

The α-arylation of enolates has become a widely used method to prepare α-aryl carboxylic acid and ketone derivatives that comprise important classes of biologically active compounds. Palladium-catalyzed α-arylation of carbonyl compounds has been developed over the past decade, but the copper-mediated α-arylation of activated methylene compounds has been known for nearly a century [Eq. (1)].[1–2] Recently,
 |
(1) |
significant improvements in the scope and reaction conditions of these copper-catalyzed Hurtley-type reactions have been achieved using a combination of copper complexes with ancillary ligands, such as 2-phenylphenol,[3] chelating Schiff bases,[4] l-prolines,[5] and 2-picolinic acid.[6]
Despite the long history of Hurtley couplings, the mechanism of this process has been little explored. Setsune et al. proposed that the active copper species in the coupling of aryl halides with diethyl malonate is a CuI enolate generated in situ and that the CuI species reacts with the aryl halide by electron transfer.[7] Buchwald et al. found that CuI and 2-phenylphenol catalyzed the arylation of diethyl malonate, but this system did not catalyze the arylation of the cyclic isopropylidene malonate and 1,3-cyclopentanedione. Accordingly, they proposed that a bidentate coordination mode through two O atoms was required for the arylation reactions to occur.[3] Thus, mechanistic proposals have been made, but no explicit structural information on the copper enolate complexes that could be intermediates in the Hurtley couplings has been reported.
We report the synthesis of a series of CuI enolate complexes supported by 1,10-phenanthroline (phen). Their structures consist of an unusual combination of one cationic CuI center ligated by two phen ligands and one free, anionic enolate unit. Kinetic data on the stoichiometric reactions of the CuI enolate complexes with iodobenzene, comparisons of the selectivity and reactivity of CuI enolate complexes containing or lacking phen ligands in reactions with different iodoarenes, reactions with substrates that probe for aryl radical intermediates, and DFT calculations all indicate that the active species is a C-bound CuI enolate complex ligated by a single phen unit, and that this intermediate reacts with the iodoarene to generate a CuIII intermediate without the intermediacy of aryl radicals. The relative reactivity of the different enolates correlates with the energy differences between the observed species and the reactive C-bound enolates, along with the barriers to oxidative addition of ArI to form aryl–CuIII enolate intermediates.
The syntheses of phen-ligated CuI complexes containing the enolates from β-dicarbonyl compounds and cyanoacetate, as well as the less stabilized enolates from α phenylesters, is outlined in Scheme 1. Treatment of copper tert-butyloxide with 2 equivalents of phen in benzene, with subsequent addition of 2,4-pentanedione, ethyl acetoacetate, diethyl malonate, dimethyl malonate, ethyl cyanoacetate, ethyl phenylacetate, and methyl phenylacetate formed the CuI complexes 1–7 in 59–82% yield.[8]
Scheme 1.

Synthesis of CuI enolate complexes 1–7.
All complexes were characterized by IR and NMR spectroscopy and elemental analysis, and complexes 1, 4, 5, and 6 were characterized by single-crystal X-ray diffraction (Figure 1).[9] The 1H NMR spectra and microanalytical data for each complex showed that the complexes contain a 2:1:1 ratio of phen/Cu/enolate. The solid-state structures of 1, 4, and 6 contain a cationic CuI center ligated by two phen ligands and a free, unligated anionic enolate unit.[10] The sp2 carbanions in the enolates (C51 in 1, C27 in 4, C28 in 6) are nearly trigonal planar, and, consistent with a conjugated enolate, the C–C bond distances are close to those in an aromatic ring.
Figure 1.

ORTEP diagrams of 1 (top left, C50–C51=1.400(3) Å, C51–C52=1.392(6) Å, 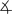 C50-C51-C52= 128.3(5)°), 4 (top right, C27–C26=1.400(3) Å, C27–C28=1.398(3) Å,
C50-C51-C52= 128.3(5)°), 4 (top right, C27–C26=1.400(3) Å, C27–C28=1.398(3) Å, 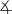 C26-C27-C28= 124.71(17)°5), (bottom left, C1–N1=1.151(4) Å, C1–C2=1.396(5) Å, C2–C3= 1.399(5) Å,
C26-C27-C28= 124.71(17)°5), (bottom left, C1–N1=1.151(4) Å, C1–C2=1.396(5) Å, C2–C3= 1.399(5) Å, 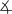 C1-C2-C3=119.9(4)°), and 6, (bottom right, C28–C29=1.432(6) Å, C27–C28=1.391(5) Å,
C1-C2-C3=119.9(4)°), and 6, (bottom right, C28–C29=1.432(6) Å, C27–C28=1.391(5) Å, 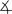 C27-C28-C29= 126.3(4)°). Thermal ellipsoids shown at 50% probability.
C27-C28-C29= 126.3(4)°). Thermal ellipsoids shown at 50% probability.
In contrast to the enolates in complexes 1, 4, and 6, the cyanoester anion in 5 is bound to copper through the N atom of the cyano group (Figure 1), and this binding creates an unusual five-coordinate, trigonal bipyramidal, formally 20 valence electron CuI site. The Cu–N bonds in the equatorial plane (1.947(3) to 2.071(3) Å) are considerably shorter than those in the axial positions (2.368(3) and 2.411 Å).
The conductivity of solutions containing the complexes was used to assess the relationship between the solid-state and solution structures. The conductivities of 1.0 mm solutions of complexes 1–7 in DMSO solvent ranged from 18.8 to 24.5 Ω–1cm2mol–1 versus 23.5 Ω–1cm2mol–1 for [NBu4]-[BPh4] and 0.3 Ω–1cm2mol–1 for ferrocene. Thus, complexes 1–7 are all predominantly salts in DMSO, including complex 5, which adopts the neutral, five-coordinate structure in the solid state.
To assess the potential of the enolate complexes 1–7 to be intermediates in the α-arylation reactions, we investigated their reactions with iodobenzene (PhI). These results are summarized in Table 1. The malonate, cyanoester, and phenylacetate complexes 3–7 reacted with PhI at 25°C to form the coupled products in 68–86% yield (100% conversion) after 12 hours, whereas the ethyl acetoacetate complex 2 required over 22 hours at 60°C for > 95% conversion and formed the arylated product in 63% yield. The acetylacetone complex 1 did not react with PhI to form the arylated product; no reaction occurred at room temperature, and multiple unidentified species formed at 60°C.
Table 1.
Reactions of copper enolate complexes with Phl.[a]
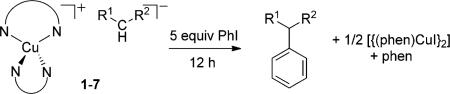 | ||||
|---|---|---|---|---|
| Complex | R1 | R2 | T [°C] | Yield [%][b] |
| 1 | MeCO | MeCO | 60 | 0 |
| 2 | MeCO | EtCO2 | 60 | 63[c] |
| 3 | EtCO2 | EtCO2 | 25 | 79 |
| 4 | MeCO2 | MeCO2 | 25 | 80 |
| 5 | CN | EtCO2 | 25 | 86 |
| 6 | Ph | EtCO2 | 25 | 68 |
| 7 | Ph | MeCO2 | 25 | 70 |
Reaction conditions: 34.4 mm of complexes, [D6]DMSO.
The yield was determined by
H NMR spectroscopy using trimethoxybenzene as an internal standard.
Reaction time: 22 h.
The reaction of diethyl malonate with 2 equivalents of PhI in the presence of 2 equivalents of Cs2CO3 catalyzed by the CuI diethyl malonate complex 3 (10 mol%) gave the diethyl arylmalonate in 73% yield after 8 hours and in 95% yield after 20 hours at 25°C.[11] The similarity of this rate to that for the stoichiometric reaction of 3 with PhI indicates that complex 3 is kinetically competent to be an intermediate in the catalytic process.
Kinetic data gained by 1H NMR spectroscopy on the reactions of 3 with varying excess quantities of PhI and added phen at 60°C showed that the reaction is first order in 3, first order in PhI, and inverse first order in phen (see the Supporting Information for data). These data imply that the reaction occurs by reversible dissociation of one phen from 3 and that the reactive CuI enolate species contains a 1:1:1 ratio of phen/Cu/enolate. This complex could be the ionic species [(phen)2Cu][Cu{CH(CO2Et)2}2] (A) or the neutral complex [(phen)Cu{CH(CO2Et)2}] (B) [Eq. (2)].
 |
(2) |
Scheme 2 summarizes the rate constants for analogous reactions of complexes 1–7 with 10 equivalents of PhI in the presence of 12 equivalents of phen. The relative reactivity follows the trend: 1, 2 < 3 < 4 < 5 < 6 ≈ 7. These data show that complexes of the more-electron-donating enolates react with the iodoarenes faster than the complexes of the less-electron-donating enolates.
Scheme 2.

Rate of reactions of enolate complexes 1–7 with PhI. [a] <5% conversion after 18 h at 60°C.
The catalytic reactions of diethyl malonate with PhI in the presence of 10 mol% of 3 were also inhibited by added phen. Without added phen, this reaction formed the product in 73% yield after 8 hours and in 95% yield after 20 hours at room temperature. The same reaction conducted with 30 mol% of added phen gave diethyl phenylmalonate in only 4% yield after 8 hours and 11% yield after 20 hours (11% conversion). The same reaction with a full equivalent of added phen gave no detectable arylated product after 8 hours. These results are consistent with the reversible dissociation of one phen ligand from 3 to generate the active intermediate (A or B) in the catalytic process.
To gain more direct information on the reactivity of the species generated by dissociation of phen, we generated this complex by treatment of the complex 3 (34.4 mm) with 1 equivalent of CuI in DMSO [Eq. (3)]. This reaction formed
| (3) |
[{(phen)CuI}2] and the ionic or neutral complex A or B containing a 1:1 ratio of phen to enolate, as indicated by 1H NMR spectroscopy. The species containing a single phen reacted with 2 equivalents of PhI to form diethyl phenylmalonate in 37% yield in less than 10 minutes at 25°C and 62% yield with 100% conversion of the copper species after 1 hour,[12] whereas complex 3 reacted with PhI under analogous reaction conditions to form the coupled product in only 23% yield after 78 minutes and 63% yield after 9 hours.
To assess further the potential of the species containing a 1:1:1 ratio of phen/Cu/enolate to be the active intermediates in the reactions of the isolated complexes with aryl halides, the ratio of products from reactions of sterically distinct o-and p-iodotoluenes with the CuI enolate complexes containing or lacking phen ligands was measured (Table 2). The reaction of a 1:1 mixture of o- and p-iodotoluene (10 equiv each) with complex 3 (34.4 mm) formed two diethyl arylmalonates in a 95:5 ratio (entry 1). The reaction of o- and p-iodotoluene with complex A or B, generated by treatment of complex 3 (34.4 mm) with 1 equivalent of CuI, gave the diethyl arylmalonates in a similar 97:3 ratio (entry 2). The similarity of this ratio is consistent with our conclusion that the species reacting with the iodoarene contains a 1:1:1 ratio of phen, enolate, and copper, as drawn from kinetic data on the reactions of isolated 3 in the presence of added phen.
Table 2.
Selectivity of Cu1 enolate complexes toward o- and p-iodo- toluene.
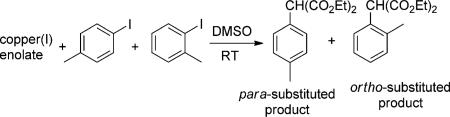 | |||||
|---|---|---|---|---|---|
| Entry Cu1 enolate | t | Yield [%] | Ratio | ||
| para-substituted product | ortho-substituted product | ||||
| 1 | 3 | 1 h | 45 | 2 | 95:5 |
| 3 h | 73 | 3 | 95:5 | ||
| 2 | A or B | < 5 min | 62 | 2 | 97:3 |
| 3 | K[CH(CO2Et)2] + Cul | 1h | 6.2 | 2.4 | 72:28 |
| 21 h | 13 | 6 | 69:31 | ||
| 4 | 2K[CH(CO2Et)2] + Cul | 2h | 23 | 10 | 70:30 |
| 23 h | 37 | 16 | 69:31 | ||
Although attempts to generate enolate complexes lacking any dative ligand did not lead to pure, isolable species (see the Supporting Information for details), we did conduct stoichio-metric reactions of the aryl iodides with the combination of copper and alkali metal enolates. The reaction of o- and p-iodotoluene with a 1:1 and 2:1 ratio of potassium diethyl malonate to CuI (34.4 mm each) without phen, formed the diethyl arylmalonates in an approximately 70:30 ratio and a total yield of only 19% after 21 hours (Table 2, entry 3) and 53% after 23 hours (entry 4), respectively. The low selectivity and the slow rate of these reactions in the absence of phen further imply that the iodoarenes react with a phen-ligated copper species during the reactions initiated with isolated phen-ligated complexes. Moreover, the lower selectivity of the enolate complex containing the putative [Cu{H-(CO2Et)2}2]– anion (entry 4) implies that the active intermediate in the reaction of 3 with iodoarenes is the neutral phen-ligated copper enolate species B, rather than the anionic [Cu{CH(CO2Et)2}2]– portion of the ionic species A.
With an understanding of the structure of the starting enolate complexes and the composition of the reactive intermediate, we sought to distinguish between reaction mechanisms involving electron transfer to generate aryl radicals proposed previously[7] and mechanisms involving the formation of CuIII intermediates by nonradical pathways more akin to those deduced for reactions of complexes with reactive anionic oxygen and nitrogen ligands.[13] To do so, we studied the reaction of complex 3 with o-(allyloxy)iodobenzene [8; Eq. (4)]. The aryl radical of 8 is known to undergo
 |
(4) |
cyclization with a rate constant of 9.6 × 109 s–1 in DMSO to form a [3-(2,3-dihydrobenzofuran)]methyl radical, which is eventually converted into 2-methyldihydrobenzofuran (11).
The reaction of 3 with 8 in DMSO at 25°C produced the arylated molonate 9 in 59% yield and ethyl 2-aryl-2-oxoacetate 10 in 14% yield after two days [Eq. (4)].[14] No detectable amount of cyclized product 11 was observed by GC/MS. The detection limit for product 11 by GC/MS was determined to be less than 0.2% of the amount of 9. Thus, any aryl radical formed from this process must react with the copper enolate complex to form the arylmalonate 9 in less than 2 × 10–13 s. Because this timescale is the lifetime of a transition state, and the organic product would be required to form by recombination of the free aryl radical with the copper enolate in less than 2 × 10–13 s, free radicals that could be formed by initial electron transfer are unlikely to be intermediates in the reaction of copper malonate complexes with aryl halides.
To assess the potential intermediacy of the arylcopper(III) complexes proposed to lie on the reaction pathway, we computed the energies for oxidative addition of PhI to the phen-ligated C-bound CuI complexes of acetylacetone, dimethyl malonate, and methyl phenylacetate anions. These three enolate complexes (1, 4, and 7) reacted with iodoarene with the most diverse rates (see Scheme 2). The free energies of activation (ΔG≠) for oxidative addition of PhI to the C-bound CuI complexes of the anions of acetylacetone, dimethyl malonate, and methyl phenylacetate to form an aryl–CuIII intermediate were calculated to be 27.2, 21.9, and 22.7 kcalmol–1, respectively, at 25°C (see 1-TS1, 4-TS1, and 7-TS1 in Figure S1 in the Supporting Information). These computational results imply that the CuIII species lie at energies that are accessible under mild reaction conditions.
However, the copper enolate complexes containing a single phen ligand could exist in the O,O-bound (or O-bound) form B1 or in the C-bound form B2 (Scheme 3; also see Table S5 in the Supporting Information). Thus, we computed the relative free energies of the C-bound species and the O-bound forms (Table S5). The O,O-bound structures containing the anions of acetylacetone and dimethyl malonate are computed to lie at energies that are lower than those of the C-bound CuI structures by 12.5 and 4.2 kcalmol–1, respectively (see 1-O,O and 4-O,O in Figure S1). Thus, the transition state for oxidative addition to the C-bound isomers of the complexes of acetyl acetonate and dimethyl malonate anions are predicted to lie 39.7 kcalmol–1 and 26.1 kcalmol–1 above the energies of the most stable species containing a 1:1:1 ratio of enolate, phen, and copper. In contrast, the more stable CuI complex containing the anion of methyl phenyl-acetate is computed to be the C-bound form. Thus, the barrier for oxidative addition of PhI to the C-bond enolate containing a 1:1:1 ratio of enolate, phen, and copper is predicted to be only 22.7 kcalmol–1 above the most stable species in this case (Figure S1). The prohibitively large barrier calculated for oxidative addition to the acetylacetonate complex is consistent with our observation of the lack of reactivity of 1 with PhI to form 3-phenyl acetylacetone. The 26.1 and 22.7 kcalmol–1 barriers computed for reaction of the malonate and phenyl-acetate complexes, respectively, agree with the moderate rate observed for the reaction of PhI with complex 4 and the faster rate observed for the reaction of iodobenzene with complex 7 (see Scheme 2).[15]
Scheme 3.

Proposed catalytic cycle with complex 3. Oxidative addition to the C-bound enolate is shown. Oxidative addition to an O-bound isomer, followed by rearrangement to the C-bound CuIII intermediate is less likely, but not ruled out by our data.
The calculated barriers for reductive elimination of α-aryl carbonyl compounds from the C-bound CuIII acetylacetone, dimethyl malonate, and methyl phenylacetate complexes are 3.5, 4.1, and 6.3 kcalmol–1, respectively (see 1-TS2, 4-TS2, and 7-TS2 in Figure S1 in the Supporting Information). These data indicate that the reductive elimination to form the C–C bond from copper complexes of these stabilized anions is much faster than reductive elimination to form the same type of C–C bond from known arylpalladium(II) enolate complexes.[16]
In summary, studies on isolated, phen-ligated enolate complexes of 1,3-dicarbonyl compounds and phenylacetates strongly indicate that the C-bound CuI enolate complex ligated by a single phen lies on the pathway for the reaction of copper enolates with iodoarenes (Scheme 3). The most stable CuI enolate complex containing two phen ligands reversibly dissociates one phen ligand to form the O,O-bound (or O-bound) enolate species B1 containing one phen ligand. We propose that this complex equilibrates with the C-bound isomer B2 and that oxidative addition of the iodoarene occurs to the C-bound isomer to form a CuIII enolate intermediate, which undergoes facile reductive elimination to release the arylated product and [{(phen)CuI}2]. Among the enolate complexes studied, the phenylacetate complex, for which the C-bound enolate is computed to be more stable than the alternative O-bound form, and for which the enolate is the most strongly electron donating, is the most reactive toward oxidative addition of iodoarenes. The acetylacetone complex, for which the O,O-chelating mode is calculated to be more favorable than the C-bound form and for which the enolate is the most weakly electron donating, is the least reactive toward addition of iodoarenes. The intermediacy of the phenligated enolate, the equilibration of species containing a free enolate anion and a C-bound enolate, and reaction by pathways lacking aryl radicals all contradict prior assertions about the mechanism of copper-catalyzed couplings of enolates[3,7] and require revisions of previous mechanistic proposals.
Supplementary Material
Footnotes
We thank the NIH (GM-58108) for support of this work.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201106719.
References
- 1.Hurtley WRH. J. Chem. Soc. 1929:1870. [Google Scholar]
- 2.a Monnier F, Taillefer M. Angew. Chem. 2009;121:7088. doi: 10.1002/anie.200804497. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2009;48:6954. [Google Scholar]; b Okuro K, Furuune M, Miura M, Nomura M. J. Org. Chem. 1993;58:7606. [Google Scholar]
- 3.Hennessy EJ, Buchwald SL. Org. Lett. 2002;4:269. doi: 10.1021/ol017038g. [DOI] [PubMed] [Google Scholar]
- 4.Cristau HJ, Cellier PP, Spindler JF, Taillefer M. Chem. Eur. J. 2004;10:5607. doi: 10.1002/chem.200400582. [DOI] [PubMed] [Google Scholar]
- 5.a Xie XA, Chen Y, Ma DW. J. Am. Chem. Soc. 2006;128:16050. doi: 10.1021/ja066991j. [DOI] [PubMed] [Google Scholar]; b Xie X, Cai G, Ma D. Org. Lett. 2005;7:4693. doi: 10.1021/ol0518838. [DOI] [PubMed] [Google Scholar]; c Jiang YW, Wu N, Wu HH, He MY. Synlett. 2005:2731. [Google Scholar]
- 6.Yip SF, Cheung HY, Zhou Z, Kwong FY. Org. Lett. 2007;9:3469. doi: 10.1021/ol701473p. [DOI] [PubMed] [Google Scholar]
- 7.a Setsune J.-i., Ueda T, Shikata K, Matsukawa K, Iida T, Kitao T. Tetrahedron. 1986;42:2647. [Google Scholar]; b Setsune J.-i., Matsukawa K, Kitao T. Tetrahedron Lett. 1982;23:663. [Google Scholar]; c Setsune J, Matsukawa K, Wakemoto H, Kitao T. Chem. Lett. 1981:367. [Google Scholar]
- 8.Complex 1 has been synthesized by a different method, but its structure remained unknown. Nast R, Lepel W-H. Chem. Ber. 1969;102:3224.
- 9.CCDC 844995 (1), 844996 (4), 844997 (5), and 844998 (6) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- 10.Related bis(phen)-ligated CuI nitrophenylmethane and nitro-methane complexes in which the enolate moieties were proposed to be present as the free anions were reported previously, but their structures were not defined by X-ray diffraction. Marsich N, Camus A, Inorg J. Nucl. Chem. 1977;39:275.
- 11.The catalytic reaction with PhBr as the electrophile gave no product under the same reaction conditions, but formed the diethyl arylmalonate in 86% yield after 15 h at 80°C.
- 12.Full conversion of the species B into diethyl phenylmalonate and free diethyl malonate after 1 h. The latter was presumably formed by protonation of B with the trace amount of water in the DMSO.
- 13.a Huffman LM, Casitas A, Font M, Canta M, Costas M, Ribas X, Stahl SS. Chem. Eur. J. 2011;17:10643. doi: 10.1002/chem.201100608. [DOI] [PubMed] [Google Scholar]; b Huffman LM, Stahl SS. Dalton Trans. 2011;40:8959. doi: 10.1039/c1dt10463b. [DOI] [PubMed] [Google Scholar]; c Casitas A, Ioannidis N, Mitrikas G, Costas M, Ribas X. Dalton Trans. 2011;40:8796. doi: 10.1039/c1dt10428d. [DOI] [PubMed] [Google Scholar]; d Yu H-Z, Jiang Y-Y, Fu Y, Liu L. J. Am. Chem. Soc. 2010;132:18078. doi: 10.1021/ja104264v. [DOI] [PubMed] [Google Scholar]; e Tye JW, Weng Z, Giri R, Hartwig JF. Angew. Chem. 2010;122:2231. doi: 10.1002/anie.200902245. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2010;49:2185. [Google Scholar]; f Giri R, Hartwig JF. J. Am. Chem. Soc. 2010;132:15860. doi: 10.1021/ja105695s. [DOI] [PMC free article] [PubMed] [Google Scholar]; g King AE, Huffman LM, Casitas A, Costas M, Ribas X, Stahl SS. J. Am. Chem. Soc. 2010;132:12068. doi: 10.1021/ja1045378. [DOI] [PubMed] [Google Scholar]; h Huffman LM, Stahl SS. J. Am. Chem. Soc. 2008;130:9196. doi: 10.1021/ja802123p. [DOI] [PubMed] [Google Scholar]; i Tye JW, Weng Z, Johns AM, Incarvito CD, Hartwig JF. J. Am. Chem. Soc. 2008;130:9971. doi: 10.1021/ja076668w. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Zhang S-L, Liu L, Fu Y, Guo Q-X. Organometallics. 2007;26:4546. [Google Scholar]
- 14.Compound 10 was likely formed through the oxidation of 9. Allen PM, Hess U, Foote CS, Baizer MM. Synth. Commun. 1982;12:123.
- 15.These isolable species are the ionic complexes 1, 4 and 7, rather than the neutral complexes on which we are able to conduct meaningful calculations. The relative stability of these ionic species and the neutral species [(phen)Cu(enolate)] is difficult to evaluate by computational methods because the ions can exist as contact, solvent-separated, or fully solvated ion pairs. At the same time, the acidity of the carbonyl compounds in DMSO (acetylacetone = 13.3, dimethyl malonate = 15.9 and methyl phenylacetate ≈ 22) shows clearly that the enolate from acetyl-acetone is more stable than that from dimethyl malonate, which is more stable than that from methyl phenylacetate. Thus the energy difference between the observed ionic enolate complex and the neutral species should follow the trend, acetylacetone > dimethyl malonate > methyl phenylacetate. These energy differences would further increase the difference of free-energy barriers between reactions of PhI with the complexes 1, 4 and 7. Thus, the relative computed barriers would remain consistent with the observed reactivity of these enolate complexes with PhI.
- 16.Culkin DA, Hartwig JF. Acc. Chem. Res. 2003;36:234. doi: 10.1021/ar0201106. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.