary areas. Endocrinology. 2008;149(7):3306–12. doi: 10.1210/en.2008-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Milner TA, McEwen BS, Hayashi S, Li CJ, Reagan LP, Alves SE. Ultrastructural evidence that hippocampal alpha estrogen receptors are located at extranuclear sites. The Journal of comparative neurology. 2001;429(3):355–71. [PubMed] [Google Scholar]
- 29.Woolley CS. Acute effects of estrogen on neuronal physiology. Annu Rev Pharmacol Toxicol. 2007;47:657–80. doi: 10.1146/annurev.pharmtox.47.120505.105219. [DOI] [PubMed] [Google Scholar]
- 30.Raz L, Khan MM, Mahesh VB, Vadlamudi RK, Brann DW. Rapid estrogen signaling in the brain. Neurosignals. 2008;16(2-3):140–53. doi: 10.1159/000111559. [DOI] [PubMed] [Google Scholar]
- 31.Levin ER. Integration of the Extranuclear and Nuclear Actions of Estrogen. Mol Endocrinol. 2005;19(8):1951–9. doi: 10.1210/me.2004-0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hammes SR, Levin ER. Extranuclear steroid receptors: nature and actions. Endocrine reviews. 2007;28(7):726–41. doi: 10.1210/er.2007-0022. [DOI] [PubMed] [Google Scholar]
- 33.Shughrue PJ, Lane MV, Merchenthaler I. Comparative distribution of estrogen receptor-alpha and -beta mRNA in the rat central nervous system. J Comp Neurol. 1997;388(4):507–25. doi: 10.1002/(sici)1096-9861(19971201)388:4<507::aid-cne1>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 34.Shughrue PJ, Merchenthaler I. Estrogen is more than just a “sex hormone”: novel sites for estrogen action in the hippocampus and cerebral cortex. Front Neuroendocrinol. 2000;21(1):95–101. doi: 10.1006/frne.1999.0190. [DOI] [PubMed] [Google Scholar]
- 35.McEwen B, Akama K, Alves S, Brake WG, Bulloch K, Lee S, Li C, Yuen G, Milner TA. Tracking the estrogen receptor in neurons: implications for estrogen-induced synapse formation. Proc Natl Acad Sci U S A. 2001;98(13):7093–100. doi: 10.1073/pnas.121146898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao C, Dahlman-Wright K, Gustafsson JA. Estrogen receptor beta: an overview and update. Nucl Recept Signal. 2008;6:e003. doi: 10.1621/nrs.06003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dechering K, Boersma C, Mosselman S. Estrogen receptors alpha and beta: two receptors of a kind? Curr Med Chem. 2000;7(5):561–76. doi: 10.2174/0929867003375010. [DOI] [PubMed] [Google Scholar]
- 38.Perez SE, Chen EY, Mufson EJ. Distribution of estrogen receptor alpha and beta immunoreactive profiles in the postnatal rat brain. Brain Res Dev Brain Res. 2003;145(1):117–39. doi: 10.1016/s0165-3806(03)00223-2. [DOI] [PubMed] [Google Scholar]
- 39.Zhang JQ, Cai WQ, Zhou DS, Su BY. Distribution and differences of estrogen receptor beta immunoreactivity in the brain of adult male and female rats. Brain Res. 2002;935(1-2):73–80. doi: 10.1016/s0006-8993(02)02460-5. [DOI] [PubMed] [Google Scholar]
- 40.Dubal DB, Zhu H, Yu J, Rau SW, Shughrue PJ, Merchenthaler I, Kindy MS, Wise PM. Estrogen receptor alpha, not beta, is a critical link in estradiol-mediated protection against brain injury. Proc Natl Acad Sci U S A. 2001;98(4):1952–7. doi: 10.1073/pnas.041483198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Merchenthaler I, Dellovade TL, Shughrue PJ. Neuroprotection by estrogen in animal models of global and focal ischemia. Ann N Y Acad Sci. 2003;1007:89–100. doi: 10.1196/annals.1286.009. [DOI] [PubMed] [Google Scholar]
- 42.Zhang QG, Raz L, Wang R, Han D, De Sevilla L, Yang F, Vadlamudi RK, Brann DW. Estrogen attenuates ischemic oxidative damage via an estrogen receptor alpha-mediated inhibition of NADPH oxidase activation. J Neurosci. 2009;29(44):13823–36. doi: 10.1523/JNEUROSCI.3574-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dai X, Chen L, Sokabe M. Neurosteroid estradiol rescues ischemia-induced deficit in the long-term potentiation of rat hippocampal CA1 neurons. Neuropharmacology. 2007;52(4):1124–38. doi: 10.1016/j.neuropharm.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 44.Miller NR, Jover T, Cohen HW, Zukin RS, Etgen AM. Estrogen can act via estrogen receptor alpha and beta to protect hippocampal neurons against global ischemia-induced cell death. Endocrinology. 2005;146(7):3070–9. doi: 10.1210/en.2004-1515. [DOI] [PubMed] [Google Scholar]
- 45.Elzer JG, Muhammad S, Wintermantel TM, Regnier-Vigouroux A, Ludwig J, Schutz G, Schwaninger M. Neuronal estrogen receptor-alpha mediates neuroprotection by 17beta-estradiol. J Cereb Blood Flow Metab. 2010;30(5):935–42. doi: 10.1038/jcbfm.2009.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arevalo MA, Santos-Galindo M, Bellini MJ, Azcoitia I, Garcia-Segura LM. Actions of estrogens on glial cells: Implications for neuroprotection. Biochim Biophys Acta. 2010;1800(10):1106–12. doi: 10.1016/j.bbagen.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 47.Azcoitia I, Santos-Galindo M, Arevalo MA, Garcia-Segura LM. Role of astroglia in the neuroplastic and neuroprotective actions of estradiol. Eur J Neurosci. 2010;32(12):1995–2002. doi: 10.1111/j.1460-9568.2010.07516.x. [DOI] [PubMed] [Google Scholar]
- 48.Dhandapani KM, Brann DW. Role of astrocytes in estrogen-mediated neuroprotection. Experimental gerontology. 2007;42(1-2):70–5. doi: 10.1016/j.exger.2006.06.032. [DOI] [PubMed] [Google Scholar]
- 49.Guo J, Krause DN, Horne J, Weiss JH, Li X, Duckles SP. Estrogen-receptor-mediated protection of cerebral endothelial cell viability and mitochondrial function after ischemic insult in vitro. J Cereb Blood Flow Metab. 2010;30(3):545–54. doi: 10.1038/jcbfm.2009.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carswell HV, Macrae IM, Gallagher L, Harrop E, Horsburgh KJ. Neuroprotection by a selective estrogen receptor beta agonist in a mouse model of global ischemia. Am J Physiol Heart Circ Physiol. 2004;287(4):H1501–4. doi: 10.1152/ajpheart.00227.2004. [DOI] [PubMed] [Google Scholar]
- 51.Donzelli A, Braida D, Finardi A, Capurro V, Valsecchi AE, Colleoni M, Sala M. Neuroprotective effects of genistein in Mongolian gerbils: estrogen receptor-beta involvement. J Pharmacol Sci. 2010;114(2):158–67. doi: 10.1254/jphs.10164fp. [DOI] [PubMed] [Google Scholar]
- 52.Wang L, Andersson S, Warner M, Gustafsson JA. Morphological abnormalities in the brains of estrogen receptor beta knockout mice. Proc Natl Acad Sci U S A. 2001;98(5):2792–6. doi: 10.1073/pnas.041617498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Funakoshi T, Yanai A, Shinoda K, Kawano MM, Mizukami Y. G protein-coupled receptor 30 is an estrogen receptor in the plasma membrane. Biochem Biophys Res Commun. 2006;346(3):904–10. doi: 10.1016/j.bbrc.2006.05.191. [DOI] [PubMed] [Google Scholar]
- 54.Matsuda K, Sakamoto H, Mori H, Hosokawa K, Kawamura A, Itose M, Nishi M, Prossnitz ER, Kawata M. Expression and intracellular distribution of the G protein-coupled receptor 30 in rat hippocampal formation. Neurosci Lett. 2008;441(1):94–9. doi: 10.1016/j.neulet.2008.05.108. [DOI] [PubMed] [Google Scholar]
- 55.Gingerich S, Kim GL, Chalmers JA, Koletar MM, Wang X, Wang Y, Belsham DD. Estrogen receptor alpha and G-protein coupled receptor 30 mediate the neuroprotective effects of 17beta-estradiol in novel murine hippocampal cell models. Neuroscience. 2010;170(1):54–66. doi: 10.1016/j.neuroscience.2010.06.076. [DOI] [PubMed] [Google Scholar]
- 56.Bologa CG, Revankar CM, Young SM, Edwards BS, Arterburn JB, Kiselyov AS, Parker MA, Tkachenko SE, Savchuck NP, Sklar LA, Oprea TI, Prossnitz ER. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat Chem Biol. 2006;2(4):207–12. doi: 10.1038/nchembio775. [DOI] [PubMed] [Google Scholar]
- 57.Zhang B, Subramanian S, Dziennis S, Jia J, Uchida M, Akiyoshi K, Migliati E, Lewis AD, Vandenbark AA, Offner H, Hurn PD. Estradiol and G1 reduce infarct size and improve immunosuppression after experimental stroke. J Immunol. 2010;184(8):4087–94. doi: 10.4049/jimmunol.0902339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Simpkins JW, Wen Y, Perez E, Yang S, Wang X. Role of nonfeminizing estrogens in brain protection from cerebral ischemia: an animal model of Alzheimer's disease neuropathology. Ann N Y Acad Sci. 2005;1052:233–42. doi: 10.1196/annals.1347.019. [DOI] [PubMed] [Google Scholar]
- 59.Simpkins JW, Yang SH, Liu R, Perez E, Cai ZY, Covey DF, Green PS. Estrogen-like compounds for ischemic neuroprotection. Stroke. 2004;35(11 Suppl 1):2648–51. doi: 10.1161/01.STR.0000143734.59507.88. [DOI] [PubMed] [Google Scholar]
- 60.Liu R, Yang SH, Perez E, Yi KD, Wu SS, Eberst K, Prokai L, Prokai-Tatrai K, Cai ZY, Covey DF, Day AL, Simpkins JW. Neuroprotective effects of a novel non-receptor-binding estrogen analogue: in vitro and in vivo analysis. Stroke. 2002;33(10):2485–91. doi: 10.1161/01.str.0000030317.43597.c8. [DOI] [PubMed] [Google Scholar]
- 61.Yi KD, Perez E, Yang S, Liu R, Covey DF, Simpkins JW. The assessment of non-feminizing estrogens for use in neuroprotection. Brain Res. 2011;1379:61–70. doi: 10.1016/j.brainres.2010.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sugawara T, Noshita N, Lewen A, Gasche Y, Ferrand-Drake M, Fujimura M, Morita-Fujimura Y, Chan PH. Overexpression of copper/zinc superoxide dismutase in transgenic rats protects vulnerable neurons against ischemic damage by blocking the mitochondrial pathway of caspase activation. J Neurosci. 2002;22(1):209–17. doi: 10.1523/JNEUROSCI.22-01-00209.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chan PH. Role of oxidants in ischemic brain damage. Stroke. 1996;27(6):1124–9. doi: 10.1161/01.str.27.6.1124. [DOI] [PubMed] [Google Scholar]
- 64.Peters O, Back T, Lindauer U, Busch C, Megow D, Dreier J, Dirnagl U. Increased formation of reactive oxygen species after permanent and reversible middle cerebral artery occlusion in the rat. J Cereb Blood Flow Metab. 1998;18(2):196–205. doi: 10.1097/00004647-199802000-00011. [DOI] [PubMed] [Google Scholar]
- 65.Fujimura M, Morita-Fujimura Y, Kawase M, Copin JC, Calagui B, Epstein CJ, Chan PH. Manganese superoxide dismutase mediates the early release of mitochondrial cytochrome C and subsequent DNA fragmentation after permanent focal cerebral ischemia in mice. J Neurosci. 1999;19(9):3414–22. doi: 10.1523/JNEUROSCI.19-09-03414.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mehta SH, Dhandapani KM, De Sevilla LM, Webb RC, Mahesh VB, Brann DW. Tamoxifen, a selective estrogen receptor modulator, reduces ischemic damage caused by middle cerebral artery occlusion in the ovariectomized female rat. Neuroendocrinology. 2003;77(1):44–50. doi: 10.1159/000068332. [DOI] [PubMed] [Google Scholar]
- 67.Mattson MP, Culmsee C, Yu ZF. Apoptotic and antiapoptotic mechanisms in stroke. Cell Tissue Res. 2000;301(1):173–87. doi: 10.1007/s004419900154. [DOI] [PubMed] [Google Scholar]
- 68.Bergamini CM, Gambetti S, Dondi A, Cervellati C. Oxygen, reactive oxygen species and tissue damage. Curr Pharm Des. 2004;10(14):1611–26. doi: 10.2174/1381612043384664. [DOI] [PubMed] [Google Scholar]
- 69.Raz L, Zhang QG, Zhou CF, Han D, Gulati P, Yang LC, Yang F, Wang RM, Brann DW. Role of Rac1 GTPase in NADPH oxidase activation and cognitive impairment following cerebral ischemia in the rat. PLoS One. 2010;5(9):e12606. doi: 10.1371/journal.pone.0012606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Abramov AY, Scorziello A, Duchen MR. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J Neurosci. 2007;27(5):1129–38. doi: 10.1523/JNEUROSCI.4468-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Simpkins JW, Yi KD, Yang SH, Dykens JA. Mitochondrial mechanisms of estrogen neuroprotection. Biochim Biophys Acta. 2010;1800(10):1113–20. doi: 10.1016/j.bbagen.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Arnold S, Beyer C. Neuroprotection by estrogen in the brain: the mitochondrial compartment as presumed therapeutic target. J Neurochem. 2009;110(1):1–11. doi: 10.1111/j.1471-4159.2009.06133.x. [DOI] [PubMed] [Google Scholar]
- 73.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87(1):245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 74.Brennan AM, Suh SW, Won SJ, Narasimhan P, Kauppinen TM, Lee H, Edling Y, Chan PH, Swanson RA. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nature neuroscience. 2009;12(7):857–63. doi: 10.1038/nn.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sorce S, Krause KH. NOX enzymes in the central nervous system: from signaling to disease. Antioxid Redox Signal. 2009;11(10):2481–504. doi: 10.1089/ars.2009.2578. [DOI] [PubMed] [Google Scholar]
- 76.Suh SW, Shin BS, Ma H, Van Hoecke M, Brennan AM, Yenari MA, Swanson RA. Glucose and NADPH oxidase drive neuronal superoxide formation in stroke. Annals of neurology. 2008;64(6):654–63. doi: 10.1002/ana.21511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cheng G, Cao Z, Xu X, van Meir EG, Lambeth JD. Homologs of gp91phox: cloning and tissue expression of Nox3, Nox4, and Nox5. Gene. 2001;269(1-2):131–40. doi: 10.1016/s0378-1119(01)00449-8. [DOI] [PubMed] [Google Scholar]
- 78.Serrano F, Kolluri NS, Wientjes FB, Card JP, Klann E. NADPH oxidase immunoreactivity in the mouse brain. Brain Res. 2003;988(1-2):193–8. doi: 10.1016/s0006-8993(03)03364-x. [DOI] [PubMed] [Google Scholar]
- 79.Vallet P, Charnay Y, Steger K, Ogier-Denis E, Kovari E, Herrmann F, Michel JP, Szanto I. Neuronal expression of the NADPH oxidase NOX4, and its regulation in mouse experimental brain ischemia. Neuroscience. 2005;132(2):233–8. doi: 10.1016/j.neuroscience.2004.12.038. [DOI] [PubMed] [Google Scholar]
- 80.Chen H, Song YS, Chan PH. Inhibition of NADPH oxidase is neuroprotective after ischemia-reperfusion. J Cereb Blood Flow Metab. 2009;29(7):1262–72. doi: 10.1038/jcbfm.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jackman KA, Miller AA, De Silva TM, Crack PJ, Drummond GR, Sobey CG. Reduction of cerebral infarct volume by apocynin requires pretreatment and is absent in Nox2-deficient mice. Br J Pharmacol. 2009;156(4):680–8. doi: 10.1111/j.1476-5381.2008.00073.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tang LL, Ye K, Yang XF, Zheng JS. Apocynin attenuates cerebral infarction after transient focal ischaemia in rats. J Int Med Res. 2007;35(4):517–22. doi: 10.1177/147323000703500411. [DOI] [PubMed] [Google Scholar]
- 83.Genovese T, Mazzon E, Paterniti I, Esposito E, Bramanti P, Cuzzocrea S. Modulation of NADPH oxidase activation in cerebral ischemia/reperfusion injury in rats. Brain Res. 2011;1372:92–102. doi: 10.1016/j.brainres.2010.11.088. [DOI] [PubMed] [Google Scholar]
- 84.Yoshioka H, Niizuma K, Katsu M, Okami N, Sakata H, Kim GS, Narasimhan P, Chan PH. NADPH oxidase mediates striatal neuronal injury after transient global cerebral ischemia. J Cereb Blood Flow Metab. 2011;31(3):868–80. doi: 10.1038/jcbfm.2010.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tang XN, Cairns B, Cairns N, Yenari MA. Apocynin improves outcome in experimental stroke with a narrow dose range. Neuroscience. 2008;154(2):556–62. doi: 10.1016/j.neuroscience.2008.03.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Brait VH, Jackman KA, Walduck AK, Selemidis S, Diep H, Mast AE, Guida E, Broughton BR, Drummond GR, Sobey CG. Mechanisms contributing to cerebral infarct size after stroke: gender, reperfusion, T lymphocytes, and Nox2-derived superoxide. J Cereb Blood Flow Metab. 2010;30(7):1306–17. doi: 10.1038/jcbfm.2010.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dubal DB, Shughrue PJ, Wilson ME, Merchenthaler I, Wise PM. Estradiol modulates bcl-2 in cerebral ischemia: a potential role for estrogen receptors. J Neurosci. 1999;19(15):6385–93. doi: 10.1523/JNEUROSCI.19-15-06385.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wu TW, Wang JM, Chen S, Brinton RD. 17Beta-estradiol induced Ca2+ influx via L-type calcium channels activates the Src/ERK/cyclic-AMP response element binding protein signal pathway and BCL-2 expression in rat hippocampal neurons: a potential initiation mechanism for estrogen-induced neuroprotection. Neuroscience. 2005;135(1):59–72. doi: 10.1016/j.neuroscience.2004.12.027. [DOI] [PubMed] [Google Scholar]
- 89.Zhao L, Brinton RD. Estrogen receptor alpha and beta differentially regulate intracellular Ca(2+) dynamics leading to ERK phosphorylation and estrogen neuroprotection in hippocampal neurons. Brain Res. 2007;1172:48–59. doi: 10.1016/j.brainres.2007.06.092. [DOI] [PubMed] [Google Scholar]
- 90.Alkayed NJ, Goto S, Sugo N, Joh HD, Klaus J, Crain BJ, Bernard O, Traystman RJ, Hurn PD. Estrogen and Bcl-2: gene induction and effect of transgene in experimental stroke. J Neurosci. 2001;21(19):7543–50. doi: 10.1523/JNEUROSCI.21-19-07543.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang QG, Wang R, Khan M, Mahesh V, Brann DW. Role of Dickkopf-1, an antagonist of the Wnt/beta-catenin signaling pathway, in estrogen-induced neuroprotection and attenuation of tau phosphorylation. J Neurosci. 2008;28(34):8430–41. doi: 10.1523/JNEUROSCI.2752-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bekinschtein P, Cammarota M, Katche C, Slipczuk L, Rossato JI, Goldin A, Izquierdo I, Medina JH. BDNF is essential to promote persistence of long-term memory storage. Proc Natl Acad Sci U S A. 2008;105(7):2711–6. doi: 10.1073/pnas.0711863105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nakajo Y, Miyamoto S, Nakano Y, Xue JH, Hori T, Yanamoto H. Genetic increase in brain-derived neurotrophic factor levels enhances learning and memory. Brain Res. 2008;1241:103–9. doi: 10.1016/j.brainres.2008.08.080. [DOI] [PubMed] [Google Scholar]
- 94.Singh M. Ovarian hormones elicit phosphorylation of Akt and extracellular-signal regulated kinase in explants of the cerebral cortex. Endocrine. 2001;14(3):407–15. doi: 10.1385/ENDO:14:3:407. [DOI] [PubMed] [Google Scholar]
- 95.Singh M, Setalo G, Jr, Guan X, Warren M, Toran-Allerand CD. Estrogen-induced activation of mitogen-activated protein kinase in cerebral cortical explants: convergence of estrogen and neurotrophin signaling pathways. J Neurosci. 1999;19(4):1179–88. doi: 10.1523/JNEUROSCI.19-04-01179.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Singer CA, Figueroa-Masot XA, Batchelor RH, Dorsa DM. The mitogen-activated protein kinase pathway mediates estrogen neuroprotection after glutamate toxicity in primary cortical neurons. J Neurosci. 1999;19(7):2455–63. doi: 10.1523/JNEUROSCI.19-07-02455.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jover-Mengual T, Zukin RS, Etgen AM. MAPK signaling is critical to estradiol protection of CA1 neurons in global ischemia. Endocrinology. 2007;148(3):1131–43. doi: 10.1210/en.2006-1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Choi YC, Lee JH, Hong KW, Lee KS. 17 Beta-estradiol prevents focal cerebral ischemic damages via activation of Akt and CREB in association with reduced PTEN phosphorylation in rats. Fundam Clin Pharmacol. 2004;18(5):547–57. doi: 10.1111/j.1472-8206.2004.00284.x. [DOI] [PubMed] [Google Scholar]
- 99.Zhang QG, Wang XT, Han D, Yin XH, Zhang GY, Xu TL. Akt inhibits MLK3/JNK3 signaling by inactivating Rac1: a protective mechanism against ischemic brain injury. J Neurochem. 2006;98(6):1886–98. doi: 10.1111/j.1471-4159.2006.04020.x. [DOI] [PubMed] [Google Scholar]
- 100.Taguchi Y, Koslowski M, Bodenner DL. Binding of estrogen receptor with estrogen conjugated to bovine serum albumin (BSA) Nucl Recept. 2004;2(1):5. doi: 10.1186/1478-1336-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Aguilar R, Bellido C, Garrido-Gracia JC, Alonso R, Sanchez-Criado JE. Estradiol and its membrane-impermeable conjugate estradiol-BSA inhibit tamoxifen-stimulated prolactin secretion in incubated rat pituitaries. Reproduction. 2006;131(4):763–9. doi: 10.1530/rep.1.00807. [DOI] [PubMed] [Google Scholar]
- 102.Razandi M, Pedram A, Greene GL, Levin ER. Cell membrane and nuclear estrogen receptors (ERs) originate from a single transcript: studies of ERalpha and ERbeta expressed in Chinese hamster ovary cells. Mol Endocrinol. 1999;13(2):307–19. doi: 10.1210/mend.13.2.0239. [DOI] [PubMed] [Google Scholar]
- 103.Harrington WR, Kim SH, Funk CC, Madak-Erdogan Z, Schiff R, Katzenellenbogen JA, Katzenellenbogen BS. Estrogen dendrimer conjugates that preferentially activate extranuclear, nongenomic versus genomic pathways of estrogen action. Mol Endocrinol. 2006;20(3):491–502. doi: 10.1210/me.2005-0186. [DOI] [PubMed] [Google Scholar]
- 104.Yang LC, Zhang QG, Zhou CF, Yang F, Zhang YD, Wang RM, Brann DW. Extranuclear estrogen receptors mediate the neuroprotective effects of estrogen in the rat hippocampus. PLoS One. 2010;5(5):e9851. doi: 10.1371/journal.pone.0009851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wang X, Wang H, Xu L, Rozanski DJ, Sugawara T, Chan PH, Trzaskos JM, Feuerstein GZ. Significant neuroprotection against ischemic brain injury by inhibition of the MEK1 protein kinase in mice: exploration of potential mechanism associated with apoptosis. J Pharmacol Exp Ther. 2003;304(1):172–8. doi: 10.1124/jpet.102.040246. [DOI] [PubMed] [Google Scholar]
- 106.Namura S, Iihara K, Takami S, Nagata I, Kikuchi H, Matsushita K, Moskowitz MA, Bonventre JV, Alessandrini A. Intravenous administration of MEK inhibitor U0126 affords brain protection against forebrain ischemia and focal cerebral ischemia. Proc Natl Acad Sci U S A. 2001;98(20):11569–74. doi: 10.1073/pnas.181213498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Henriksson M, Stenman E, Vikman P, Edvinsson L. MEK1/2 inhibition attenuates vascular ETA and ETB receptor alterations after cerebral ischaemia. Exp Brain Res. 2007;178(4):470–6. doi: 10.1007/s00221-006-0753-7. [DOI] [PubMed] [Google Scholar]
- 108.Sawe N, Steinberg G, Zhao H. Dual roles of the MAPK/ERK1/2 cell signaling pathway after stroke. J Neurosci Res. 2008;86(8):1659–69. doi: 10.1002/jnr.21604. [DOI] [PubMed] [Google Scholar]
- 109.Razandi M, Pedram A, Merchenthaler I, Greene GL, Levin ER. Plasma membrane estrogen receptors exist and functions as dimers. Mol Endocrinol. 2004;18(12):2854–65. doi: 10.1210/me.2004-0115. [DOI] [PubMed] [Google Scholar]
- 110.Pedram A, Razandi M, Sainson RC, Kim JK, Hughes CC, Levin ER. A conserved mechanism for steroid receptor translocation to the plasma membrane. J Biol Chem. 2007;282(31):22278–88. doi: 10.1074/jbc.M611877200. [DOI] [PubMed] [Google Scholar]
- 111.Evinger AJ, 3rd, Levin ER. Requirements for estrogen receptor alpha membrane localization and function. Steroids. 2005;70(5-7):361–3. doi: 10.1016/j.steroids.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 112.Nishio M, Kuroki Y, Watanabe Y. Subcellular localization of estrogen receptor beta in mouse hippocampus. Neurosci Lett. 2004;355(1-2):109–12. doi: 10.1016/j.neulet.2003.10.064. [DOI] [PubMed] [Google Scholar]
- 113.Azcoitia I, Sierra A, Garcia-Segura LM. Localization of estrogen receptor beta-immunoreactivity in astrocytes of the adult rat brain. Glia. 1999;26(3):260–7. [PubMed] [Google Scholar]
- 114.Gorosito SV, Lorenzo AG, Cambiasso MJ. Estrogen receptor alpha is expressed on the cell-surface of embryonic hypothalamic neurons. Neuroscience. 2008;154(4):1173–7. doi: 10.1016/j.neuroscience.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 115.Marin R, Diaz M, Alonso R, Sanz A, Arevalo MA, Garcia-Segura LM. Role of estrogen receptor alpha in membrane-initiated signaling in neural cells: interaction with IGF-1 receptor. J Steroid Biochem Mol Biol. 2009;114(1-2):2–7. doi: 10.1016/j.jsbmb.2008.12.014. [DOI] [PubMed] [Google Scholar]
- 116.Kelly MJ, Ronnekleiv OK. Control of CNS neuronal excitability by estrogens via membrane-initiated signaling. Mol Cell Endocrinol. 2009;308(1-2):17–25. doi: 10.1016/j.mce.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bondar G, Kuo J, Hamid N, Micevych P. Estradiol-induced estrogen receptor-alpha trafficking. J Neurosci. 2009;29(48):15323–30. doi: 10.1523/JNEUROSCI.2107-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chaban VV, Lakhter AJ, Micevych P. A membrane estrogen receptor mediates intracellular calcium release in astrocytes. Endocrinology. 2004;145(8):3788–95. doi: 10.1210/en.2004-0149. [DOI] [PubMed] [Google Scholar]
- 119.Dhandapani KM, Wade FM, Mahesh VB, Brann DW. Astrocyte-derived transforming growth factor-{beta} mediates the neuroprotective effects of 17{beta}-estradiol: involvement of nonclassical genomic signaling pathways. Endocrinology. 2005;146(6):2749–59. doi: 10.1210/en.2005-0014. [DOI] [PubMed] [Google Scholar]
- 120.Garcia-Ovejero D, Azcoitia I, Doncarlos LL, Melcangi RC, Garcia-Segura LM. Glia-neuron crosstalk in the neuroprotective mechanisms of sex steroid hormones. Brain Res Brain Res Rev. 2005;48(2):273–86. doi: 10.1016/j.brainresrev.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 121.Madak-Erdogan Z, Kieser KJ, Kim SH, Komm B, Katzenellenbogen JA, Katzenellenbogen BS. Nuclear and extranuclear pathway inputs in the regulation of global gene expression by estrogen receptors. Mol Endocrinol. 2008;22(9):2116–27. doi: 10.1210/me.2008-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kow LM, Pfaff DW. The membrane actions of estrogens can potentiate their lordosis behavior-facilitating genomic actions. Proc Natl Acad Sci U S A. 2004;101(33):12354–7. doi: 10.1073/pnas.0404889101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Vasudevan N, Pfaff DW. Membrane-initiated actions of estrogens in neuroendocrinology: emerging principles. Endocr Rev. 2007;28(1):1–19. doi: 10.1210/er.2005-0021. [DOI] [PubMed] [Google Scholar]
- 124.Rocca WA, Grossardt BR, Shuster LT. Oophorectomy, menopause, estrogen treatment, and cognitive aging: clinical evidence for a window of opportunity. Brain Res. 2011;1379:188–98. doi: 10.1016/j.brainres.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Simpkins JW, Rajakumar G, Zhang YQ, Simpkins CE, Greenwald D, Yu CJ, Bodor N, Day AL. Estrogens may reduce mortality and ischemic damage caused by middle cerebral artery occlusion in the female rat. J Neurosurg. 1997;87(5):724–30. doi: 10.3171/jns.1997.87.5.0724. [DOI] [PubMed] [Google Scholar]
- 126.Simpkins JW, Singh M. More than a decade of estrogen neuroprotection. Alzheimers Dement. 2008;4(1 Suppl 1):S131–6. doi: 10.1016/j.jalz.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 127.Henderson VW. Estrogen-containing hormone therapy and Alzheimer's disease risk: understanding discrepant inferences from observational and experimental research. Neuroscience. 2006;138(3):1031–9. doi: 10.1016/j.neuroscience.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 128.Henderson VW. Cognitive changes after menopause: influence of estrogen. Clin Obstet Gynecol. 2008;51(3):618–26. doi: 10.1097/GRF.0b013e318180ba10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Espeland MA, Rapp SR, Shumaker SA, Brunner R, Manson JE, Sherwin BB, Hsia J, Margolis KL, Hogan PE, Wallace R, Dailey M, Freeman R, Hays J. Conjugated equine estrogens and global cognitive function in postmenopausal women: Women's Health Initiative Memory Study. JAMA. 2004;291(24):2959–68. doi: 10.1001/jama.291.24.2959. [DOI] [PubMed] [Google Scholar]
- 130.Shumaker SA, Legault C, Rapp SR, Thal L, Wallace RB, Ockene JK, Hendrix SL, Jones BN, 3rd, Assaf AR, Jackson RD, Kotchen JM, Wassertheil-Smoller S, Wactawski-Wende J. Estrogen plus progestin and the incidence of dementia and mild cognitive impairment in postmenopausal women: the Women's Health Initiative Memory Study: a randomized controlled trial. JAMA. 2003;289(20):2651–62. doi: 10.1001/jama.289.20.2651. [DOI] [PubMed] [Google Scholar]
- 131.Wassertheil-Smoller S, Hendrix SL, Limacher M, Heiss G, Kooperberg C, Baird A, Kotchen T, Curb JD, Black H, Rossouw JE, Aragaki A, Safford M, Stein E, Laowattana S, Mysiw WJ. Effect of estrogen plus progestin on stroke in postmenopausal women: the Women's Health Initiative: a randomized trial. JAMA. 2003;289(20):2673–84. doi: 10.1001/jama.289.20.2673. [DOI] [PubMed] [Google Scholar]
- 132.Sherwin BB. The critical period hypothesis: can it explain discrepancies in the oestrogen-cognition literature? Journal of neuroendocrinology. 2007;19(2):77–81. doi: 10.1111/j.1365-2826.2006.01508.x. [DOI] [PubMed] [Google Scholar]
- 133.Craig MC, Murphy DG. Estrogen therapy and Alzheimer's dementia. Ann N Y Acad Sci. 2010;1205:245–53. doi: 10.1111/j.1749-6632.2010.05673.x. [DOI] [PubMed] [Google Scholar]
- 134.Sherwin BB. Estrogen therapy: is time of initiation critical for neuroprotection? Nat Rev Endocrinol. 2009;5(11):620–7. doi: 10.1038/nrendo.2009.193. [DOI] [PubMed] [Google Scholar]
- 135.Wu WW, Adelman JP, Maylie J. Ovarian hormone deficiency reduces intrinsic excitability and abolishes acute estrogen sensitivity in hippocampal CA1 pyramidal neurons. J Neurosci. 2011;31(7):2638–48. doi: 10.1523/JNEUROSCI.6081-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Smith CC, Vedder LC, Nelson AR, Bredemann TM, McMahon LL. Duration of estrogen deprivation, not chronological age, prevents estrogen's ability to enhance hippocampal synaptic physiology. Proc Natl Acad Sci U S A. 2010;107(45):19543–8. doi: 10.1073/pnas.1009307107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Daniel JM, Hulst JL, Berbling JL. Estradiol replacement enhances working memory in middle-aged rats when initiated immediately after ovariectomy but not after a long-term period of ovarian hormone deprivation. Endocrinology. 2006;147(1):607–14. doi: 10.1210/en.2005-0998. [DOI] [PubMed] [Google Scholar]
- 138.Bohacek J, Daniel JM. The beneficial effects of estradiol on attentional processes are dependent on timing of treatment initiation following ovariectomy in middle-aged rats. Psychoneuroendocrinology. 2010;35(5):694–705. doi: 10.1016/j.psyneuen.2009.10.010. [DOI] [PubMed] [Google Scholar]
- 139.Rivera CM, Grossardt BR, Rhodes DJ, Rocca WA. Increased mortality for neurological and mental diseases following early bilateral oophorectomy. Neuroepidemiology. 2009;33(1):32–40. doi: 10.1159/000211951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Rocca WA, Grossardt BR, Geda YE, Gostout BS, Bower JH, Maraganore DM, de Andrade M, Melton LJ., 3rd Long-term risk of depressive and anxiety symptoms after early bilateral oophorectomy. Menopause. 2008;15(6):1050–9. doi: 10.1097/gme.0b013e318174f155. [DOI] [PubMed] [Google Scholar]
- 141.Rocca WA, Bower JH, Maraganore DM, Ahlskog JE, Grossardt BR, de Andrade M, Melton LJ., 3rd Increased risk of cognitive impairment or dementia in women who underwent oophorectomy before menopause. Neurology. 2007;69(11):1074–83. doi: 10.1212/01.wnl.0000276984.19542.e6. [DOI] [PubMed] [Google Scholar]
- 142.Rocca WA, Grossardt BR, Maraganore DM. The long-term effects of oophorectomy on cognitive and motor aging are age dependent. Neurodegener Dis. 2008;5(3-4):257–60. doi: 10.1159/000113718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Rocca WA, Grossardt BR, Shuster LT. Oophorectomy, menopause, estrogen, and cognitive aging: the timing hypothesis. Neurodegener Dis. 2010;7(1-3):163–6. doi: 10.1159/000289229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Suzuki S, Brown CM, Dela Cruz CD, Yang E, Bridwell DA, Wise PM. Timing of estrogen therapy after ovariectomy dictates the efficacy of its neuroprotective and antiinflammatory actions. Proc Natl Acad Sci U S A. 2007;104(14):6013–8. doi: 10.1073/pnas.0610394104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Jesmin S, Hattori Y, Sakuma I, Liu MY, Mowa CN, Kitabatake A. Estrogen deprivation and replacement modulate cerebral capillary density with vascular expression of angiogenic molecules in middle-aged female rats. J Cereb Blood Flow Metab. 2003;23(2):181–9. doi: 10.1097/01.WCB.0000043341.09081.37. [DOI] [PubMed] [Google Scholar]
- 146.Pinna C, Cignarella A, Sanvito P, Pelosi V, Bolego C. Prolonged ovarian hormone deprivation impairs the protective vascular actions of estrogen receptor alpha agonists. Hypertension. 2008;51(4):1210–7. doi: 10.1161/HYPERTENSIONAHA.107.106807. [DOI] [PubMed] [Google Scholar]
- 147.Zhang QHD, Yang F, Wang R, Brann DW. Increased Alzheimer's disease-related protein induction and hypersensitivity in the hippocampal CA3 region following cerebral ischemia in long term estrogen-deprived rats. Annual Meeting of the Society for Neuroscience. 2010 [Google Scholar]
- 148.LaCroix AZ, Chlebowski RT, Manson JE, Aragaki AK, Johnson KC, Martin L, Margolis KL, Stefanick ML, Brzyski R, Curb JD, Howard BV, Lewis CE, Wactawski-Wende J. Health outcomes after stopping conjugated equine estrogens among postmenopausal women with prior hysterectomy: a randomized controlled trial. JAMA. 2011;305(13):1305–14. doi: 10.1001/jama.2011.382. [DOI] [PMC free article] [PubMed] [Google Scholar]