

Abstract

The single-step synthesis of fused tricyclic pyridazino[1,2-a] indazolium ring systems is described. Structural details revealed by crystallography explain the unexpected reactivity. The method is applied to the gram scale synthesis of nigeglanine hydrobromide.
Indazole derivatives are a versatile class of compounds that have found use in biology, catalysis and medicinal chemistry.1 Although rare in nature,2 indazoles exhibit a variety of biological activities such as HIV protease inhibition,3 antiarrhythmic and analgesic activities,4 anti-tumor activity,5 and anti-hypertensive properties.6 Indazolium ions have found additional uses as precursors to N-heterocyclic carbenes (NHCs) with organo-catalytic activity7 and they are strong donor ligands for transition metal complexes.8 While a variety of capable synthetic methods exist for indazole synthesis,9 we report a new precursor for indazole formation – 2-formyl dialkylanilines. With hydroxylamine, this precursor allows for easy entry to the fused tricyclic pyridazino[1,2-a]indazolium ring system seen in the indazole natural products. This reaction invloves an exchange of the N–O bond of the oxime for the N–N bond of the heterocycle and was applied to the gram scale synthesis of nigeglanine hydrobromide (1).
During our investigation of ortho-hydroxyaryloxime antidotes for organophosphorous nerve agent sensing we found that treatment of aldehyde 2 with hydroxylamine hydrochloride in ethanol at 65 °C gave the expected oxime (3) only as the minor product (1:4, Figure 1). The major product was identified by single crystal X-ray diffraction as the substituted N-methyl indazole 4 (Figure 1, bottom right).
Figure 1.
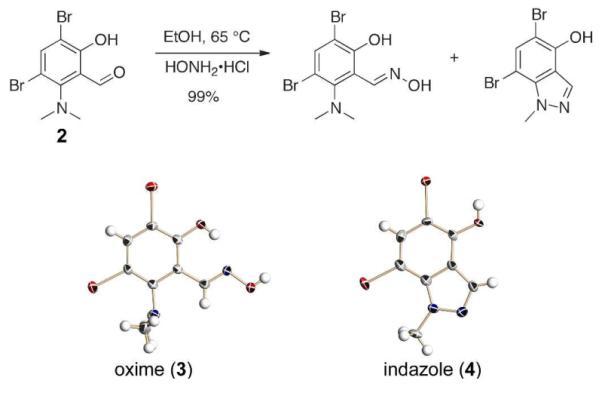
Initial reaction discovery (top). Crystal structure of oxime 3 (bottom left). Crystal structure of indazole 4 (bottom right).
The oxime product (3) can be obtained in better yield at room temperature, and its crystal structure (Figure 1, bottom left) reveals the reason for the unexpected formation of 4. The dimethylamino group is twisted out of the aromatic ring plane to avoid steric clashes with the adjacent bulky bromine atom. Although phenols react with activated oximes to form isoxazoles,10 the nitrogen of the twisted aniline is more basic and its lone pair electrons are predisposed to attack the oxime. This reaction results in N–N bond formation as water is lost. Demethylation, presumably by the halide, completes the sequence.
In general indazoles, or heterocycles, are synthesized by N–C bond forming reactions but examples of indazoles formed from creating N–N bonds have also been reported. Electrophilic amination of this type has been exploited previously by Hassner11 and more recently by Stambuli.9c,d In both examples an activating agent — DCC in the former and MsCl in the later — was used to promote attack of the oxime nitrogen to form the N–N bond. In our approach, no such activating agent is required.
We used 2-pyrrolidinyl benzaldehyde (5) to determine the nature of the nucleophile involved in the dealkylation step. Increased temperatures were required for the reaction to proceed, and after four hours at 150 °C the three expected products were obtained: the oxime (6), the N-alkyl indazole product (8), and the tricyclic indazolium (9). NMR and MS analysis (see SI) confirms that chloride dealkylates the quaternary nitrogen of the former pyrrolidine, giving rise to 8. The proposed mechanism for this transformation is shown in Scheme 1. In specific cases, spirocyclic quaternary nitrogen atoms of the type shown in the scheme (7) have been isolated as betaines (1,1-disubstituted indazol-3-ylio oxides) and and are known to dealkylate in a similar manner.12 By extending the reaction time it is possible to obtain the tricyclic indazolium product exclusively and in high yield.
Scheme 1.

Proposed mechanism for the synthesis of fused indazolium ring systems.
We optimized reaction conditions (1.2 equiv. NH2OH·HCl, EtOH, 150 °C, 18 h, sealed tube) and explored the scope of this cyclization cascade with a series of substituted 2-pyrrolidinyl benzaldehydes. These were obtained from commercially available 2-fluorobenzaldehydes and pyrrolidine (See SI). As shown in Scheme 2, both electron donating and withdrawing groups are tolerated. The strongly donating pyrrolidinyl group (16) gave the highest yield. Halogens Br (11), Cl (14), and F (10) were also well tolerated. The use of methoxy derivatives in place of the free phenols is recommended due to decomposition of the latter during the cyclization reaction. Mild electron withdrawing groups such as trifluoromethyl (18) and carboxylate (17) also form products but in lower yields. Substitution with a nitro group (19) on the otherhand gives a different product: dehydration of the oxime is favored, and the nitrile 19 is formed (see SI for X-ray structure of 19).
Scheme 2.

Scope of the reaction with substituted 2-pyrrolidinyl benzaldehydes. Isolated yields are reported.
Besides altering the substitutents on the aromatic ring, we varied the aniline ring size to further test the scope of the reaction (See SI, Table S-1). Increasing the ring size of the aniline from five (pyrrolidine) to six (piperidine) gave the chloroalkyl indazole in moderate yield, showing that formation of the seven membered ring is unfavored even at high temperatures. We were unable to synthesize aniline starting materials with smaller ring sizes, having observed rapid decomposition in the SNAr reaction with azetidine and aziridine, which prevented these anilines from being investigated. We also explored the synthesis of C3 substituted indazole products from acetophenone or benzophenone substrates. These substrates provided the N-alkylchloride indazole along with the usual ketoxime. Ring-closure to the tricyclic indazolium products was observed only in trace amounts (<2%).
To highlight our method we set out to synthesize a member of the indazole natural product family— nigeglanine. Isolated from nigella glandulifera,2c nigeglanine is one member of a small family of four indazole natural products, all of which contain the tricyclic pyridazino[1,2-a]indazolium ring system. Located largely in southwest and western China, the whole herb has been used as a folk remedy for a variety of aliments such as cold, cough, insomnia, and bronchial asthma. Nigeglanine and related nigellicine, have been synthesized previously by Kelly and co-workers.13 Their synthesis of nigeglanine proceeds by transformation of an isatin to give an advanced indazole intermediate, followed by alkylation of the indazole with 1,4,-dibromobutane, and subsequent decarboxylation and deprotection to give nigeglanine as the HBr salt (1) in 12 steps and 13 % overall yield.
Our synthesis began from commercially available 3-fluoro-5-methylphenol (20) (Scheme 3). The phenol was alkylated with cesium carbonate and methyl iodide in DMF to give 21. We expected the methoxy and fluorine functions to direct ortho lithiation,14 which occurred at the desired position and led to formylation with DMF to afford 22 as a single regioisomer. We synthesised the aniline using pyrrolidine under SNAr conditions (DSMO, potassium carbonate, 100 °C) to give 23 in good yield. The cyclization cascade of this compound went smoothly on the gram scale to yield methylated nigeglanine 24. Deprotection of the methoxy group was carried out with Kelly’s reported method, using BBr3 in DCM to give 1.6 grams of the natural product nigeglanine as the HBr salt (1), in 5 steps and ~47 % overall yield. Pentane diffusion into a methanol/chloroform solution of 1 provided single crystals suitable for X-ray diffraction. The pale yellow bars crystallized as a non-merohedral twin15 and refinement confirmed the structure of 1.
Scheme 3.

Gram scale synthesis and crystal structure ofnegeglanine hydrobromide (1). Counter anion is omitted for clarity.
In summary we report the serendipitous discovery of a new precursor for the one step preparation of indazoles, and crystal structures of oxime 2 and the corresponding indazole 3 that provide clues to the reaction mechanism. The reaction conditions were generalized to afford several examples of the tricyclic pyridazino[1,2-a]indazolium ring system (9-18), and extended to the gram scale synthesis of nigeglanine hydrobromide 1. We note in passing that many of these indazolium ions are fluorescent with colors of blue (9), blue/green (1), and purple (15). As this method quickly affords indazollium analogs of the natural product, their fluorescence may provide an opportunity for cellular imaging studies in the future.
Supplementary Material
Acknowledgment
We are grateful to the Skaggs Institute and the NIGMS (GM 27953) for financial support. A.C.S. is a Skaggs Pre-doctoral Fellow and ARCS scholar. A Ruth L. Kirschstein Postdoctoral fellowship for O.B.B. was provided by NIH (F32GM087068). We would also like to thank Dr. Curtis E. Moore (UCSD) for assistance with solution of the X-ray structures.
Footnotes
Supporting Information Available: General information and full experimental details for all compounds discussed. Cif files for all crystal structures along with tables of relevant structure data are included. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Schmidt A, Beutler A, Snovydovych B. Eur. J. Org. Chem. 2008:4073–4095. [Google Scholar]
- (2)(a).Atta-ur-Rahman MS, He CH, Clardy J. Tetrahedron Lett. 1985;26:2759–2762. [Google Scholar]; (b) Atta-ur-Rahman MS, Malik S, Hasan S, Choudhary MI, Ni C, Clardy J. Tetrahedron Lett. 1995;36:1993–1996. [Google Scholar]; (c) Liu Y, Yang J, Liu Q. Chem. Pharm. Bull. 2004;52:454–455. doi: 10.1248/cpb.52.454. [DOI] [PubMed] [Google Scholar]; (d) Ali Z, Ferreira D, Carvalho P, Avery MA, Khan IA. J. Nat. Prod. 2008;71:1111–1112. doi: 10.1021/np800172x. [DOI] [PubMed] [Google Scholar]
- (3)(a).Han W, Pelletier JC, Hodge CN. Bioorg. Med. Chem. Lett. 1998;8:3615–3620. doi: 10.1016/s0960-894x(98)00659-3. [DOI] [PubMed] [Google Scholar]; (b) Patel M, Rodgers JD, McHugh RJ, Jr., Johnson BL, Cordova BC, Klaba RM, Bacheler LT, Erickson-Viitanen S, Ko SS. Bioorg. Med. Chem. Lett. 1999;9:3217–3220. doi: 10.1016/s0960-894x(99)00564-8. [DOI] [PubMed] [Google Scholar]; (c) Sun J-H, Teleha CA, Yan J-S, Rodgers JD, Nugiel DA. J. Org. Chem. 1997;62:5627–5629. [Google Scholar]
- (4).Mosti L, Menozzi G, Fossa P, Filippelli W, Gessi S, Rinaldi B, Falcone G. Arzneim.-Forsch./Drug Res. 2000;50:963–972. doi: 10.1055/s-0031-1300319. [DOI] [PubMed] [Google Scholar]
- (5)(a).Jakupec MA, Reisner E, Eichinger A, Pongratz M, Arion VB, Galanski M, Hartinger CG, Keppler BK. J. Med. Chem. 2005;48:2831–2837. doi: 10.1021/jm0490742. [DOI] [PubMed] [Google Scholar]; (b) Showalter HDH, Angelo MM, Berman EM, Kanter GD, Ortwine DF, Ross-Kesten SG, Sercel AD, Turner WR, Werbel LM, Worth DF, Elslager EF, Leopald WR, Shillis JL. J. Med. Chem. 1988;31:1527–1539. doi: 10.1021/jm00403a009. [DOI] [PubMed] [Google Scholar]
- (6).Goodman KB, Cui H, Dowdell SE, Gaitanopoulos DE, Ivy RL, Sehon CA, Stavenger RA, Wang GZ, Viet AQ, Xu W, Ye G, Semus SF, Evans C, Fries HE, Jolivette LJ, Kirkpatrick RB, Dul E, Khandekar SS, Yi T, Jung DK, Wright LL, Smith GK. J. Med. Chem. 2007;50:6–9. doi: 10.1021/jm0609014. [DOI] [PubMed] [Google Scholar]
- (7)(a).Schmidt A, Snovydovych B, Habeck T, Drottboom P, Gjikaj M, Adam A. Eur. J. Org. Chem. 2007:4909–4916. doi: 10.1021/jo062391r. [DOI] [PubMed] [Google Scholar]; (b) Schmidt A, Snovydovych B, Gjikaj M. Synthesis. 2008;17:2798–2804. [Google Scholar]
- (8).Jothibasu R, Huynh HV. Chem. Commun. 2010;46:2986–2988. doi: 10.1039/b925977e. [DOI] [PubMed] [Google Scholar]
- (9)(a).Wheeler RC, Baxter E, Campbell IB, Macdonald SJF. Org. Process Res. Dev. 2011;15:565–569. [Google Scholar]; (b) Spiteri C, Keeling S, Moses JE. Org. Lett. 2010;12:3368–3371. doi: 10.1021/ol101150t. [DOI] [PubMed] [Google Scholar]; (c) Counceller CM, Eichman CC, Wray BC, Stambuli JP. Org. Lett. 2008;10:1021–1023. doi: 10.1021/ol800053f. [DOI] [PubMed] [Google Scholar]; (d) Wray BC, Stambuli JP. Org. Lett. 2010;12:4576–4579. doi: 10.1021/ol101899q. [DOI] [PubMed] [Google Scholar]; (e) Inamoto K, Katsuno M, Yoshino T, Arai Y, Hiroya K, Sakamoto T. Tetrahedron. 2007;63:2695–2711. [Google Scholar]; (f) Lukin K, Hsu MC, Fernando D, Leanna MR. J. Org. Chem. 2006;71:8166–8172. doi: 10.1021/jo0613784. [DOI] [PubMed] [Google Scholar]; (g) Jin T, Yamamoto Y. Angew. Chem., Int. Ed. 2007;46:3323–3325. doi: 10.1002/anie.200700101. [DOI] [PubMed] [Google Scholar]; (h) Stadlbauer W. Sci. Synth. 2002;12:227–324. [Google Scholar]
- (10).Dale TJ, Sather AC, Rebek J., Jr Tetrahedron Lett. 2009;50:6173–6175. [Google Scholar]
- (11).Hassner A, Michelson MJ. J. Org. Chem. 1962;27:298–301. [Google Scholar]
- (12)(a).Ruiz JR, Arán VJ, Stud M. Tetrahedron Lett. 1988;29:697–700. [Google Scholar]; (b) Arán VJ, Asensio JL, Ruiz J, Stud M. J. Chem. Perkin Trans. 1. 1993:1119–1127. [Google Scholar]; (c) Waldron NM, Montevalli M, Azam S, Dasopoulos PC. J. Chem. Soc., Chem. Commun. 1995:81–82. [Google Scholar]; (d) Waldron NM, Raza M. J. Chem. Soc., Perkin Trans. 1. 1996:271–276. [Google Scholar]
- (13).Elliott EL, Bushell SM, Cavero M, Tolan B, Kelly TR. Org. Lett. 2005;7:2449–2451. doi: 10.1021/ol050769m. [DOI] [PubMed] [Google Scholar]
- (14).Marzi E, Mongin F, Spitaleri A, Schlosser M. Eur. J. Org. Chem. 2001:2911–2915. [Google Scholar]
- (15).TWINABS - Bruker AXS scaling for twinned crystals - Version 2012/1
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.