

Abstract
Oxidative protein folding in the endoplasmic reticulum (ER) requires strict regulation of redox homeostasis. Disruption of the lumenal redox balance induces an integrated ER stress response that is associated with reduced protein translation, increased chaperone activity, and ultimately cell death. Imexon is a small molecule chemotherapeutic agent that has been shown to bind glutathione (GSH) and induce oxidative stress in tumor cells, however the mechanism of cytotoxicity is not well understood. This In this report, we investigate the effects of imexon on the integrated ER stress response in pancreatic carcinoma cells. Acute exposure to imexon induces an ER stress response characterized by accumulation of the oxidized form of the oxidoreductase Ero1α, phosphorylation of eIF2alpha and inhibition of protein synthesis. An RNA interference chemo-sensitization screen identified the eukaryotic translation initiation factor eIF2B5 as a target that enhanced imexon-induced growth inhibition of MiaPaCa-2 pancreatic cancer cells, but did not significantly augment the effects of imexon on protein synthesis. Concurrent reduction of intracellular thiols with N-acetyl cysteine reversed imexon activity, however co-treatment with superoxide scavengers had no effect, suggesting thiol binding may be a primary component of the oxidative effects of imexon. Moreover, the data suggest that disruption of the redox balance in the ER is a potential therapeutic target.
Keywords: endoplasmic reticulum, oxidative stress, imexon, pancreatic
Introduction
Imexon is a thiol-reactive small molecule that has shown preclinical activity in pancreatic cancer [1, 2] and was evaluated in patients with advanced pancreatic cancer in combination with gemcitabine [3]. The mechanism of action is believed to involve binding to reduced sulfhydryls such as cysteine and glutathione [4]. In cell line models, the biological consequences of this binding include a decrease in glutathione levels, the accumulation of reactive oxygen species, loss of the mitochondrial membrane potential, cytochrome C leakage from the mitochondria and finally, activation of caspases 3 and 9 of the intrinsic pathway of apoptosis, and caspase 8 of the extrinsic pathway of apoptosis [5 – 8]. In human pancreatic cancer cells similar effects were seen involving the accumulation of ROS, loss of the mitochondrial membrane potential, cell cycle arrest in G2 phase and activation of caspases 3, 8 and 9 leading to apoptosis [1]. Pancreatic cancer cells treated with imexon also showed a marked reduction in overall protein translation including reduced synthesis of the tumor survival protein hypoxia-inducible factor-1α [9]. The mechanism for the global inhibition of protein synthesis by imexon in pancreatic cancer cells was not established.
Whereas the previous mechanistic studies focused largely on the effects of imexon-induced ROS on the mitochondria, no studies had been performed on possible oxidative effects in the endoplasmic reticulum (ER), an organelle which is known to be highly dependant on proper redox balance to allow for proper protein folding [10]. Disulfide bond formation in the ER is catalyzed by the transfer of electrons from substrate proteins to oxidized PDI, resulting in the reduction of conserved thioredoxin-motifs (CXXC). Oxidized PDI is then regenerated by the thiol oxidase flavoprotein Ero1α. Protein assembly and folding in the ER requires coordinated oxidation and reduction processes, and we hypothesized that those redox processes could be disrupted by the pro-oxidant effects of imexon. In the current study, we provide evidence that imexon interferes with redox balance in the ER, inducing an ER stress response characterized by reduced protein translation and inhibition of cell growth.
Materials and methods
Cell Lines
Three human pancreatic cancer cell lines were obtained from low passage seed stocks at the American Type Culture Collection (Manassas, VA). A range of differentiation phenotypes was used, including the poorly differentiated BxPC3 (CRL-1687), the relatively gemcitabine-resistant Panc-1 cells (CRL-1469), and the largely undifferentiated MiaPaCa-2 cells (CRL-1420). All cell lines were maintained at 37°C in humidified 5% CO2 in RPMI-1640 supplemented with penicillin (100 U/mL), streptomycin (100 μg/mL) and L-glutamine (2 mM) (Invitrogen, Carlsbad, CA) and 10% bovine calf serum (Hyclone, Rockford, IL). Cell line identity was verified by short tandem repeat (STR) analysis by the University of Arizona Genomics Core (UAGC, Tucson, AZ). Imexon (4-imino-1,3-diazabicyclo[3.1.0.]-hexan-one) was provided by the Pharmaceutical Resources Branch of the National Cancer Institute (Bethesda, MD) under a Rapid Access to Intervention in Development (RAID) grant to R. Dorr.
siRNA synthetic lethal screen
An siRNA human library comprising four siRNA duplexes for each of 21,000 target genes was obtained from Dharmacon (Lafayette, CO). Transfection efficiency and target gene silencing were optimized using a panel of 9 non-coding, and 4 cell death inducing sequences (siTOX, siKIF11, siPLK-1, siCOPB2) [11]. For each target, the four siRNA duplexes provided in the library were combined and arrayed in a 384-well format. MiaPaCa-2 cells were transfected with the siRNA library for 48h using Lipofectamine RNAiMax (Invitrogen, Carlsbad, CA), at which time 90 μM imexon (72h IC10) was added and the plates were incubated an additional 48h. Genes identified in the high throughput screening were validated using Dharmacon ON-TARGET plus SMARTpool siRNAs, which were distinct sequences from those used in the screening process. Non-targeting siRNA sequences were also utilized to ensure no off-target effects. The efficiency of RNA interference (RNAi) was measured by immunoblotting.
Growth inhibition analysis
Cell survival for the siRNA screening experiments were calculated by the conversion of resazurin to resorufin by metabolically active cells resulting in a fluorescent product (em. 490) [12] (Promega, Inc. Madison, WI). Confirmatory growth inhibition assays with eIF2b silencing were done using the methyl-thiazolyl-diphenyl-tetrazolium bromide (MTT) assay as previously described [13]. Cell growth inhibition data are expressed as percent survival, compared to untreated cells. The IC50 is defined as the drug concentration required to produce 50% growth inhibition.
Colony-forming cell assay
Colony-forming assays were performed on MiaPaca-2 cells transfected with eIF2B5 or non-targeting siRNA (Dharmacon ON-TARGET plus SmartPool siRNA), followed by exposure to imexon for an additional 72h. Imexon was removed, and cells were then plated in MethoCult (StemCell Technologies, Inc., Vancouver, Canada) following the manufacturer’s instructions and incubated for 7–10d, and colony formation (> 50 cells) was monitored by manual colony counting using a scoring grid. Data are expressed as percent of control, where the control is mock transfected (no siRNA) cells (Mean ± SEM, n=3). Statistical significance was evaluated using ANOVA, with post-ANOVA Tukey’s analysis.
Protein synthesis assay
Total protein synthesis was analyzed by 14C valine incorporation studies. Cells were plated in 96-well plates, transfected with non-targeting siRNA control (si CT) or si eIF2B5 overnight and exposed to imexon for a total of 24h, with 0.5 μCi/ml of 14C valine (Amersham Biosciences, Piscataway, NJ) added during the last 6h of imexon exposure. Cells with incorporated radioactivity were harvested using a Packard FilterMate Harvester (Perkin Elmer, Waltham, MA). Data are expressed as percent of control, where the control is untreated cells transfected with siCT (Mean ± SEM, n=7). Data were analyzed by a two sample Wilcoxon rank sum test. Analysis of the dose effect within each siRNA treatment group was performed by ANOVA, with Dunnett’s correction for multiple comparisons against a control.
Reversal of oxidative effects of imexon by N-acetyl cysteine (NAC)
MiaPaCa-2 cells were pre-treated overnight with 10 mM NAC, followed by incubation with 0, 250 or 500 μM of imexon for a total of 24h. During this 24h period, NAC was either maintained (“continuous”) or washed out and replaced with fresh media (“pre-treated only”). Alternatively, imexon was added 2h prior to the addition of NAC (“post imexon”). Protein synthesis was measured by incorporation of 500 μCi/ml of 3H-leucine (Perkin Elmer, Waltham, MA) for 18h. Data are expressed as percent of control, where the control is untreated cells (Mean ± SEM, n = 20). Statistical significance was evaluated using ANOVA.
Reversal of oxidative effects of imexon by superoxide scavengers
MiaPaCa-2 cells were pre-treated for 2h with the superoxide scavengers, 100 U/ml superoxide dismutase (PEG-SOD, Sigma Chemical Co, St Louis, MO), 200 U/ml pegylated catalase (PEG-CAT, Sigma Chemical Co.), or 100 μM 1-oxyl-2,2,6,6-tetramethyl-4-hydroxy-piperidine (Tempol, Axxora, LLC, San Diego, CA), followed by incubation with 0, 100, 250 or 500 μM of imexon for a total of 24h. Protein synthesis was measured by incorporation of 3H-leucine (Perkin Elmer, Waltham, MA) for 18h. Data are expressed as percent of control, where the control is untreated cells (Mean ± SEM, n = 18). Statistical significance was evaluated using ANOVA.
Measurement of ER stress
Transcriptional activity of the endoplasmic reticulum stress element (ERSE) was measured using the Cignal ERSE Reporter Luciferase Assay (SA Bioscience Qiagen, Frederick, MD). The ERSE reporter assay is a mixture of an ERSE-responsive luciferase construct and a constitutively expressed Renilla luciferase construct (40:1). Luciferase activity was measured using Promega’s Dual Luciferase assay kit, and the results are expressed as a ratio of the ERSE firefly reporter luciferase to Renilla luciferase. MiaPaCa-2, Panc-1, or BxPC3 cells were transfected in 96-well plates with an ERSE reporter, a negative control, or a positive control for 24h. The cells were then exposed for an additional 8h with increasing concentrations of imexon. Positive control ER stress-inducing agents used included thapsigargin (100 nM), tunicamycin (2 μg/ml), or hydrogen peroxide (0.2 mM). Data are expressed as mean ± SEM (n=6–9), with each experiment standardized to untreated control cells.
Immunoblotting
Pancreatic cells were washed with ice cold PBS, and lysed with M-PER protein lysis buffer (Thermo Scientific, Inc, Rockford, IL) plus protease inhibitors aprotinin, soybean trypsin inhibitor, leupeptin, sodium orthovanadate, and PMSF. Samples were centrifuged to remove insoluble particles, and protein was quantitated by BCA assay (Bio-Rad, Hercules, CA). Samples were separated by SDS-PAGE, transferred to PVDF membranes (Bio-Rad) and probed with antibodies from Cell Signaling (Davers, MA), with the exception of β-actin, which was from Sigma (St. Louis, MO).
Ero-1α Redox Immunoblotting
MiaPaCa-2 cells were treated with increasing concentrations of imexon for 6h and lysed using M-PER, as described. Proteins were separated on a 4–20% non-reducing polyacrylamide gel. Cells were also treated with the thiol reductant, dithiothreitol (DTT), or the oxidant diamide, to identify the differences in the electrophoretic mobility of fully reduced (OX1) versus fully oxidized (OX2) Ero1α species.
Results
Imexon induces oxidative stress in the ER
Oxidative protein folding in the ER relies on a series of direct thiol-disufide exchange reactions. We postulated that imexon could induce ER stress by interfering with the redox cycling of the key ER oxidoreductases, PDI and ER oxidase (Ero1α). Ero1α activity is regulated by intramolecular disulfide bonds, and the oxidation status can be determined by the electrophoretic mobility of the reduced (OX1) versus the oxidized (OX2) form on a non-reducing acrylamide gel [14]. To determine if the redox status of Ero1α could be altered by imexon treatment, we examined the distribution of oxidized and reduced forms of these proteins in MiaPaCa-2 cells. Electrophoretic mobility on a non-reducing gel demonstrated a dose dependent shift in the migration of Ero1α from a primarily reduced form to a highly oxidized form (Fig. 1). However, there was no significant change in the level of Ero1α expression following incubation with imexon. Control treatments of DTT and diamide are used to identify the fully reduced (OX1) and fully oxidized (OX2) species.
Fig. 1.

Accumulation of oxidized Ero1α by imexon treated MiaPaCa-2 cells. MiaPaCa-2 cells were incubated with the indicated concentration of imexon for 6h, and separated on a non-reducing acrylamide gel. Diamide treatment was used to identify fully oxidized (OX2) Ero1α (lane 7), while DTT was used to identify fully reduced (OX1) Ero1α (lane 8).
Imexon induces an integrated stress response
Inhibition of protein translation initiation through phosphorylation of eIF2alpha and reduced activity of the eIF2 initiation complex is one of the hallmarks of the integrated ER-stress response [15]. To determine if imexon treatment induces an integrated ER stress response, we first examined activation of the cis-acting ER Stress Response Element (ERSE). Pancreatic cell lines were transfected with an ERSE dual-luciferase reportergene (24h) and incubated with the indicated concentrations of imexon for an additional 8h (Fig. 2). The data are expressed as a ratio of ERSE reporter luciferase to Renilla luciferase, and standardized to untreated cells. We identified an approximate 2-fold increase in ERSE reporter activity in the three cell lines upon exposure to imexon. This reached statistical significance in the Panc-1 and BxPC3 cell lines at concentrations of 500 μM and 1000 μM imexon (Fig. 2). These data were validated using three well- characterized ER stress-inducing agents; thapsigargin, tunicamycin, and H202, which each demonstrated a 3 to 5-fold increase in ERSE activity (data not shown).
Fig. 2.

Imexon activates an ER stress response. (A) Panc-1, (B) BxPC3, and (C) MiaPaCa-2 were transfected with an ERSE reporter, a negative control, or a positive control for 24h, at which time the cells were washed and then exposed to drugs for an additional 8h with increasing concentrations of imexon. Data shown are the mean ± SEM, n=9 (for MiaPaCa-2 and Panc-1) and n=6 (for BxPC3). *p < 0.05, **p < 0.01.
To investigate the effects of imexon on the expression of additional ER-resident stress response proteins, the three pancreatic cancer cell lines were incubated with increasing concentrations of imexon for 24h. As shown in Fig. 3, all 3 cell lines demonstrated increased expression of the oxidoreductase protein disulfide isomerase (PDI), but as in Fig. 1, no change in the expression of Ero1α. In contrast to the effects of tunicamycin, imexon did not increase levels of the molecular chaperone BiP (Grp78). This suggests that the ER stress response activated by imexon is not likely a secondary effect of misfolded protein accumulation, but rather, may be directly related to ER protein redox status.
Fig. 3.

Effect of imexon on ER oxidative response proteins. Pancreatic cell lines were incubated with the indicated concentration of imexon for 24h, and cell lysates were separated by SDS-PAGE and analyzed by immunoblotting. PDI, Ero1α, BiP, and β-actin are 57kDa, 60 kDa, 78 kDa, and 42 kDa, respectively.
ER associated proteins participate in the imexon response
To identify gene products that may contribute to imexon-induced ER stress and growth inhibition, we conducted an RNA interference synthetic lethal screen in MiaPaCa-2 cells. An siRNA array was used to individually silence 21,000 genes, and cell survival was assayed by resorufin fluorescence after 48h exposure to imexon. Array plates were incubated with or without minimally toxic concentrations of imexon (72h IC10 of 90 μM). Molecular targets were identified as those resulting in at least 50% change in response to imexon, calculated as the ratio of % survival in the [siRNA + imexon] treatment population to % survival in [siRNA only] treatment. Genes whose silencing resulted in greater than 50% cell death in the [siRNA only] treatment group were eliminated from consideration. Of the remaining 18,512 genes silenced, only 20 known genes demonstrated a cell death ratio less than 50%, signifying an increased contribution to cell growth inhibition by the siRNA + imexon combination (Table 1). Three general classes of gene products emerged in this analysis: 1) the serpin family of extracellular peptidase inhibitors; 2) G-coupled receptor proteins; and 3) gene products associated with protein translation. The most significant enhancement of imexon cytotoxicity was seen with silencing of eIF2B5, a guanine nucleotide exchange factor (GEF), which is a key regulator of translation initiation.
Table 1.
Gene products whose silencing resulted in greater than 50% cell death in the presence of 90 μM imexon (IC10)
| Gene Symbol | Gene Name | siRNA (% survival) | siRNA+imexon (% survival) | Ratio siRNA +imexon siRNA |
|---|---|---|---|---|
| EIF2B5 | Eukaryotic translation initiation factor 2B, subunit 5 | 78.2 | 27.7 | 35.4 |
| SLC9A3R2 | Solute carrier family 9 (sodium/hydrogen exchanger), member 3 regulator 2 | 53.7 | 19.4 | 36.0 |
| ARHGAP26 | Rho GTPase activating protein 26 | 73.1 | 31.0 | 42.5 |
| SERPINC1 | serpin peptidase inhibitor, clade C, member 1 | 85.9 | 36.6 | 42.6 |
| OR2AE1 | olfactory receptor, family 2, subfamily AE, member 1 | 101.7 | 43.4 | 42.7 |
| ARHGEF5 | Rho guanine nucleotide exchange factor (GEF) 5 | 89.2 | 38.8 | 43.8 |
| KDR | kinase insert domain receptor | 87.5 | 38.1 | 43.5 |
| SERPINE1 | serpin peptidase inhibitor, clade E, member 1 | 66.1 | 28.8 | 43.5 |
| SET | SET nuclear oncogene | 82.4 | 36.2 | 43.9 |
| OR1L1 | olfactory receptor, family 1, subfamily L, member 1 | 105.7 | 47.2 | 44.7 |
| SERPINE2 | serpin peptidase inhibitor, clade E, member 2 | 55.1 | 25.2 | 45.7 |
| FBX08 | F-box protein 8 | 65.4 | 30.1 | 46.0 |
| ZNF200 | zinc finger protein 200 | 102.2 | 47.2 | 46.2 |
| SP7 | Sp7 transcription factor | 36.5 | 17.0 | 46.7 |
| ZFP36L1 | zinc finger protein 36, C3H type-like 1 | 76.8 | 36.0 | 46.9 |
| OR51G2 | olfactory receptor, family 51, subfamily G, member 2 | 85.1 | 40.4 | 47.5 |
| WASH1 | WAS protein family homolog 1 | 90.7 | 43.7 | 48.2 |
| MANF | mesencephalic astrocyte-derived neurotrophic factor | 60.0 | 29.4 | 49.0 |
| CYTH1 | cytohesin 1 | 55.1 | 27.3 | 49.6 |
| MRLP10 | mitochondrial ribosomal protein L10 | 65.4 | 32.5 | 49.8 |
To validate the effects of eIF2B5 gene silencing on pancreatic cell response to imexon, we examined growth inhibition in three pancreatic cell lines by MTT assay. The MiaPaCa-2, Panc-1, and BxPC3 cells were transfected with eIF2B5 siRNA, followed by incubation with increasing concentrations of imexon for 72h. The data demonstrate that reduction of eIF2B5 expression significantly inhibits the growth of all three pancreatic cell lines, and this effect is enhanced with the addition of imexon (Fig. 4A). We also examined the ability of MiaPaca-2 cells to form colonies after eIF2B5 silencing and subsequent exposure to imexon. As was observed with the MTT assay, there was a 70% reduction in colony formation in cells with silenced eIF2B5, and an additional 10–20% reduction in colony formation with the addition of imexon (Fig. 4B). However, these latter results did not reach statistical significance. Western blot analysis of eiF2B5 protein expression in pancreatic cell lines following 24h transfection with si eIF2B5 is shown in Fig. 4C. These results confirm a near total knock-down of eIF2B5 expression.
Fig. 4.
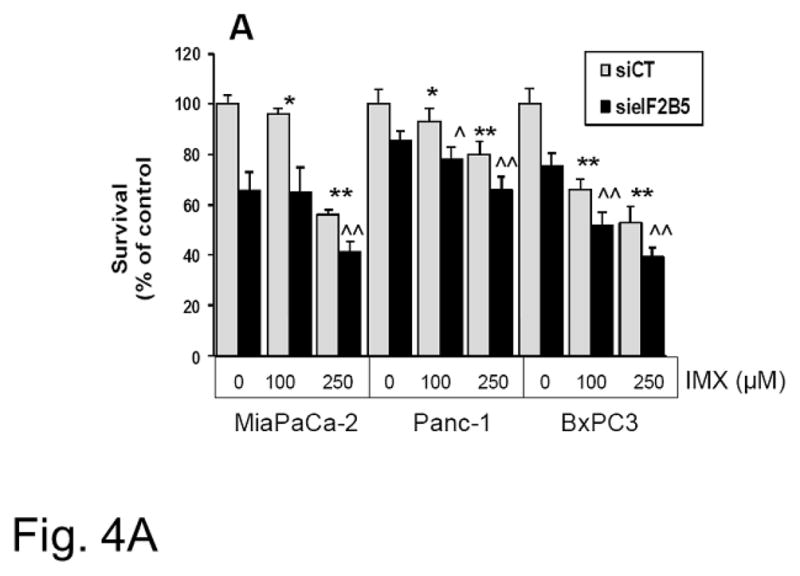


Growth inhibitory effects of imexon in pancreatic cell lines with eIF2B5 silencing. Pancreatic cells were transfected with siRNA to eIF2B5 or a non-targeting sequence (si CT) followed by 72h incubation with imexon (IMX). A) Growth inhibition was analyzed by MTT dye metabolism. Data are presented as % of control, where 100% is si CT with no imexon (Mean ± SD, n=8).
Effect of IMX in si CT: *p < 0.05; **p < 0.01;
Effect of IMX in si elF2B5: ^p < 0.05, ^^p < 0.01;
The effect of si elF2B5 vs. si CT is significant at all concentrations of IMX (p < 0.01).
B) Growth inhibition was analyzed by colony-forming assay. Data are presented as % of control, where control cells are mock transfected with transfection reagent but no siRNA (Mean ± SEM, n=3). Effect of IMX: *p < 0.05
C) Western blot analysis of eIF2B5 protein expression in pancreatic cell lines following 24h transfection with siRNA. eIF2B5 and β-actin are 85 kDa and 42kDa, respectively.
To investigate the role of eIF2B5 in imexon-mediated protein synthesis inhibition, we next examined protein synthesis in the MiaPaCa-2 cell line with or without silencing of eIF2B5. As shown in Fig. 5A, siRNA silencing of eIF2B5 inhibits protein synthesis by 70% (p = 0.0017). In the absence of eIF2B5 silencing, comparable inhibition of translation required exposure to 500 μM imexon for 24h. Moreover, the addition of 250 or 500 μM imexon to cells with reduced eIF2B5 expression further inhibited protein translation by only 8.5 and 5.2% respectively. This suggests that the effects of imexon on protein synthesis are mediated within the eIF2 pathway. To address this question, we examined the direct effects of imexon on the expression eIF2B5 and other proteins associated with translation initiation. MiaPaCa-2, Panc-1, and BxPC3 cells were incubated with imexon for 24h, and analyzed by Western blot. In all three cell lines, imexon did not significantly alter the levels of eIF2B5, however there was a dose-dependent increase in the phosphorylation of eIF2alpha, as well as an increase in the levels of GTP exchange protein eIF2B2 (Fig. 6). It is important to note that protein synthesis inhibition (Fig. 5A) was analyzed following 24h of drug exposure, which would produce minimal cell growth inhibition, whereas growth inhibitory effects (Fig. 4A) were analyzed after 72h of incubation with imexon.
Fig. 5.



Fig. 5A Effects of imexon and eIF2B5 silencing on protein expression. MiaPaCa-2 cells were transfected with eIF2B5 siRNA or a non-targeting sequence (si CT) and examined for protein synthesis. Data are expressed as the percent of control, where the control is untreated cells transfected with a non-targeting siRNA (Mean ± SD, n=8). Imexon significantly inhibited protein synthesis in both si CT and si eIF2B5 transfected cells (*p ≤ 0.05). Knockdown of eiF2B5 also inhibits protein synthesis and is augmented by 250 μM IMX (^p = 0.0017). There is no significant augmentation of synthesis inhibition by 500 μM IMX (p = 0.75, si elF2B5 vs. si CT).
Fig. 5B Effect of NAC supplementation on protein synthesis inhibition by imexon. MiaPaCa-2 cells were pre-treated with 10 mM NAC overnight. NAC was either maintained (“continuous”) or the NAC removed (“pre-treated only”), in the presence of varying concentrations of imexon for 24h. Alternatively, the addition of NAC was delayed until 2h after imexon exposure (“post imexon”). Protein synthesis was measured by 3H-leucine incorporation. Data are expressed as percent of control, where the control is untreated cells (Mean ± SEM, n = 20). *p<0.05, **p<0.01
Fig 5C. Effect of ROS scavengers on protein synthesis inhibition by imexon. MiaPaCa-2 cells were pre-treated for 2h with the 100 U/ml of pegylated superoxide dismutase (PEG-SOD), 200 U/ml of pegylated catalase (PEG-CAT), or 100μM of 1-oxyl-2,2,6,6-tetramethyl-4-hydroxy- piperidine (tempol) before the addition of imexon for a total of 24h. Protein synthesis was measured by 3H-leucine incorporation. Data are expressed as percent of control, where the control is untreated cells (Mean ± SEM, n = 18).
Fig. 6.

Effect of imexon on proteins associated with ER translational control. The pancreatic cell lines MiaPaCa-2, Panc-1, or BxPC3 were incubated with the indicated concentration of imexon for 24h. Cell lysates were separated by SDS-PAGE and analyzed by immunoblotting. Data shown are representative of 3 independent experiments. eiF2B5, eiF2B2, p-EIF2α, eIF2Bα, and β-actin are 85 kDa, 39 kDa, 38 KDa, 38kDa, and 42 kDa, respectively.
Previous studies have demonstrated inhibition of protein synthesis in cells treated with imexon, but a molecular mechanism for the inhibition was not identified [9, 16]. To determine if the oxidative activity of imexon is responsible for the observed inhibition of protein translation, we examined new protein synthesis under three conditions: pre-treatment with the reducing agent N-acetyl cysteine (NAC) and continuous exposure during imexon incubation period; pre-treatment with NAC and wash out prior to imexon incubation; addition of NAC 2h after imexon treatment. Imexon mediated inhibition of protein synthesis was reversed only when NAC was present during the imexon incubation period (Fig. 5B). Furthermore, adding NAC 2h post imexon resulted in a partial reversal of imexon’s oxidative effects, demonstrating that NAC is not simply binding to imexon and preventing it’s uptake into the cell. These data were statistically significant only at the highest concentrations of imexon.
We also examined the effect of the ROS scavengers, PEG-SOD, PEG-CAT and tempol on imexon induced inhibition of protein synthesis (Fig. 5C). Although there was a trend toward reversal of protein synthesis inhibition by PEG-CAT, especially at the 250 μM imexon dose, the difference was not statistically significant between treatment groups (p=0.26846, p=0.38651, and p=0.74059 at 100, 250 and 500 μM imexon, respectively). Thus none of these ROS scavengers, except NAC, significantly reversed protein synthesis inhibition caused by imexon.
Protein synthesis is mediated by a series of eukaryotic translation initiation factors (eIFs) that are responsive to a variety of physiological conditions and cell stresses. One of the primary regulators of protein synthesis is the interaction between the classic G-protein, eIF2α and the guanine nucleotide exchange factor (GEF) eIF2B. Phosphorylation of eIF2α in response to ER stress increases its affinity for eIF2B thereby reducing the nucleotide exchange function and resulting in decreased translation initiation [17]. The current results show that imexon induces the phosphorylation of eIF2α and reduces protein synthesis to approximately the same extent as the silencing of the catalytic subunit of the eIF2B GEF, eIF2B5. These effects were not synergistic, suggesting a common pathway for the inhibitory activity.
The ER is the primary site of folding and post-translational modification of newly synthesized proteins. Protein folding in the ER requires the formation of disulfide bonds in a relatively oxidizing environment. This is accomplished via thiol-disulfide exchange reactions catalyzed by thiol-disulfide oxidoreductases, including protein disulfide isomerases (PDI) and the endoplasmic reticulum oxidase 1 (Ero1) family [18, 19]. The rate limiting step in oxidative protein folding is generally considered to be the terminal FAD-dependent electron transfer by Ero1α and regeneration of the fully oxidized PDI enzyme [19]. In order to catalyze these oxidative reactions, the active site disulfides in these enzymes need to be reduced, highlighting the intricate balance of oxidation and reduction that must be maintained for proper protein folding. Our data demonstrate that imexon maintains Ero1α in the oxidized, and thereby, inactive state, suggesting that disruption of the redox status of the ER may be part of imexon’s mechanism of action. Since previous studies have shown that imexon readily reacts with both cysteine and GSH in vitro [4], it is possible that imexon is directly binding intracellular GSH leading to a loss of reducing equivalents in the ER, and interfering with redox homeostasis.
In spite of the relatively low ratio of GSH:GSSG in the lumen of the ER (reported to range from 1:1 to 1:3), GSH has been proposed as the primary source of reducing equivalents for ER redox homeostasis [20, 21]. Previous work by Molteni et al. [22] and Chakravarthi et al [23] demonstrated the critical role of GSH as an antagonist of Ero1α. In these studies, depletion of GSH either via cell membrane permeabilization or inhibition of γ-glutamyl cysteine synthetase increased the rate of protein oxidation and lead to an accumulation of oxidized PDI and Ero1α. We cannot speculate as to whether an imexon-GSH conjugate is formed within the ER lumen, or if cytotosolic GSH depletion by imexon is sufficient to reduce the transport of GSH into the ER. Nonetheless, these data suggest that oxidative stress within the ER is induced by exposure to imexon.
The widespread introduction of RNA interference technology has provided an opportunity to systematically identify targets for drug response and resistance [24]. Several recent studies have highlighted the use of RNA interference chemosensitization screens to identify novel targets with chemotherapeutic agents [25, 26, 27]. For example, Giroux et al used a human kinome library to identify kinases that support the survival of pancreatic cell lines, and a subset that contribute to gemcitabine resistance [26]. In a similar study, Azorsa et al used siRNA’s to identify the checkpoint kinase 1 (CHK1) as a sensitizing target in pancreatic cancer cells exposed to gemcitabine [25]. Using a 21,000 member genome wide siRNA library, we identified molecular pathways associated with protein synthesis as potential mediators of imexon-induced growth inhibition in human pancreatic carcinoma. We initially hypothesized that this screen might highlight anti-oxidant response pathways. In contrast, the major class of gene products whose inhibition significantly enhanced the cytotoxic activity of imexon comprised genes involved in protein synthesis, and particularly eIF2B5.
Previous studies have established imexon as a pro-oxidant that can deplete intracellular thiols and induce oxidative stress [4, 28]. In addition, we have previously demonstrated that pancreatic cancer cell lines incubated with imexon display a marked inhibition of protein translation and degradation prior to commitment to cell death [9,16]. The new findings show that the addition of NAC blocks the effects of imexon on protein synthesis, suggesting that oxidative effects of the drug may mediate the reduction in protein synthesis. The effect of imexon on eIF2α is identical to that of H202, which typically inhibits translation by the phosphorylation of eIF2α [29]. However, none of the enzymatic inhibitors of H202 significantly blocked imexon’s effect on protein synthesis. This indirectly suggests that the oxidative stress induced by imexon may be mediated by cellular oxidants other than H202 and superoxide, but still causing phosphorylation of eIF2α.
In summary, the current findings extend the oxidizing effects of imexon from the mitochondria to the ER compartment. The data further suggest that the alteration in ER redox status may mediate the inhibitory effects of imexon on protein synthesis. However, the degree that ER protein oxidation and reduced protein synthesis contribute to overall imexon-induced tumor cell growth inhibition is not known. Importantly, the concentrations of imexon used to inhibit protein translation in the current experiments are within the range of concentrations that can be achieved in cancer patients [30]. For example, at the maximally tolerated phase I dose of 875 mg/m2/day, imexon peak plasma levels of 715 μM were routinely obtained.”
Acknowledgments
We would like to thank Geoffrey V. Grandjean for technical expertise in performing siRNA gene silencing studies.
Grant support
NIH CA017094 (RTD), NIH CA023074 (D Alberts), NIH CA077204 (GP), NIH CA120613 (GP)
Footnotes
Disclosures: Dr. Dorr is a corporate officer and unpaid consultant to AmpliMed Corporation (Tucson, AZ) and holds stock in AmpliMed Corporation. He is also an inventor that has established University of Arizona patents for imexon and other cyanoaziridine analogs that are licensed to AmpliMed Corporation. Dr. Landowski has received research laboratory funding from AmpliMed Corporation (Tucson, AZ).
Contributor Information
Elena V. Sheveleva, Email: lena.shevelev@gmail.com.
Terry H. Landowski, Email: tlandowski@azcc.arizona.edu.
Betty K. Samulitis, Email: bsamulitis@azcc.arizona.edu.
Geoffrey Bartholomeusz, Email: gbarthol@mdanderson.org.
Garth Powis, Email: gpowis@mdanderson.org.
Robert T. Dorr, Email: bdorr@azcc.arizona.edu.
References
- 1.Dorr RT, Raymond MA, Landowski TH, Roman NO, Fukushima S. Induction of apoptosis and cell cycle arrest by imexon in human pancreatic cancer cell lines. Int J Gastrointest Cancer. 2005;36:15–28. doi: 10.1385/IJGC:36:1:015. [DOI] [PubMed] [Google Scholar]
- 2.Roman NO, Samulitis BK, Wisner L, Landowski TH, Dorr RT. Imexon enhances gemcitabine cytotoxicity by inhibition of ribonucleotide reductase. Cancer Chemother Pharm. 2011;67:183–92. doi: 10.1007/s00280-010-1306-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cohen SJ, Zalupski MM, Modiano MR, Conkling P, Patt YZ, Davis P, et al. A phase I study of imexon plus gemcitabine as first-line therapy for advanced pancreatic cancer. Cancer Chemother Pharmacol. 2010;66:287–94. doi: 10.1007/s00280-009-1162-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iyengar BS, Dorr RT, Remers WA. Chemical basis for the biological activity of imexon and related cyanoaziridines. J Med Chem. 2004;47:218–23. doi: 10.1021/jm030225v. [DOI] [PubMed] [Google Scholar]
- 5.Dvorakova K, Payne CM, Tome ME, Briehl MM, McClure T, Dorr RT. Induction of oxidative stress and apoptosis in myeloma cells by the aziridine-containing agent imexon. Biochem Pharmacol. 2000;60:749–58. doi: 10.1016/s0006-2952(00)00380-4. [DOI] [PubMed] [Google Scholar]
- 6.Dvorakova K, Waltmire CN, Payne CM, Tome ME, Briehl MM, Dorr RT. Induction of mitochondrial changes in myeloma cells by imexon. Blood. 2001;97:3544–51. doi: 10.1182/blood.v97.11.3544. [DOI] [PubMed] [Google Scholar]
- 7.Dvorakova K, Payne CM, Landowski TH, Tome ME, Halperin DS, Dorr RT. Imexon activates an intrinsic apoptosis pathway in RPMI8226 myeloma cells. Anticancer Drugs. 2002;13:1031–42. doi: 10.1097/00001813-200211000-00007. [DOI] [PubMed] [Google Scholar]
- 8.Evens AM, Prachand S, Shi B, Paniaqua M, Gordon LI, Gartenhaus RB. Imexon-induced apoptosis in multiple myeloma tumor cells is caspase-8 dependent. Clin Cancer Res. 2004;10:1481–91. doi: 10.1158/1078-0432.ccr-1058-03. [DOI] [PubMed] [Google Scholar]
- 9.Samulitis BK, Landowski TH, Dorr RT. Inhibition of protein synthesis by imexon reduces HIF-1alpha expression in normoxic and hypoxic pancreatic cancer cells. Invest New Drugs. 2009;27:89–98. doi: 10.1007/s10637-008-9149-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sevier CS. New insights into oxidative folding. J Cell Biol. 2010;188:757–58. doi: 10.1083/jcb.201002114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bartholomeusz G, Cherukuri P, Kingston J, Cognet L, Lemos R, Leeuw TK, et al. In vivo therapeutic silencing of hypoxia-inducible factor 1 alpha (HIF-1α) using single-walled carbon nanotubes noncovalently coated with siRNA. Nano Res. 2009;2:279–91. doi: 10.1007/s12274-009-9026-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O’Brien J, Wilson I, Orton T, Pognan F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur J Biochem. 2000;267:5421–6. doi: 10.1046/j.1432-1327.2000.01606.x. [DOI] [PubMed] [Google Scholar]
- 13.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 14.Benham AM, Cabibbo A, Fassio A, Bulleid N, Sitia R, Braakman I. The CXXCXXC motif determines the folding, structure and stability of human Ero1-Lalpha. EMBO J. 2000;19:4493–502. doi: 10.1093/emboj/19.17.4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–33. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 16.Hersh EM, Gschwind CR, Taylor CW, Dorr RT, Taetle R, Salmon SE. Antiproliferative and antitumor activity of the 2-cyanoaziridine compound imexon on tumor cell lines and fresh tumor cells in vitro. J Natl Cancer Inst. 1992;84:1238–44. doi: 10.1093/jnci/84.16.1238. [DOI] [PubMed] [Google Scholar]
- 17.Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. 2005;307:935–9. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- 18.Ellgaard L, Ruddock LW. The human protein disulphide isomerase family: substrate interactions and functional properties. EMBO Rep. 2005;6:28–32. doi: 10.1038/sj.embor.7400311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hatahet F, Ruddock LW. Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxid Redox Signal. 2009;11:2807–50. doi: 10.1089/ars.2009.2466. [DOI] [PubMed] [Google Scholar]
- 20.Hwang C, Sinskey AJ, Lodish HF. Oxidized redox state of glutathione in the endoplasmic reticulum. Science. 1992;257:1496–502. doi: 10.1126/science.1523409. [DOI] [PubMed] [Google Scholar]
- 21.Bass R, Ruddock LW, Klappa P, Freedman RB. A major fraction of endoplasmic reticulum-located glutathione is present as mixed disulfides with protein. J Biol Chem. 2004;279:5257–62. doi: 10.1074/jbc.M304951200. [DOI] [PubMed] [Google Scholar]
- 22.Molteni SN, Fassio A, Ciriolo MR, Filomeni G, Pasqualetto E, Fagioli C, et al. Glutathione limits Ero1-dependent oxidation in the endoplasmic reticulum. J Biol Chem. 2004;279:32667–73. doi: 10.1074/jbc.M404992200. [DOI] [PubMed] [Google Scholar]
- 23.Chakravarthi S, Bulleid NJ. Glutathione is required to regulate the formation of native disulfide bonds within proteins entering the secretory pathway. J Biol Chem. 2004;279:39872–9. doi: 10.1074/jbc.M406912200. [DOI] [PubMed] [Google Scholar]
- 24.Iorns E, Lord CJ, Turner N, Ashworth A. Utilizing RNA interference to enhance cancer drug discovery. Nat Rev Drug Discov. 2007;6:556–68. doi: 10.1038/nrd2355. [DOI] [PubMed] [Google Scholar]
- 25.Whitehurst AW, Bodemann BO, Cardenas J, Ferguson D, Girard L, Peyton M, et al. Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature. 2007;446:815–19. doi: 10.1038/nature05697. [DOI] [PubMed] [Google Scholar]
- 26.Azorsa DO, Gonzales IM, Basu GD, Choudhary A, Arora S, Bisanz KM, et al. Synthetic lethal RNAi screening identifies sensitizing targets for gemcitabine therapy in pancreatic cancer. J Transl Med. 2009;7:43. doi: 10.1186/1479-5876-7-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giroux V, Iovanna J, Dagorn JC. Probing the human kinome for kinases involved in pancreatic cancer cell survival and gemcitabine resistance. FASEB J. 2006;20:1982–91. doi: 10.1096/fj.06-6239com. [DOI] [PubMed] [Google Scholar]
- 28.Samulitis BK, Landowski TH, Dorr RT. Correlates of imexon sensitivity in human multiple myeloma cell lines. Leuk Lymphoma. 2006;47:97–109. doi: 10.1080/10428190500266210. [DOI] [PubMed] [Google Scholar]
- 29.Grant CM. Regulation of translation by hydrogen peroxide. Antioxid Redox Signal. 2011;15:191–203. doi: 10.1089/ars.2010.3699. [DOI] [PubMed] [Google Scholar]
- 30.Dragovich T, Gordon M, Mendelson D, Wong L, Modiano M, Chow HH, Samulitis B, O’ Day S, Grenier K, Hersh E, Dorr R. Phase I trial of imexon in patients with advanced malignancy. J Clin Oncol. 2007;25:1779–84. doi: 10.1200/JCO.2006.08.9672. [DOI] [PubMed] [Google Scholar]