

Abstract
Human blood neutrophils rolling on E- or P-selectin reduced their rolling velocity when intercellular adhesion molecule (ICAM)–1 was available. Similar to mouse neutrophils, this was dependent on P-selectin glycoprotein ligand 1 (PSGL1), αLβ2 integrin, the Src family tyrosine kinase FGR and spleen tyrosine kinase SYK. Blocking phospholipase C or p38 MAP kinase attenuated, but did not abolish the velocity reduction. To test expression of integrin activation epitopes, we adapted an immobilized reporter assay and developed a new homogeneous microfluidics-based reporter antibody binding assay. Rolling on E- or P-selectin induced the extension reporter epitopes KIM127 and NKI-L16, but not the high affinity reporter epitope monoclonal antibody (mAb) 24. This enabled rolling neutrophils to bind to immobilized extension reporter, but not activation reporter antibodies and allowed binding of soluble KIM127 during rolling. We conclude that human neutrophil rolling on E- or P-selectin induces the extended αLβ2 integrin conformation through signaling triggered by PSGL-1 engagement.
Introduction
E-selectin or P-selectin binding to human neutrophils has long been known to induce activation as demonstrated by phosphorylation of p38 MAP kinase.1–3 In Ficoll-isolated human neutrophils, this process leads to arrest from rolling, polarization, and firm adhesion to endothelial monolayers under flow.4–7 The proximal signal transduction pathway leading from E-selectin binding to integrin-dependent adhesion is unknown. Whether neutrophil integrins assume the extended or high affinity conformation is also unknown.
Mouse neutrophils in whole blood reduce their rolling velocity on E-selectin when intercellular adhesion molecule 1 (ICAM1) is also available.8 This requires P-selectin glycoprotein ligand 1 (PSGL1),8,9 a cell surface expressed O-glycan that is a ligand for all 3 selectins.10 Slow rolling probably involves extension of the integrin LFA-1, which requires the Src family tyrosine kinase Fgr, the immunoreceptor tyrosine-based activation motif (ITAM)–containing adapter molecules DAP-12 and FcRγ,11 and Syk.8 Binding of isolated human neutrophils to E-selectin increases intracellular calcium levels,7 which is blocked by phospholipase C inhibition. Indeed, PLCγ2 was recently shown to be activated downstream of Syk after integrin engagement.12 We found little evidence for LFA-1 activation during mouse neutrophil rolling on P-selectin, but McEver et al reported that rolling on P-selectin can trigger LFA-1 activation.9
Like other integrins,13,14 LFA-1 undergoes dramatic conformational changes when activated.15 Fluorescence resonance energy transfer (FRET) studies show that the cytoplasmic and transmembrane domains of the αL and β2 subunits of LFA-1 move apart,16 forcing the extracellular domain of LFA-1 into the extended conformation.15 Conformational unbending of α4β1 integrin was also shown more directly using FRET between a fluorophore on α4subunit and an acceptor in the lipid bilayer.17 Using an allosteric inhibitor that stabilizes LFA-1 in the extended conformation and prevents it from assuming the high affinity conformation suggests that rolling on E-selectin may induce the extended, but not high affinity conformation.8 However, definitive evidence for this is difficult to obtain in mice, because no reporter antibodies are available that can distinguish between extended and high affinity LFA-1.15
KIM127 is a mouse anti–human monoclonal antibody (mAb) that recognizes an epitope near the genu of the β2 subunit of human LFA-1 that is only accessible when LFA-1 is extended.15,18,19 Similarly, NKI-L16 recognizes an epitope near the genu of the αL subunit that is only accessible when LFA-1 is extended.20,21 mAb 24 sees an epitope in a loop near the metal ion–dependent adhesion site (MIDAS) of the I-like domain in the β2-subunit of LFA-122 and can be induced by Mn2+.22,23 mAb 24 binding is characteristic of the high-affinity state of LFA-1. The epitope of mAb 24 is likely formed as a result of interaction between the αL subunit I domain and the β2 subunit I–like domain leading to the open, high-affinity state of LFA-1.24
Studies with human lymphocytes have suggested that immobilized chemokines induce the extended form of LFA-1,25 which may then convert to the high affinity conformation upon ligand binding. In mice, E-selectin–dependent LFA-1 activation provides a significant alternative pathway enabling neutrophil recruitment even in the absence of chemokine signaling.8,11,26 The present study was undertaken to test whether extended LFA-1 is induced when human neutrophils roll on E- or P-selectin.
Methods
Antibodies, recombinant proteins, and other reagents
Recombinant human E-selectin-Fc, P-selectin Fc, and ICAM-1-Fc were obtained from R&D Systems. The SYK-inhibitor piceatannol was purchased from A.G. Scientific. The Src-inhibitor PP2 and p38 MAPK-inhibitor SB203580 were purchased from EMD Biosciences. The TS 1/22 mAb were purchased from Thermo Fisher Scientific. The mAb 24 to human β2-subunit was a gift from N. Hogg (Cancer Research Institute, London, United Kingdom).27 The NKI-L16 mAb to human αL-subunit was a gift from C. G. Figdor (University Medical Center St Radboud, Nijmegen, The Netherlands).20 The KIM127 mAb to human β2-subunit19 was purified at the Lymphocyte Culture Center at the University of Virginia from hybridoma supernatant (ATCC). fMLP was purchased from Sigma-Aldrich. The isotype control Abs were purchased from Biolegend. Anti–E-selectin mAB (IBIG-E4) and phycoerythrin (PE)–labeled anti–mouse immunoglobulin G (IgG) antibody were purchased from R&D Systems. PE-labeled anti–L-selectin antibody (DREG-56) or anti-CD11b antibody (ICRF44) were purchased from BD PharMingen.
Human blood
Heparinized whole human blood was obtained from healthy human donors after informed consent, as approved by the Institutional Review Board of La Jolla Institute of Allergy & Immunology and the University of Muenster in accordance with the Declaration of Helsinki.
Flow cytometry
For detection of LFA-1 activation epitopes, whole human blood with or without fMLP were incubated for 5 minutes at 37°C with 5 μg/mL isotype control antibody or integrin reporter antibodies. Cells were washed and fixed with 1% paraformaldehyde for 10 minutes at 4°C. Red blood cells (RBCs) in whole blood were lysed with RBC lysis buffer (eBioscience). Then, cells were stained with PE-labeled goat F(ab′)2 anti–mouse IgG antibody for 20 minutes at 4°C. For detection of cell activation, whole human blood with or without fMLP was incubated for 10 minutes at 37°C, washed, and fixed with 1% paraformaldehyde for 10 minutes at 4°C. After the RBC lysis, cells were stained with PE-labeled anti–L-selectin antibody or anti-CD11b antibody for 20 minutes at 4°C.
Blood-perfused microflow chamber
To investigate the integrin activation status of human neutrophils, we established the whole human blood chamber assay based on a microflow chamber system.8 Briefly, rectangular glass capillaries (20 × 200 μm) were coated with E-selectin (5 μg/mL)/P-selectin (20 μg/mL) alone or in combination with ICAM-1 (3 μg/mL and 5 μg/mL, respectively; R&D Systems) for 2 hours and then blocked for 1 hour using casein (Thermo Fisher Scientific). One side of the chamber was connected to a PE 50 tubing (Becton Dickinson) and used to control the wall shear stress in the capillary as described.8 The other side of the chamber was inserted into a syringe filled with heparinized whole blood. One representative field of view was recorded for 1 minute using an SW40/0.75 objective and a digital camera (Sensicam QE; Cooke Corporation).
For the antibody-coated chamber, rectangular glass capillaries were filled with protein G (500 μg/mL; EMD Biosciences) for 2 hours and washed with phosphate-buffered saline (PBS). Then capillaries were filled with the indicated concentrations of E-selectin or P-selectin in combination with TS1/22, KIM127, or NKI-L16 (25 μg/mL) for 1 hour and blocked for 1 hour with 10% casein (Fisher Scientific). The chamber was connected at one side to a syringe filled with human whole blood. The other side of the chamber was connected to a PE 50 tubing and used to control the wall shear stress, which was calculated as described.8 Capillaries were perfused with human whole blood for 2 minutes, washed with Hanks buffered salt solution (HBSS) containing 1mM Ca2+ and 1mM Mg2+ for 30 seconds, fixed with 2% paraformaldehyde for 2 minutes, stained with Wright-Giemsa stain solution for 3 hours, and washed with distilled water for 1 minute. The number of neutrophils in each flow chamber was counted and expressed per field of view. In some experiments, the capillaries were treated with 5 μg/mL IBIG-E4 antibody for 20 minutes, or whole human blood was incubated with piceatannol (200μM at 37°C for 60 minutes), PP2 (10μM at 37°C for 10 minutes), or SB203580 (20mM at 37°C for 30 minutes) before the perfusion. For the Mn2+ stimulation assay, human neutrophils were isolated from heparin-anti–coagulated whole blood from healthy donors by polymorphprep (Accurate Chemical) according to the manufacturer's instruction. Mn2+ were added to the cell suspension (1 × 107 cells/mL) to the final concentration of 1mM and immediately perfused through the capillary. The capillaries were treated in the same way as whole human blood capillaries, the number of neutrophils on the flow chambers was counted, and the number per one field of view was calculated.
Homogeneous binding assay
For the homogeneous binding assay (ie, the continuous real-time measurement without separation of free antibody),28 the isotype control antibodies or reporter antibodies were labeled with the APEX Alexa Fluor 594 Antibody Labeling kit (Invitrogen) according to the manufacturer's instructions. Rectangular glass capillaries (20 × 200 μm) were filled with protein G (500 μg/mL) for 2 hours, washed with PBS, filled with E-selectin (20 μg/mL) for 1 hour, blocked for 30 minutes with 1% casein (Thermo Fisher Scientific), blocked for 30 minutes with human serum, and blocked for 1 hour with 10% casein.
The whole human blood was incubated with fluorochrome-conjugated reporter antibodies for 3 minutes and immediately perfused through the capillary without the separation of free antibodies. In some experiments, after reaching a plateau, interleukin-8 (IL-8, 100 ng/mL; PeproTech) was added to whole blood. Microscopy was conducted with a Zeiss Axioskop (Carl Zeiss) with a saline immersion objective (SW 40/0.75). Images were recorded with a digital camera (Sensicam QE; Cooke Corporation) with enhancer (model VS4-1845; Video Scope International). For the qualitative analysis, the pixel density was obtained using ImageJ Version 1.43u software (National Institutes of Health), and the background intensity was subtracted.
Selectin engagement assay
Isolated human blood neutrophils were suspended in PBS (containing 1 mM each CaCl2 and MgCl2) and incubated on E-selectin–coated Petri dishes at 65 rpm for 10 minutes as previously described.11 Neutrophils were lysed with RIPA buffer.11 Lysates were boiled with sample buffer or incubated with Sepharose A/G beads (Santa Cruz Biotechnology) and anti-FGR or anti-SYK (both from Santa Cruz Biotechnology) antibody for 4 hours on ice. Beads were washed 4 times, and bound proteins were eluted by adding boiling sample buffer.
Cell lysates and immunoprecipitates were run on 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotted using antibodies against phosphotyrosine (4G10; Millipore), p38 MAP kinase, phospho-p38 MAP kinase, phospho-Src (PY416; all from Cell Signaling Technology) and developed using the enhanced chemiluminescence (ECL) system (GE Healthcare).
Cell line, plasmid, and transfection
U937 cells were cultured as previously described.29 A constitutively active FGR (mutation of Y511 to F) was generated by polymerase chain reaction (PCR) from wild-type FGR cDNA and the following primers (forward primer: 5′-CCGAGACTTGAGGGCAGC-3′; reverse primer: 5′-GATCGAATTCTATGTCTGGTCTCCAGGCTGGAACTGTGG-3′), confirmed by direct DNA sequencing, and subcloned into the retroviral vector pMIG-W. Transient transfection of U937 cells was performed with Amaxa Nucleofector (Amaxa Biosystems). After 24 hours, detection of LFA-1 activation epitopes was performed by flow cytometry.
Statistics
Statistical analysis was performed with Prism and included 1-way analysis of variance and t test where appropriate. All data are presented as mean plus or minus SEM. P values less than .05 were considered significant.
Results
PSGL-1 engagement triggers LFA-dependent slow rolling
To test the effects of selectin-dependent rolling on LFA-1 activation by human neutrophils, we adapted the microfluidic whole blood perfusion system originally developed for mouse blood.8,11 Human neutrophils rolled on E-selectin (Figure 1A) or P-selectin (Figure 1B) at a velocity of approximately 5 μm/s, which was reduced to approximately 3 μm/s when ICAM-1 was added to the substrate. Specificity controls showed that this reduction in rolling velocity was entirely due to LFA-1, because a mAb to LFA-1, but not to Mac-1 (αMβ2 integrin) completely reverted rolling velocity to baseline (Figure 1C). After perfusion of whole blood, the capillaries were rinsed with HBSS, fixed, stained, and the number of neutrophils was counted. More than 90% of cells were neutrophils.
Figure 1.

E-selectin and P-selectin induce LFA-1–dependent slow rolling in human neutrophils. Whole human heparinized blood was perfused through flow chambers coated with E-selectin with or without ICAM-1 or P-selectin with or without ICAM-1. Rolling velocity on E-selectin (A) or P-selectin (B) with or without ICAM-1–coated flow chambers. PL-1 is a PSGL-1 blocking mAb. (C-D) Whole human blood was treated with anti–LFA-1 or anti-Mac-1 antibody (10 μg/mL for 20 minutes at room temperature[RT]) before perfusion through (C) E-selectin or E-selectin plus ICAM-1–coated flow chambers or (D) P-selectin or P-selectin plus ICAM-1–coated flow chambers. #P < .05 from column without antibodies. *P < .05 from selectin column. †P < .05 from selectin plus ICAM-1 column. Data presented are mean ± SEM and are representative of 3 independent experiments.
We next tested the behavior of human blood neutrophils on P-selectin, because we had previously observed that mouse neutrophils showed a much smaller reduction in rolling velocity on P-selectin and ICAM-1 compared with E-selectin and ICAM-1.8 Human neutrophils rolled on P-selectin at a similar velocity (5 μm/s) as on E-selectin (Figure 1B). Similar to what was observed on E-selectin, the reduction in rolling velocity on P-selectin and ICAM-1 was completely prevented by a mAb to LFA-1, but not Mac-1 (Figure 1D).
Mouse neutrophils require PSGL-1 for slow rolling on E-selectin, which was demonstrated by complete abolition of slow rolling in PSGL-1 knockout mice.8 Since PSGL-1–deficient humans have not been described, we resorted to PL-1, a mAb to PSGL-1 that binds near the N-terminus and blocks P-selectin binding to PSGL-1.30,31 As expected, this antibody completely blocked human neutrophil rolling on P-selectin (Figure 1B). Unexpectedly, monvalent Fab fragments of this antibody also prevented the velocity decrease when neutrophils rolled on E-selectin coimmobilized with ICAM-1 (Figure 1A). This suggests that E-selectin may engage the N-terminus of PSGL-1 during rolling to induce signaling. As was shown previously, PL-1 was unable to block rolling on E-selectin, because E-selectin has other binding sites on PSGL-132 and also binds to other ligands such as CD44.33
Src family kinases, SYK, phospholipase C, and p38 are involved in integrin activation after E-selectin and P-selectin binding to PSGL-1
To test whether the signaling pathways engaged by human neutrophils are similar to those used by mouse neutrophils, we used kinase and phospholipase inhibitors. PP2, an inhibitor of Src family tyrosine kinases, completely blocked the reduction in rolling velocity seen on E-selectin when ICAM-1 was added (Figure 2A), as did piceatannol, an inhibitor of SYK (Figure 2B). The phospholipase C inhibitor U73122 partially inhibited slow rolling on E-selectin and ICAM-1 (Figure 2C), as did the p38 MAPkinase inhibitor SB203580 (Figure 2D). Adding an LFA-1 blocking mAb to SB203580-treated neutrophils in whole blood completely restored their rolling velocity to the level seen on E-selectin alone, confirming that the p38 pathway was only partially involved in slow rolling. Virtually the same effects were seen on P-selectin and ICAM-1 (Figure 2A-D).
Figure 2.

E-selectin and P-selectin signaling in human neutrophils. (A-D) Whole human heparinized blood was treated with (A) Src family kinase inhibitor PP2 (15μM for 30 minutes at RT), (B) SYK inhibitor piceatannol (25μM at 30 minutes at RT), (C) PLC inhibitor U-73122 (10μM for 30 minutes at RT), or (D) p38 MAPK inhibitor SB203580 (10μM for 30 minutes at RT), or SB203580 (10μM) plus anti–LFA-1 antibody (10 μg/mL for 20 minutes at RT), then perfused through flow chambers coated with E-selectin with or without ICAM-1 or P-selectin with or without ICAM-1. (E) Human purified neutrophils were plated on blocked (control), E-selectin–coated, or P-selectin–coated wells, and incubated under shear (65 rpm) for 10 minutes and lysed. Lysates were immunoprecipitated with anti-FGR antibody, followed by immunoblotting with a pan-Src antibody that recognizes PY416. (F) U937 cells were transfected with a constitutively active FGR or with empty vector. The binding of KIM127 or mAb 24 was analyzed by flow cytometry. Data presented are mean ± SEM and are representative of 3 independent experiments. (G-H) Human purified neutrophils were plated on blocked (control), E-selectin–coated, or P-selectin–coated wells and incubated under shear (65 rpm) for 10 minutes and lysed. (G) Lysates were immunoprecipitated with anti-SYK antibody, followed by immunoblotting with phosphotyrosine antibody 4G10. (H) Lysates were immunoblotted with antibody to detect phosphorylated p38 MAPK (phospho p38) or total p38. Data are representative of 3 independent experiments.
To investigate the activation of key signaling molecules, we used immunoprecipitation and analysis by phosphotyrosine immunoblotting. As expected, FGR was found to become phosphorylated at Y416, a residue in the kinase domain that reflects activation, when human neutrophils were incubated on E-selectin or P-selectin under shear stress conditions (Figure 2E). After stimulation, HCK and LYN were also phosphorylated at Y416 (data not shown). To test this further, we added a gain-of-function experiment. When the human monocyte cell line U937 was transiently transfected with a constitutively active form of FGR, the cells expressed the LFA-1 β2 extension epitope KIM127 but not the activation epitope mAb 24 (Figure 2F). Taken together, these data suggest that FGR activation is necessary and sufficient to induce LFA-1 extension.
In mouse neutrophils, Syk is downstream of Fgr activation.11 Therefore, we tested SYK phosphorylation in human neutrophils exposed to E-selectin or P-selectin under shear conditions. In both cases, SYK phosphorylation was induced (Figure 2G). p38MAPK was also strongly phosphorylated (Figure 2H).
LFA-1 activation epitope expression in resting and fMLP-stimulated human neutrophils
To test the activation status of LFA-1 in rolling human neutrophils, we investigated whole blood human neutrophils for expression of LFA-1 activation epitopes. Although LFA-1 was strongly expressed on whole blood neutrophils as detected by the pan–LFA-1 antibody TS1/22 that binds to LFA-1 independent of conformation (Figure 3A), neither KIM-127 (Figure 3B) nor NKI-L16 (Figure 3C) extension epitopes nor the mAb 24 activation epitope were expressed on resting neutrophils in whole blood (Figure 3D, depicted as thin lines). As a positive control, we briefly activated neutrophils with fMLP, an agonist of the formyl peptide G protein-coupled receptor (GPCR). This induced strong expression of KIM-127 (Figure 3B), NKI-L16 (Figure 3C), and mAb 24 on all neutrophils (Figure 3D). As expected,34 this treatment also induced up-regulation of Mac-1 surface expression and shedding of L-selectin (Figure 3E-F).
Figure 3.

Induction of LFA-1 activation epitopes by fMLP. Whole blood neutrophils were stained with the pan-LFA-1 mAb TS1-22 (A), the LFA-1 extension reporter mAbs KIM127 (B), or NKI-L16 (C), or the high affinity reporter mAb 24 (D) with or without the fMLP stimulation for 5 minutes. Whole blood neutrophils were stained with anti–Mac-1 antibody (E) or anti–L-selectin antibody (F) before or after fMLP stimulation for 10 minutes. Data are representative of at least 10 independent experiments.
Selectin-mediated LFA-1 extension requires Src family kinases and SYK
To directly test the activation status of LFA-1 in rolling neutrophils, we adapted an immobilized reporter assay (Figure 4A) previously used to test chemokine-induced activation of isolated human lymphocytes.25 Whole human blood was perfused at 6 dyn/cm2 through 20 × 200 μm rectangular capillaries coated with selectin and reporter antibodies (Figure 4B), flushed, fixed, and stained to reveal adherent neutrophils. The E-selectin concentration used for coating (6.6 μg/mL) was chosen to optimize signal-to-noise (supplemental Figure 1, available on the Blood Web site; see the Supplemental Materials link at the top of the online article). Under these conditions, no adherent neutrophils were seen when the flow chamber was coated with isotype control or pan-LFA-1 mAb TS1/22 only (Figure 4C). Approximately 5 adherent neutrophils per one field of view were found on E-selectin alone or E-selectin combined with isotype control, probably reflecting the rolling cells that happened to be caught during flushing and fixing, because this was completely abrogated by IBIG-E4, a blocking antibody to E-selectin (Figure 4C). When E-selectin was present, human neutrophils were able to readily adhere to the pan–LFA-1 antibody TS1/22 (19.0 ± 2.5 cells/field of view; Figure 4C).
Figure 4.

Establishment of a whole human blood immobilized reporter assay. (A) View of the whole human blood flow chamber coated with the LFA-1 activation reporter antibodies. (B) Cells rolling in the microflow chambers visualized by transillumination. Scale bar = 100 μm. (C) Flow chambers coated with isotype control, TS1-22 and/or E-selectin were perfused with heparinized human whole blood for 2 minutes at 5.94 dyn/cm2, washed with HBSS with 1mM Ca2+ and 1mM Mg2+ for 30 seconds, fixed with 2% paraformaldehyde, and stained with Wright-Giemsa stain solution and number of neutrophils counted. Some flow chambers were incubated with E-selectin blocking antibody IBIG-E4 before perfusion, and number per field of view was calculated. Data are presented as mean ± SEM and are representative of 3 independent experiments.
In this same assay, human neutrophils rolling on E-selectin were also able to bind to the extension reporter antibodies KIM127 and NKI-L16, but not to the activation reporter mAb 24 (Figure 5A). As a positive control, we induced high affinity LFA-1 by incubating neutrophils with 1mM Mn2+ (Figure 5B), which enabled them to accumulate on E-selectin coimmobilized with mAb 24. Neutrophil adhesion was also induced when the flow chamber was coated with E-selectin, CXCL1, and mAb 24, but not when E-selectin, CXCL1, and an isotype control antibody were present (7.6 ± 1.6 cells/field of view vs 0.3 ± 0.44 cells/field of view; P < .05). Similar results were found on P-selectin (Figure 5D-E).
Figure 5.

Rolling on E-selectin or P-selectin evokes extended, but not high affinity LFA-1, in human neutrophils. (A) Whole blood was perfused through flow chambers coated with E-selectin and isotype antibody, KIM127, NKI-L16, mAb 24, or TS1-22 for 2 minutes at 5.94 dyn/cm2. (B) Isolated human neutrophils were stimulated with 1mM Mn2+ and immediately perfused through flow chambers coated with E-selectin and isotype antibody, mAb 24 or TS1-22 for 2 minutes at 5.94 dyn/cm2. (C) Whole blood was treated with piceatannol (200μM at 37°C for 60 minutes), PP2 (10μM at 37°C for 10 minutes), or SB203580 (20mM at 37°C for 30 minutes) before the perfusion through the flow chamber coated with E-selectin and isotype antibody or KIM127. (D) Whole blood was perfused through flow chambers coated with P-selectin and isotype antibody, KIM127, NKI-L16, mAb 24, or TS1-22 for 2 minutes at 5.94 dyn/cm2. (E) Purified human neutrophils were stimulated with 1mM Mn2+ and immediately perfused through flow chambers coated with P-selectin and isotype antibody, mAb 24, or TS1-22 for 2 minutes at 5.94 dyn/cm2. Number of neutrohils was counted after the Wright-Giemsa staining. Data are normalized to E-selectin or P-selectin and KIM127 (= 100%; panels A, C, and D) or E-selectin or P-selectin and TS1-22 (= 100%; panels B and E) and presented as mean ± SEM. *P < .05 from isotype control. †P < .05 from KIM127. Data are representative of at least 3 independent experiments.
To confirm some of the signaling pathways in this assay, we incubated whole human blood with inhibitors. The SYK inhibitor piceatannol and the Src inhibitor PP2 each inhibited accumulation when KIM127 was coimmobilized with E-selectin (Figure 5C), confirming that Src and SYK activation are indeed necessary for expression of this epitope. Similar to the findings in the rolling velocity assay (Figure 2), inhibiting p38 MAPkinase with SB 203580 had a partial effect (Figure 5C).
Rolling neutrophils gradually induce integrin extension
Although the immobilized reporter assay provides a robust readout of LFA-1 extension, it does not reveal the time course during which LFA-1 is activated in rolling neutrophils. To address this issue, we developed a homogeneous binding assay (ie, the continuous real-time measurement without separation of free antibody),28 in which fluorescently labeled LFA-1 antibodies were added to whole human blood and epitope expression was examined using a very sensitive image intensifier and camera system. As expected, the isotype control showed no signal (data not shown). In contrast, KIM127 binding increased systematically (Figure 6A), reaching a plateau at 50 seconds (Figure 6B; supplemental Video 1). After reaching a plateau, IL-8 (100 ng/mL) was added to whole blood. However, no significant change was detected (KIM127 intensity, before IL-8 7.3 ± 1.3, after IL-8 8.6 ± 1.1, P < .05; Figure 6C). The mAb 24 epitope was not detectable in rolling neutrophils (data not shown), but was readily induced by IL-8 (Figure 6C). The pan-LFA-1 antibody TS1/22 served as a positive control (stained all rolling cells, binding did not increase with rolling time, data not shown; supplemental Video 2).
Figure 6.
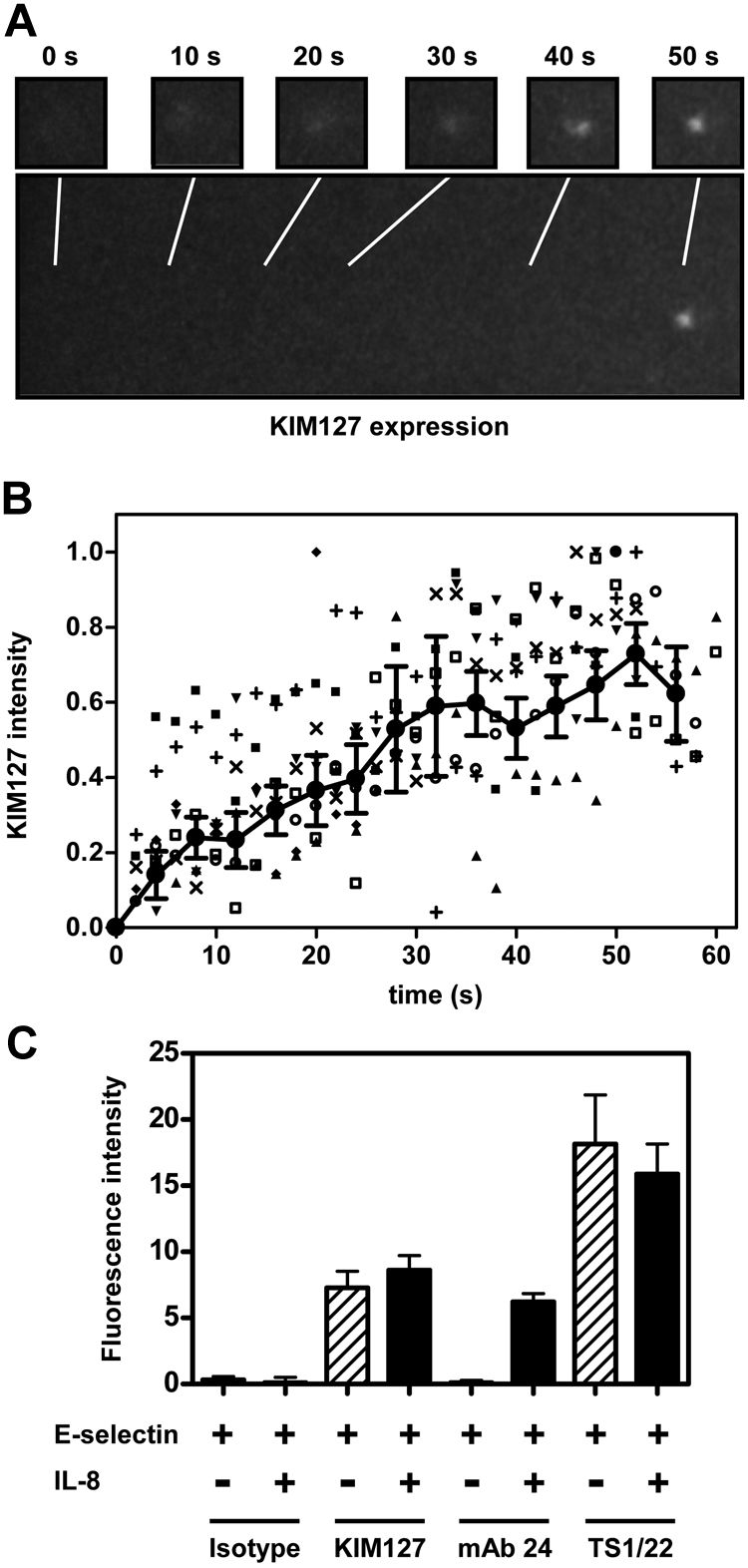
Real-time imaging of the LFA-1 activation in whole human blood flow chambers. (A-B) Whole blood was treated with Alexa Fluor 594–conjugated KIM127 (5 μg/mL). After 5 minutes, whole blood was perfused through flow chambers coated with E-selectin. Fluorescence intensity was detected by the epifluorescence microscope with the enhancer. (B) Fluorescence intensity reflecting KIM127 binding was measured at 2-second intervals for representative cells (■, □, ▼, ♦, ○, ▲, +, ×) and normalized to the maximal intensity (= 1). Data were averaged at 4-second intervals (indicated by ● joined by line; mean ± SEM). Data representative of 2 independent experiments. (C) Whole blood was treated with Alexa Fluor 594–conjugated antibody (5 μg/mL) and perfused through flow chambers coated with E-selectin. After saturation, IL-8 was added to whole blood, and the change of the Alexa Fluor 594 intensity was measured. Data are presented as mean ± SEM and are representative of 3 independent experiments.
Discussion
The present data establish that human neutrophils express the extended conformation of LFA-1 while rolling on E- or P-selectin. Signaling is PSGL-1–dependent and involves SYK activation through the Src family tyrosine kinase FGR. FGR activation is not only necessary, but also sufficient to extend LFA-1 in neutrophils. Selectin-dependent neutrophil activation is slower than its chemokine-dependent counterpart, reaching half-maximal activation within 27.5 plus or minus 2.4 seconds, which is consistent with previous in vivo studies in a mouse model of inflammation.35 By contrast, chemokine activation of arrest is almost instantaneous in lymphocytes25 and in neutrophils.36
In this paper, we show that whole blood human neutrophils do not express LFA-1 activation epitopes. The discovery that PSGL-1 engagement induces LFA-1 activation was made possible by using neutrophils in whole blood rather than isolated neutrophils. Neutrophil isolation by Percoll or Ficoll leads to increased Mac-1 expression on the cell surface and decreased ability to roll.37,38 In the newly developed human whole blood flow assay, we demonstrate a clear difference in rolling velocity on E-selectin versus E-selectin/ICAM-1, whereas isolated human neutrophils rolled at similar velocities on L cells transfected with E-selectin alone or in combination with ICAM-1.2 Then, we show that PSGL-1 engagement by E-selectin or P-selectin leads to a reduction of the rolling velocity of human neutophils by forcing LFA-1 in the extended conformation. As isolated neutrophils are already activated, the differences in the rolling velocity were probably missed in earlier studies.
Induction of β2-integrin activation epitopes has been studied previously. In a study of isolated human lymphocytes, Alon et al demonstrated that immobilized chemokines can induce immediate lymphocyte arrest while rolling on L-selectin ligand and ICAM-1.25 They used KIM127, NKI-L16, mAb 24, 327A, and 327C antibodies to monitor LFA-1 activation. Some baseline expression of mAb 24 and NKI-L16 is seen in isolated human peripheral blood lymphocytes,25 which differs from our findings using whole blood neutrophils. Our data extend these previous findings from lymphocytes to neutrophils and to the selectin-induced rather than chemokine-induced activation.
PSGL-1 signals through a cascade involving FGR-mediated phosphorylation of the ITAM adapters DAP12 and FcRγ, which in turn recruit SYK to assemble a signalosome that activates downstream pathways.8,11 By contrast, chemokines signal through GPCRs, where neutrophil arrest chemokines couple through Gαi2.39 The signaling pathway for integrin activation downstream of GPCRs probably involves PLC-β,40 release of intracellular free calcium, the guanosine nucleotide exchange factor (GEF) CalDAG-GEFI,41 and its target Rap1.42 Our data suggest that PLC is also partially involved in the PSGL-1–dependent pathway of LFA-1 activation, because the PLC inhibitor U73122 partially attenuated the reduction of rolling velocity when ICAM-1 was added to the selectin substrate.
Our data does not address which isoform of PLC is involved in neutrophil LFA-1 activation. Recently, PLC-γ2 was shown to be involved in integrin outside-in signaling downstream of Syk.12 The integrin outside-in signaling pathway shares many similarities with the PSGL-1 pathway, including the DAP12, FcRγ, and Syk signaling intermediates.43,44 Therefore, it is plausible that PLC-γ2 may be involved in PSGL-1 signaling downstream of SYK. Unlike the PSGL-1 signaling pathway, which is strictly FGR-dependent, the integrin pathway also involves 2 other Src kinases, Hck and Lyn.43
The present report is not the first to suggest that E- or P-selectin binding activates neutrophils. P-selectin binding to PSGL-1 evokes phosphorylation of Nef-associated factor 1 and downstream signaling to PI3Kδ in mouse neutrophils.45 A large body of work by Scott Simon's laboratory has established that E-selectin engagement can activate β2 integrins.2 His group also demonstrated that inhibitors of p38 MAPK reduce integrin activation. Inhibition of p38 MAPK by a pharmacologic inhibitor decreases the number of neutrophils attaching to L cells expressing E-selectin and ICAM-1, and the ability of neutrophils to attach to albumin-coated latex beads after cross-linking of E-selectin ligands.2 Furthermore, increased expression and clustering of the β2 integrin activation epitope 327C by cross-linking E-selectin ligands with E-selectin-IgG followed by secondary antibody was also inhibited by blocking p38 MAPK.46 The current paper adds details to the signaling pathway and demonstrates that selectin signaling happens in whole blood at physiologically relevant shear stresses.
Alon suggested that chemokine-mediated integrin activation induces the extended conformation and that ligand binding under tension (pulling force) induces the high affinity form.25,47 The present data and previous work8,11 suggest that PSGL-1 engagement induces extended LFA-1 integrin, but fails to induce the high affinity conformation. This may mean that a signaling element is missing from this pathway that the chemokine receptor pathway provides. The nature of this element remains unknown. Although the selectivity of the used mAbs is supported by the literature, their interaction still remains a surrogate for the different states of the receptor, and it cannot be excluded that their binding may be induced by as yet undefined changes in the receptor, other than only an extended or an activated state.
The distal signaling of integrin activation involves talin-148 and kindlin-3.49,50 Whether one or both of these molecules bind LFA-1 upon PSGL-1 ligation remains to be determined.
In conclusion, the present data establishes PSGL-1–dependent LFA-1 extension in human neutrophils. Taken together with data in mouse inflammation models,8,11 this validates PSGL-1 as a target for anti-inflammatory interventions. Blocking PSGL-1 interaction with P- or E-selectin will not only limit leukocyte rolling, but also activation. The clinical indications for which such an intervention might be useful remain to be determined.
Supplementary Material
Acknowledgments
We thank Clare L. Abram and Clifford A. Lowell for providing the constitutively active FGR construct, Nancy Hogg for providing mAb 24, and Carl G. Figdor for providing mAb NKI-L16.
This work was supported by grants from the National Institutes of Health (HL58108 to K.L.), the German Research Foundation (AZ 428/3-1 to A.Z.), and the Interdisciplinary Clinical Research Center (IZKF Muenster, Germany, Za2/001/10 to A.Z.). Y.K. was supported by a Postdoctoral Fellowship grant from the Japan Society for the Promotion of Science (JSPS).
Note added in proof.
While this manuscript was under review, another relevant paper appeared. Yago T, Shao B, Miner JJ, et al. E-selectin engages PSGL-1 and CD44 through a common signaling pathway to induce integrin {alpha}L{beta}2-mediated slow leukocyte rolling. Blood. 2010;116(3):485-494.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: Y.K. designed and performed research, analyzed data, and wrote the paper; O.S. performed experiments and helped analyze the data; H.Z. performed experiments; K.L. designed the study, analyzed data, interpreted the results, and wrote parts of the manuscript; and A.Z. designed the study, analyzed the data, interpreted the results, and wrote parts of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for O.S. is Department of Anesthesiology and Intensive Care Medicine, University of Cologne, Cologne, Germany.
Correspondence: Alexander Zarbock, Department of Anesthesiology and Critical Care Medicine, University of Muenster, Albert-Schweitzer Str 33, 48149 Muenster, Germany; e-mail: zarbock@uni-muenster.de.
References
- 1.Evangelista V, Manarini S, Sideri R, et al. Platelet/polymorphonuclear leukocyte interaction: P-selectin triggers protein-tyrosine phosphorylation-dependent CD11b/CD18 adhesion: role of PSGL-1 as a signaling molecule. Blood. 1999;93(3):876–885. [PubMed] [Google Scholar]
- 2.Simon SI, Hu Y, Vestweber D, Smith CW. Neutrophil tethering on E-selectin activates beta 2 integrin binding to ICAM-1 through a mitogen-activated protein kinase signal transduction pathway. J Immunol. 2000;164(8):4348–4358. doi: 10.4049/jimmunol.164.8.4348. [DOI] [PubMed] [Google Scholar]
- 3.Totani L, Piccoli A, Manarini S, et al. Src-family kinases mediate an outside-in signal necessary for β2 integrins to achieve full activation and sustain firm adhesion of polymorphonuclear leucocytes tethered on E-selectin. Biochem J. 2006;396(1):89–98. doi: 10.1042/BJ20051924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Green CE, Schaff UY, Sarantos MR, Lum AF, Staunton DE, Simon SI. Dynamic shifts in LFA-1 affinity regulate neutrophil rolling, arrest, and transmigration on inflamed endothelium. Blood. 2006;107(5):2101–2111. doi: 10.1182/blood-2005-06-2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sarantos MR, Raychaudhuri S, Lum AF, Staunton DE, Simon SI. Leukocyte function-associated antigen 1-mediated adhesion stability is dynamically regulated through affinity and valency during bond formation with intercellular adhesion molecule-1. J Biol Chem. 2005;280(31):28290–28298. doi: 10.1074/jbc.M501662200. [DOI] [PubMed] [Google Scholar]
- 6.Schaff UY, Sarantos MR, Ting H, Simon SI. Optical and fluorescence detection of neutrophil integrin activation. Methods Mol Biol. 2007;412:203–210. doi: 10.1007/978-1-59745-467-4_13. [DOI] [PubMed] [Google Scholar]
- 7.Schaff UY, Yamayoshi I, Tse T, Griffin D, Kibathi L, Simon SI. Calcium flux in neutrophils synchronizes β2 integrin adhesive and signaling events that guide inflammatory recruitment. Ann Biomed Eng. 2008;36(4):632–646. doi: 10.1007/s10439-008-9453-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zarbock A, Lowell CA, Ley K. Spleen tyrosine kinase Syk is necessary for E-selectin-induced α(L)β(2) integrin-mediated rolling on intercellular adhesion molecule-1. Immunity. 2007;26(6):773–783. doi: 10.1016/j.immuni.2007.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miner JJ, Xia L, Yago T, et al. Separable requirements for cytoplasmic domain of PSGL-1 in leukocyte rolling and signaling under flow. Blood. 2008;112(5):2035–2045. doi: 10.1182/blood-2008-04-149468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McEver RP, Cummings RD. Role of PSGL-1 binding to selectins in leukocyte recruitment. J Clin Invest. 1997;100(11 Suppl):S97–103. [PubMed] [Google Scholar]
- 11.Zarbock A, Abram CL, Hundt M, Altman A, Lowell CA, Ley K. PSGL-1 engagement by E-selectin signals through Src kinase Fgr and ITAM adapters DAP12 and FcR gamma to induce slow leukocyte rolling. J Exp Med. 2008;205(10):2339–2347. doi: 10.1084/jem.20072660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jakus Z, Simon E, Frommhold D, Sperandio M, Mocsai A. Critical role of phospholipase Cgamma2 in integrin and Fc receptor-mediated neutrophil functions and the effector phase of autoimmune arthritis. J Exp Med. 2009;206(3):577–593. doi: 10.1084/jem.20081859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takagi J, Petre BM, Walz T, Springer TA. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell. 2002;110(5):599–511. doi: 10.1016/s0092-8674(02)00935-2. [DOI] [PubMed] [Google Scholar]
- 14.Xiong JP, Stehle T, Diefenbach B, et al. Crystal structure of the extracellular segment of integrin alpha Vbeta3. Science. 2001;294(5541):339–345. doi: 10.1126/science.1064535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luo BH, Carman CV, Springer TA. Structural Basis of Integrin Regulation and Signaling. Annu Rev Immunol. 2007;25:619–47. doi: 10.1146/annurev.immunol.25.022106.141618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim M, Carman CV, Springer TA. Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science. 2003;301(5640):1720–1725. doi: 10.1126/science.1084174. [DOI] [PubMed] [Google Scholar]
- 17.Chigaev A, Waller A, Zwartz GJ, Buranda T, Sklar LA. Regulation of cell adhesion by affinity and conformational unbending of alpha4beta1 integrin. J Immunol. 2007;178(11):6828–6839. doi: 10.4049/jimmunol.178.11.6828. [DOI] [PubMed] [Google Scholar]
- 18.Beglova N, Blacklow SC, Takagi J, Springer TA. Cysteine-rich module structure reveals a fulcrum for integrin rearrangement upon activation. Nat Struct Biol. 2002;9(4):282–287. doi: 10.1038/nsb779. [DOI] [PubMed] [Google Scholar]
- 19.Robinson MK, Andrew D, Rosen H, et al. Antibody against the Leu-CAM beta-chain (CD18) promotes both LFA-1- and CR3-dependent adhesion events. J Immunol. 1992;148(4):1080–1085. [PubMed] [Google Scholar]
- 20.Keizer GD, Visser W, Vliem M, Figdor CG. A monoclonal antibody (NKI-L16) directed against a unique epitope on the alpha-chain of human leukocyte function-associated antigen 1 induces homotypic cell-cell interactions. J Immunol. 1988;140(5):1393–1400. [PubMed] [Google Scholar]
- 21.Xie C, Shimaoka M, Xiao T, Schwab P, Klickstein LB, Springer TA. The integrin alpha-subunit leg extends at a Ca2+-dependent epitope in the thigh/genu interface upon activation. Proc Natl Acad Sci U S A. 2004;101(43):15422–15427. doi: 10.1073/pnas.0406680101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu C, Shimaoka M, Zang Q, Takagi J, Springer TA. Locking in alternate conformations of the integrin alphaLbeta2 I domain with disulfide bonds reveals functional relationships among integrin domains. Proc Natl Acad Sci U S A. 2001;98(5):2393–2398. doi: 10.1073/pnas.041618598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dransfield I, Cabanas C, Craig A, Hogg N. Divalent cation regulation of the function of the leukocyte integrin LFA-1. J Cell Biol. 1992;116(1):219–226. doi: 10.1083/jcb.116.1.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stanley P, Smith A, McDowall A, Nicol A, Zicha D, Hogg N. Intermediate-affinity LFA-1 binds alpha-actinin-1 to control migration at the leading edge of the T cell. Embo J. 2008;27(1):62–75. doi: 10.1038/sj.emboj.7601959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shamri R, Grabovsky V, Gauguet JM, et al. Lymphocyte arrest requires instantaneous induction of an extended LFA-1 conformation mediated by endothelium-bound chemokines. Nat Immunol. 2005;6(5):497–506. doi: 10.1038/ni1194. [DOI] [PubMed] [Google Scholar]
- 26.Smith ML, Olson TS, Ley K. CXCR2- and E-selectin-induced neutrophil arrest during inflammation in vivo. J Exp Med. 2004;200(7):935–939. doi: 10.1084/jem.20040424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cabanas C, Hogg N. Ligand intercellular adhesion molecule 1 has a necessary role in activation of integrin lymphocyte function-associated molecule 1. Proc Natl Acad Sci U S A. 1993;90(12):5838–5842. doi: 10.1073/pnas.90.12.5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sklar LA, Edwards BS, Graves SW, Nolan JP, Prossnitz ER. Flow cytometric analysis of ligand-receptor interactions and molecular assemblies. Annu Rev Biophys Biomol Struct. 2002;31:97–119. doi: 10.1146/annurev.biophys.31.082901.134406. [DOI] [PubMed] [Google Scholar]
- 29.Ramos CL, Huo Y, Jung U, et al. Direct demonstration of P-selectin- and VCAM-1-dependent mononuclear cell rolling in early atherosclerotic lesions of apolipoprotein E-deficient mice. Circ Res. 1999;84(11):1237–1244. doi: 10.1161/01.res.84.11.1237. [DOI] [PubMed] [Google Scholar]
- 30.Li F, Erickson HP, James JA, Moore KL, Cummings RD, McEver RP. Visualization of P-selectin glycoprotein ligand-1 as a highly extended molecule and mapping of protein epitopes for monoclonal antibodies. J Biol Chem. 1996;271(11):6342–6348. doi: 10.1074/jbc.271.11.6342. [DOI] [PubMed] [Google Scholar]
- 31.Thatte A, Ficarro S, Snapp KR, et al. Binding of function-blocking mAbs to mouse and human P-selectin glycoprotein ligand-1 peptides with and without tyrosine sulfation. J Leukoc Biol. 2002;72(3):470–477. [PubMed] [Google Scholar]
- 32.Li F, Wilkins PP, Crawley S, Weinstein J, Cummings RD, McEver RP. Post-translational modifications of recombinant P-selectin glycoprotein ligand-1 required for binding to P- and E-selectin. J Biol Chem. 1996;271(6):3255–3264. [PubMed] [Google Scholar]
- 33.Hidalgo A, Peired AJ, Wild MK, Vestweber D, Frenette PS. Complete identification of E-selectin ligands on neutrophils reveals distinct functions of PSGL-1, ESL-1, and CD44. Immunity. 2007;26(4):477–489. doi: 10.1016/j.immuni.2007.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kishimoto TK, Jutila MA, Berg EL, Butcher EC. Neutrophil Mac-1 and MEL-14 adhesion proteins inversely regulated by chemotactic factors. Science. 1989;245(4923):1238–1241. doi: 10.1126/science.2551036. [DOI] [PubMed] [Google Scholar]
- 35.Kunkel EJ, Dunne JL, Ley K. Leukocyte arrest during cytokine-dependent inflammation in vivo. J Immunol. 2000;164(6):3301–3308. doi: 10.4049/jimmunol.164.6.3301. [DOI] [PubMed] [Google Scholar]
- 36.Ley K, Baker JB, Cybulsky MI, Gimbrone MA, Jr, Luscinskas FW. Intravenous interleukin-8 inhibits granulocyte emigration from rabbit mesenteric venules without altering L-selectin expression or leukocyte rolling. J Immunol. 1993;151(11):6347–6357. [PubMed] [Google Scholar]
- 37.Kuijpers TW, Tool AT, van der Schoot CE, et al. Membrane surface antigen expression on neutrophils: a reappraisal of the use of surface markers for neutrophil activation. Blood. 1991;78(4):1105–1111. [PubMed] [Google Scholar]
- 38.Glasser L, Fiederlein RL. The effect of various cell separation procedures on assays of neutrophil function. A critical appraisal. Am J Clin Pathol. 1990;93(5):662–669. doi: 10.1093/ajcp/93.5.662. [DOI] [PubMed] [Google Scholar]
- 39.Zarbock A, Deem TL, Burcin TL, Ley K. Galphai2 is required for chemokine-induced neutrophil arrest. Blood. 2007;110(10):3773–3779. doi: 10.1182/blood-2007-06-094565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Z, Jiang H, Xie W, Zhang Z, Smrcka AV, Wu D. Roles of PLC-beta2 and -beta3 and PI3Kgamma in chemoattractant-mediated signal transduction. Science. 2000;287(5455):1046–1049. doi: 10.1126/science.287.5455.1046. [DOI] [PubMed] [Google Scholar]
- 41.Bergmeier W, Goerge T, Wang HW, et al. Mice lacking the signaling molecule CalDAG-GEFI represent a model for leukocyte adhesion deficiency type III. J Clin Invest. 2007;117(6):1699–1707. doi: 10.1172/JCI30575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Katagiri K, Hattori M, Minato N, Irie S, Takatsu K, Kinashi T. Rap1 is a potent activation signal for leukocyte function-associated antigen 1 distinct from protein kinase C and phosphatidylinositol-3-OH kinase. Mol Cell Biol. 2000;20(6):1956–1969. doi: 10.1128/mcb.20.6.1956-1969.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mocsai A, Abram CL, Jakus Z, Hu Y, Lanier LL, Lowell CA. Integrin signaling in neutrophils and macrophages uses adaptors containing immunoreceptor tyrosine-based activation motifs. Nat Immunol. 2006;7(12):1326–1333. doi: 10.1038/ni1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mocsai A, Zhou M, Meng F, Tybulewicz VL, Lowell CA. Syk is required for integrin signaling in neutrophils. Immunity. 2002;16(4):547–558. doi: 10.1016/s1074-7613(02)00303-5. [DOI] [PubMed] [Google Scholar]
- 45.Wang HB, Wang JT, Zhang L, et al. P-selectin primes leukocyte integrin activation during inflammation. Nat Immunol. 2007;8(8):882–892. doi: 10.1038/ni1491. [DOI] [PubMed] [Google Scholar]
- 46.Green CE, Pearson DN, Camphausen RT, Staunton DE, Simon SI. Shear-dependent capping of L-selectin and P-selectin glycoprotein ligand 1 by E-selectin signals activation of high-avidity beta2-integrin on neutrophils. J Immunol. 2004;172(12):7780–7790. doi: 10.4049/jimmunol.172.12.7780. [DOI] [PubMed] [Google Scholar]
- 47.Alon R, Ley K. Cells on the run: shear-regulated integrin activation in leukocyte rolling and arrest on endothelial cells. Curr Opin Cell Biol. 2008;20(5):525–532. doi: 10.1016/j.ceb.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tadokoro S, Shattil SJ, Eto K, et al. Talin binding to integrin beta tails: a final common step in integrin activation. Science. 2003;302(5642):103–106. doi: 10.1126/science.1086652. [DOI] [PubMed] [Google Scholar]
- 49.Moser M, Bauer M, Schmid S, et al. Kindlin-3 is required for beta2 integrin-mediated leukocyte adhesion to endothelial cells. Nat Med. 2009;15(3):300–305. doi: 10.1038/nm.1921. [DOI] [PubMed] [Google Scholar]
- 50.Moser M, Legate KR, Zent R, Fassler R. The tail of integrins, talin, and kindlins. Science. 2009;324(5929):895–899. doi: 10.1126/science.1163865. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}