

Abstract
BACKGROUND
Chronic alcohol abuse is a comorbid variable of Acute Respiratory Distress Syndrome (ARDS). Previous studies showed that, in the lung, chronic alcohol consumption increased oxidative stress and impaired alveolar macrophage (AM) function. NADPH oxidases (Nox) are the main source of reactive oxygen species (ROS) in AMs. Therefore, we hypothesized that chronic alcohol consumption increases AM oxidant stress through modulation of Nox1, Nox2 and Nox4 expression.
METHODS
AMs were isolated from male C57BL/6J mice, aged 8-10 weeks, which were treated ± ethanol in drinking water (20% w/v, 12 weeks). MH-S cells, a mouse AM cell line, were treated ± ethanol (0.08%, 3 days) for in vitro studies. Selected cells were treated with apocynin (300 μM), a Nox1 and Nox2 complex formation inhibitor, or were transfected with Nox siRNAs (20-35 nM), prior to ethanol exposure. Human AMs were isolated from alcoholic and control patients’ bronchoalveolar lavage fluid. Nox mRNA levels (qRT-PCR), protein levels (western blot and immunostaining), oxidative stress (DCFH-DA and Amplex Red analysis), and phagocytosis (S. aureus internalization) were measured.
RESULTS
Chronic alcohol increased Nox expression and oxidative stress in mouse AMs in vivo and in vitro. Experiments using apocynin and Nox siRNAs demonstrated that ethanol-induced Nox4 expression, oxidative stress, and AM dysfunction were modulated through Nox1 and Nox2 upregulation. Further, Nox1, Nox2 and Nox4 protein levels were augmented in human AMs from alcoholics compared with controls.
CONCLUSIONS
Ethanol induces AM oxidative stress initially through upregulation of Nox1 and Nox2 with downstream Nox4 upregulation and subsequent impairment of AM function.
Keywords: ethanol, alveolar macrophage, NADPH oxidases, oxidative stress
INTRODUCTION
Acute respiratory distress syndrome (ARDS) is a severe form of lung injury with a 26% mortality rate (1). Disease pathogenesis is characterized by the development of pulmonary edema and inflammation in response to trauma, sepsis, or aspiration which results in activation of systemic proinflammatory cascades (2). Chronic alcohol abuse is a co-morbid variable associated with increased ARDS susceptibility (3). Specifically, the incidence of ARDS in alcoholic patients was 43%, compared with 22% in non-alcoholic patients (4). Chronic alcohol ingestion predisposes patients to develop ARDS through multiple mechanisms, of which, enhanced oxidative stress plays a key role (5-8). Available evidence suggests that chronic alcohol consumption reduces levels of the critical antioxidant glutathione (GSH) in bronchoalveolar lavage (BAL) fluid (9) and alveolar macrophages (AMs) (10). Further, clinical and animal studies show that GSH depletion in the alveolar space is associated with chronic oxidative stress and AM dysfunction (8, 10-12). AMs play an important role in innate and acquired immunity (13) because of their ability to phagocytose and clear apoptotic cells and infectious particles (14). Previous investigations demonstrate in animal models that the ability of AMs to bind and internalize inactive Staphylococcus aureus is impaired with chronic ethanol ingestion (11) and that treatment with a GSH precursor improves AM phagocytosis in vivo (10). Collectively, these studies demonstrate that chronic alcohol ingestion causes reduced GSH and oxidant stress in the alveolar space leading to AM dysfunction (15). However, the specific mechanisms by which ethanol cause AM dysfunction have yet to be clearly defined.
Reactive oxygen species (ROS) mediate complex physiological processes such as cell signaling and apoptosis (16, 17) and play critical roles in the pathogenesis of various diseases. NADPH oxidases (Nox) (18) within AMs are the main source of ROS generation in the lungs under physiologic conditions (19). In AMs, the primary ROS generated by Nox proteins is superoxide, a reactive species that is essential to the respiratory burst involved in the killing of microbes after phagocytosis (18).
Nox proteins are multi-component, membrane-associated enzymes that use NADPH as an electron donor to catalyze the reduction of molecular oxygen to superoxide and hydrogen peroxide (20). Nox1 (21-23), Nox2 (21-23), and Nox4 (21-23) are expressed in the human lung. p22phox, a transmembrane subunit, interacts with these active and inactive Noxes (24-26). Nox1 is primarily activated by interactions with the cytosolic subunits NoxO1, NoxA1 and GTP-Rac (27, 28). However, NoxO1 and NoxA1 can be replaced by p47phox and p67phox, respectively (28, 29). Nox2 activation involves association with p47phox, p67phox, p40phox and GTP-Rac (30) and is responsible for respiratory burst in alveolar macrophages (23). Nox4 associates with p22phox to produce ROS (31) and has been implicated in various physiological processes, including cellular senescence (32), differentiation (33-36) and oxygen sensing (37). Although Nox4 is constitutively active, its expression and/or activity can be increased through several pathways, including: angiotensin II binding to the angiotensin II type 1 receptor, insulin activation of the insulin receptor, TGFβ1 binding to the TGFβR (20), and Poldip2 association with p22phox (38). In the human lung, Nox1, Nox2 and Nox4 constitute critical sources of ROS generation in response to ethanol exposure in mouse embryos (39). Furthermore, chronic ethanol exposure increased the expression of these Noxes in the mouse lung (23). Taken together, these findings suggest that NADPH oxidases may play an important role in ethanol-induced oxidative stress and pathogenesis of lung injury (23).
The objective of this study is to define the molecular mechanisms by which chronic alcohol ingestion mediates oxidant stress in AMs. We hypothesize that chronic alcohol consumption augments oxidant stress in AMs through modulation of Nox expression. The investigations presented herein demonstrate that ethanol induces Nox1 and Nox2 expression in the AM which, in turn, enhance Nox4 expression, resulting in intracellular production of superoxide and hydrogen peroxide.
MATERIALS & METHODS
Mouse model of chronic ethanol consumption
All animal studies were performed in accordance with NIH guidelines outlined in the Guide for the Care and Use of Laboratory Animals, as described in protocols reviewed and approved by the Emory University Institutional Animal Care and Use Committee. Male C57BL/6J mice (Jackson Laboratory, Bar Harbor, ME), aged 8-10 weeks, were given either no ethanol or ethanol (5-20% w/v, starting at 5% and increasing by 5% increments each week for two weeks to help the animals acclimate to this intervention). Ethanol-fed mice then received 20% w/v for the following ten weeks in their drinking water (n=5 per group). This method replicates blood alcohol levels following chronic ethanol consumption in human subjects, as assessed by studies using pair-fed ethanol-treated female or male C57BL/6J or BALB/c mice (40, 41). Male Nox2 knockout mice (Jackson Laboratory), generated as previously described on a C57BL/6J background (42), were fed ethanol using the same protocol. Following sacrifice, tracheas from all mice were cannulated, and bronchoalveolar lavage (BAL) fluid was collected via tracheotomy. Mouse alveolar macrophages (mAM) were then isolated from the fluid by centrifugation at 8000 rpm for 5 min. The cell pellet was resuspended in RPMI-1640 medium containing 2% fetal bovine serum (FBS) and 1% penicillin/streptomycin. After staining with Diff-Quik (Dade Behring, Newark, DE) and counting with a hemocytometer, the cell population was determined by differential staining to be ~95% alveolar macrophages (15). The macrophages were then plated overnight in RPMI-1640 medium containing 2% FBS and 1% penicillin/streptomycin at 37°C in 5% CO2 atmosphere, prior to beginning experiments.
MH-S and mAM cell culture and ethanol stimulation
The mouse alveolar macrophage cell line, MH-S (ATCC, Manassas, VA), was used as a model system for studying effects of ethanol in vitro. Cells were cultured in RPMI-1640 media containing 10% FBS and 1% penicillin/streptomycin. 24 hrs after plating, MH-S cells were cultured in media containing 2% FBS in a modular incubation chamber (Billups-Rothenberg, Inc., Del Mar, CA). Selected cells were treated with 0.08% ethanol for 1-3 days as indicated. Selected cells were also treated with 300 μM apocynin (Sigma-Aldrich, Inc., St. Louis, MO), an inhibitor of cytosolic p47phox translocation to the cell membrane (43), for the duration of ethanol exposure.
Mouse alveolar macrophages isolated from wildtype, Nox1 knockout (Jackson Laboratory), and Nox2 knockout (gift from Dr. Kathy Griendling, Emory University) were used to study the effects of ethanol exposure ex vivo. Cells were cultured in RPMI-1640 media containing 5% FBS and 1% penicillin/streptomycin. 24 hrs after plating, these mAMs were cultured in media containing 2% FBS in a modular incubation chamber (Billups-Rothenberg, Inc.). Selected cells were treated with 0.08% ethanol for 3 days or as indicated. These studies were also performed in accordance with NIH guidelines outlined in the Guide for the Care and Use of Laboratory Animals, as described in protocols reviewed and approved by the Emory University Institutional Animal Care and Use Committee.
Estimation of cellular ROS production
mAMs from control and ethanol-fed mice, or untreated and ethanol-treated MH-S and mAM cells, were cultured in RPMI-1640 media containing 2% FBS for 24h, prior to the start of the experiment. Intracellular ROS production in mAM or MH-S cells were determined using 2’,7’-dichlorofluorescein-diacetate (DCFH-DA) dye as described previously (44). In brief, cells were incubated with 5 μM DCFH-DA in RPMI at 37°C for 30 min in the dark, and then washed with phosphate-buffered saline 3 times to remove excess dye. Fluorescence was measured using FluoView (Olympus, Melville, NY) via quantitative digital analysis. ROS production values are expressed as mean ± SEM, relative to average control values. H2O2 released into media collected from mAM or MH-S cells was determined using the Amplex Red assay (Invitrogen, Carlsbad, CA), according to the manufacturer’s protocol. In brief, cells were incubated with 500 μL Amplex Red (20 μM) and horseradish peroxidase (0.1 U/ml) at 37°C for 30 minutes. The reaction mixtures were then measured for fluorescence in duplicate (excitation 540 nm, emission 590 nm), and H2O2 concentrations were calculated utilizing standard curves generated with reagent H2O2. Cell cultures were then lysed in 100 μL cell lysis buffer, and centrifuged at 12,000 rpm for 10 min. Supernatant protein concentrations were determined using a BCA assay. H2O2 concentrations were normalized to cellular protein concentrations and are expressed as mean ± SEM, relative to average control values.
RNA isolation and quantitative RT-PCR (qRT-PCR)
mAMs were isolated from BAL fluid of control and ethanol-fed mice, and total RNA was extracted using TRIzol reagent (Invitrogen). Cultured MH-S and mAM cells were treated with or without ethanol (0.08%) for three days, followed by RNA extraction. mRNA expression was determined and quantified using specific mRNA primers given in Table I. Using the iScript One-Step RT-PCR kit with SYBR Green (Bio-Rad, Hercules, CA), real-time qRT-PCR of total RNA (100 ng) was performed using the Applied Biosystems ABI Prism 7500 version 1.4 sequence detection system under the following conditions: cDNA synthesis at 50°C for 10 min, iScript reverse transcriptase inactivation at 95°C for 5 min, and PCR for 40 cycles entailing 95°C for 10 s, followed by annealing at 60°C for 1 min and detection. Values are expressed as the relative expression of mRNA normalized to 9s mRNA.
Table I.
Mouse and human primer sequences to measure mRNA expression using quantitative RT-PCR.
| Primer | Forward Sequence (5’-3’) | Reverse Sequence (5’-3’) |
|---|---|---|
| Nox1 | CGCTCCCAGCAGAAGGTCGTGATTACCAAG | GGAGTGACCCCAATCCCTGCCCCAACCA |
| Nox2 | GTCACACCCTTCGCATCCATTCTCAAGTCAGT | CTGAGACTCATCCCAGCCAGTGAGGTAG |
| Nox4 | CTGGAGGAGCTGGCTCGCCAACGAAG | GTGATCATGAGGAATAGCACCACCACCATGCAG |
| p22phox | AACGAGCAGGCGCTGGCGTCCG | CACAGTGGTATTTCGGCGCC |
| p47phox | CCAGCACTATGTGTACATGT | AAGGAGATGTTCCCCATTGA |
| p67phox | GCCTTCTGGTAAGCACCTAA | GAGGCCTTCAGCGAGGTGCA |
| 9s | GAGCTAGCCTCTGCCAGAGG | TCCAAGCCTCAAGACAGGAA |
Western blot analysis for MH-S cells
Proteins were isolated from MH-S cells using a cell lysis buffer consisting of 2.5 mM EDTA, 20 mM Tris pH 7.4, 100 mM NaCl, 1 mM Na3VO4, 1% Triton X-100, 10 mM NaF, 1% sodium deoxycholate, 0.1% SDS, 2.5 mM sodium pyrophosphate, 1 mM β-glycerolphosphate, and 1 tablet/10 ml EDTA-free complete protease inhibitor cocktail (Roche, Indianapolis, IN). Whole cell extracts prepared from untreated and ethanol-treated MH-S cells (40 μg/lane) were resolved in 4-12% bis-tris polyacrylamide gels (Invitrogen), followed by transfer to nitrocellulose membranes. Membranes were probed with primary antibodies for Nox1 (Santa Cruz Biotechnology, Santa Cruz, CA; 1:100), Nox2 (Abcam, Cambridge, MA; 1:1000), Nox4 (gift from Dr. David Lambeth, Emory University; 1:2500), or GAPDH (Santa Cruz Biotechnology; 1:100). Proteins were visualized by incubation with peroxidase-coupled anti-rabbit or anti-goat IgG (1:2000) in the presence of LumiGlo reagent while exposing in a Bio-Rad Chemidoc XRS/HQ. Densitometric analysis was performed using Bio-Rad Quantity One (Version 4.5.0) software. Values are expressed as the relative expression of protein normalized to GAPDH protein.
Phagocytosis
Phagocytic ability of MH-S cells treated with or without ethanol, in the presence or absence of apocynin, and mAMs treated with or without ethanol (0.08%) ex vivo for 3 days was assessed as previously described (15). In brief, cells were incubated with 106 particles of pH-sensitive pHrodo Staphylococcus aureus BioParticles conjugate (Invitrogen) for 2 hours and then fixed with 4% paraformaldehyde. Phagocytosis of bacteria and microbial killing by lysosomes was analyzed using an Olympus confocal microscope containing an argon/krypton laser. Cells from 10 fields per experimental condition were assessed using quantitative digital fluorescence imaging software (Olympus FluoView 300, Version 4.3). To measure S. aureus internalization, laser confocal microscopy was performed at 50% of cell depth using identical background and gain settings. MH-S cells with internalized bacteria were considered positive for phagocytosis. Phagocytosis was quantified by phagocytic index, which is calculated from the percentage of phagocytic cells multiplied by the relative fluorescence units of S. aureus per cell.
Confocal Immunostaining of hAMs
Alcoholic patients (n=5) and non-alcoholic control subjects (n=5) were recruited from the Substance Abuse Treatment Program at the Atlanta Veterans Affairs (VA) Medical Center. The Short Michigan Alcoholism Screening Test (SMAST) questionnaire was administered to each patient, and those with a score of >3 were enrolled in the study. Other inclusion criteria were daily or almost daily alcohol abuse, where the last alcoholic drink was <8 days prior to bronchoscopy. Patients were excluded if they: primarily abused substances other than alcohol, currently had other medical problems requiring ongoing active management (other than alcohol abuse), were HIV positive, were >55 years old, and had abnormal chest radiographs. Alcoholic patients were recruited and matched with healthy controls for age, race, gender and smoking status. Prior to implementation, this project was reviewed and approved by both the Emory University Institutional Review Board and the Atlanta VA Medical Center Research and Development Committee.
Fiberoptic bronchoscopy was performed in control and chronic alcoholic patients using topical anesthesia, and bronchoalveolar lavage was performed in the right middle lobe, as previously described (45). Sterile saline (150 ml) was suffused and withdrawn by suction in three 50 ml aliquots. The BAL fluid was passed through sterile gauze and centrifuged at 8000 rpm for 5 min. The cell pellet of alveolar macrophages (purity of AMs ~90% as measured by Diff-Quik (Dade Behring) staining and cell counting (15)) was resuspended in RPMI-1640 medium containing 2% FBS and 1% penicillin/streptomycin and cultured for 24 h. Total RNA was isolated from these hAMs, and mRNA expression was determined and quantified using specific mRNA primers for Nox1, Nox2, and Nox4, given as homologous mouse and human sequences in Table I, as described above. Selected hAMs cells were fixed to chamber slides with 4% paraformaldehyde. Cells were incubated with primary antibodies for Nox1 (Santa Cruz Biotechnology; 1:100), Nox2 (Abcam; 1:100) and Nox4 (Santa Cruz Biotechnology; 1:100), followed by incubation with fluorescent TRITC-labeled secondary antibodies. Fluorescence was measured using FluoView (Olympus) via quantitative digital analysis. Values are expressed as mean ± SEM relative fluorescent units (RFU) per cell.
Transient transfection of MH-S cells
Expression of Nox1, Nox2 or Nox4 was attenuated using respective siRNAs or control siRNA (Qiagen, Valencia, CA). At ~50% confluence, MH-S cells were incubated with the transfection reagent, Gene Silencer (Genlantis, San Diego, CA) and Nox1 (20 nM), Nox2 (20 nM) or Nox4 siRNAs (35 nM) for 4 hr in serum-free RPMI-1640 media, following the manufacturer’s recommendations. Control siRNA concentrations were adjusted accordingly for comparison. For example, 20 nM of Nox1 and Nox2 siRNA were compared to 20 nM of control siRNA, and 35 nM of Nox4 siRNA was compared to 35 nM of control siRNA. However, because no significant difference was observed between 20 nM and 35 nM control siRNA, these results were combined and are presented as a single siRNA value. Complete media containing 10% FBS with or without ethanol was then added to the cells for 3 days. Nox1, Nox2 and Nox4 mRNA and protein levels in transiently transfected MH-S cells were measured using real-time PCR and Western blot analysis.
Statistical Analysis
Data are represented as means ± SEM. Statistical significance was calculated using one-way ANOVA followed by Tukey-Kramer test to detect differences between individual groups using GraphPad Prism version 5 (GraphPad, San Diego, CA). p<0.05 was considered statistically significant.
RESULTS
Chronic alcohol exposure increased ROS generation in mAMs in vivo and in vitro
mAMs were isolated from control and ethanol-fed mice (20% w/v ethanol for 12 weeks) to evaluate the effects of ethanol on AM oxidative stress in vivo. For in vitro experiments, MH-S cells were treated with or without ethanol (0.08%) for 3 days. In both in vivo and in vitro experiments, alveolar macrophage oxidative stress was determined in response to ethanol using DCFH-DA fluorescence to measure reactive oxygen species (ROS) production, and Amplex Red to determine hydrogen peroxide generation. ROS production by AMs from ethanol-fed mice increased by 4.7-fold (Con = 3.2 × 105 relative fluorescence units and EtOH = 1.5×106 relative fluorescence units) (Fig. 1A) and hydrogen peroxide generation by 3.9-fold (Con = 1.2 μM and EtOH = 4.6 μM) (Fig. 1B), compared with AMs control animals. Similarly, compared with untreated MH-S cells, ethanol stimulated cells exhibited increased generation of ROS by 1.8-fold (None = 4.4 × 105 relative fluorescence units and EtOH = 0.8×106 relative fluorescence units) (Fig. 1C) and hydrogen peroxide by 2.8-fold (None = 1.1μM and EtOH = 3.1 μM) (Fig. 1D). These findings indicated that ethanol stimulated ROS generation in mAMs both in vivo and in vitro.
Figure 1.
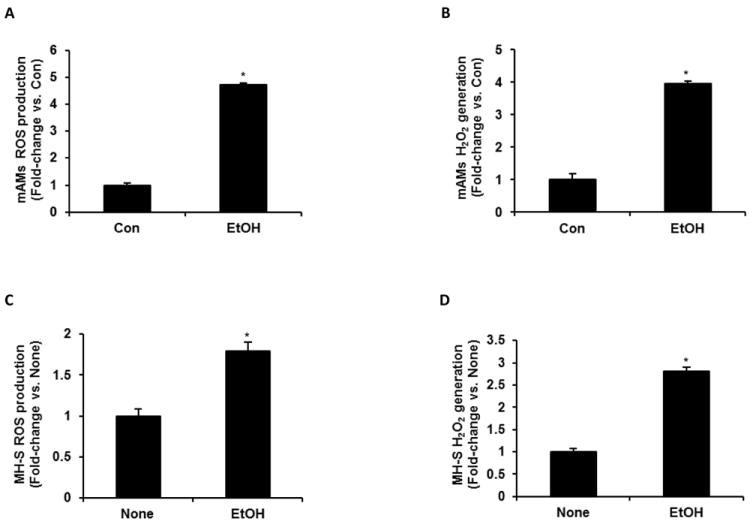
Ethanol induced ROS generation in mAMs. In mAMs collected from control (Con) and ethanol-fed (EtOH) mice (n=5, in duplicate), ROS production was measured by DCFH-DA fluorescence assay (A), and H2O2 generation was measured by Amplex Red assay (B). In cultured MH-S cells that were either untreated (None) or ethanol-treated (EtOH, 0.08%) for 3 days (n=3 independent experiments, in duplicate), ROS production (C) and H2O2 generation (D) were measured. H2O2 values were normalized to cellular protein concentration. All values are expressed as mean ± SEM, relative to control or no treatment. *p<0.05, EtOH vs. Con or None.
Chronic ethanol exposure increased NADPH oxidase (Nox) expression
Since previous studies showed that NADPH oxidases are major producers of ROS (20), we determined the expression of AM Nox subunits after ethanol treatment. Compared with AMs isolated from control-fed mice, AMs isolated from ethanol-fed mice exhibited increased mRNA expression levels of Nox1 (3.8-fold), Nox2 (5.6-fold), Nox4 (4.1-fold), p22phox (3.2-fold), p47phox (2.5-fold) and p67phox (2.8-fold) (Fig. 2A). As shown in Fig. 2B, similar results were seen in ethanol treated MH-S cells, where ethanol increased the mRNA expression of Nox1 (3.0-fold), Nox2 (5.5-fold), Nox4 (6.4-fold), and p22phox (7.0-fold), as well as the regulatory subunits of Nox1 and Nox2, p47phox (4.8-fold) and p67phox (4.9-fold). As shown in Fig. 2C, ethanol-induced increases in NADPH oxidase mRNA expression were associated with similar increases in protein levels. Ethanol also increased Nox1, Nox2 and, more dramatically, Nox4 protein levels in ethanol treated MH-S cells (Fig. 2C). Collectively, these data indicated that chronic ethanol exposure enhanced NADPH oxidase mRNA and protein expression in mouse alveolar macrophages, and highlights a mechanism by which ethanol contributed to increased ROS and oxidative stress in alveolar macrophage.
Figure 2.
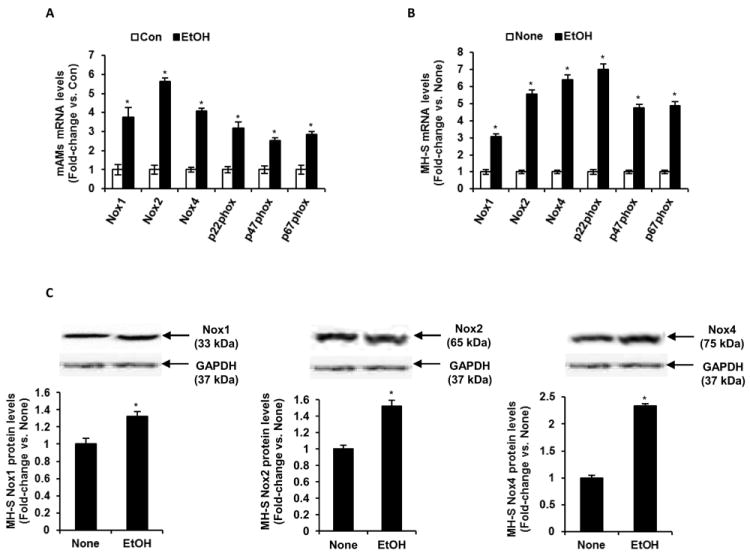
Ethanol induced Nox mRNA and protein expression levels in vivo and in vitro. A, mAMs were collected from control (Con) and ethanol-fed (EtOH) mice. mRNA levels of Nox1, Nox2, Nox4, p22phox, p47phox and p67phox in these mAMs were measured (n=5). B, Cultured MH-S cells were either untreated (None) or ethanol-treated (EtOH, 0.08%) for 3 days. mRNA levels of Nox1, Nox2, Nox4, p22phox, p47phox and p67phox in these MH-S cells were measured (n=3 independent experiments). All mRNA values were measured by qRT-PCR, normalized to 9s mRNA, and expressed as mean ± SEM, relative to no treatment or control. C, Protein expression of Nox1, Nox2 and Nox4 in cultured MH-S cells either untreated (None) or ethanol-treated (EtOH, 0.08%) for 3 days. Protein values in these MH-S cells were assessed by Western blotting and densitometric analysis, normalized to GAPDH protein levels (n=3 independent experiments), and expressed as mean ± SEM, relative to no treatment. *p<0.05, EtOH vs. Con or None.
Apocynin inhibited ethanol-induced Nox expression, oxidative stress, and AM dysfunction in vitro
To further examine potential interrelationships between ethanol-induced alterations in NADPH oxidase subunits, time course studies following ethanol stimulation were performed. The mRNA expression of Nox1 and Nox2 in MH-S cells increased after 6 hours of ethanol exposure, whereas Nox4 mRNA levels increased after 12 hours (Fig. 3A). Further, in mAMs treated with ethanol ex vivo, Nox1 and Nox2 mRNA expression also increased after 6 hours and Nox4 mRNA levels increased after 12 hours (Fig. 3B). These results suggested that ethanol-induced Nox1 and/or Nox2 induction could contribute to Nox4 induction. Although our data demonstrate that ethanol exposure for 12 hours was sufficient to increase Nox expression, subsequent studies were performed using ethanol exposure for 3 days to more accurately model prolonged exposure to elevated levels of alcohol in patients with a history of chronic alcohol abuse.
Figure 3.
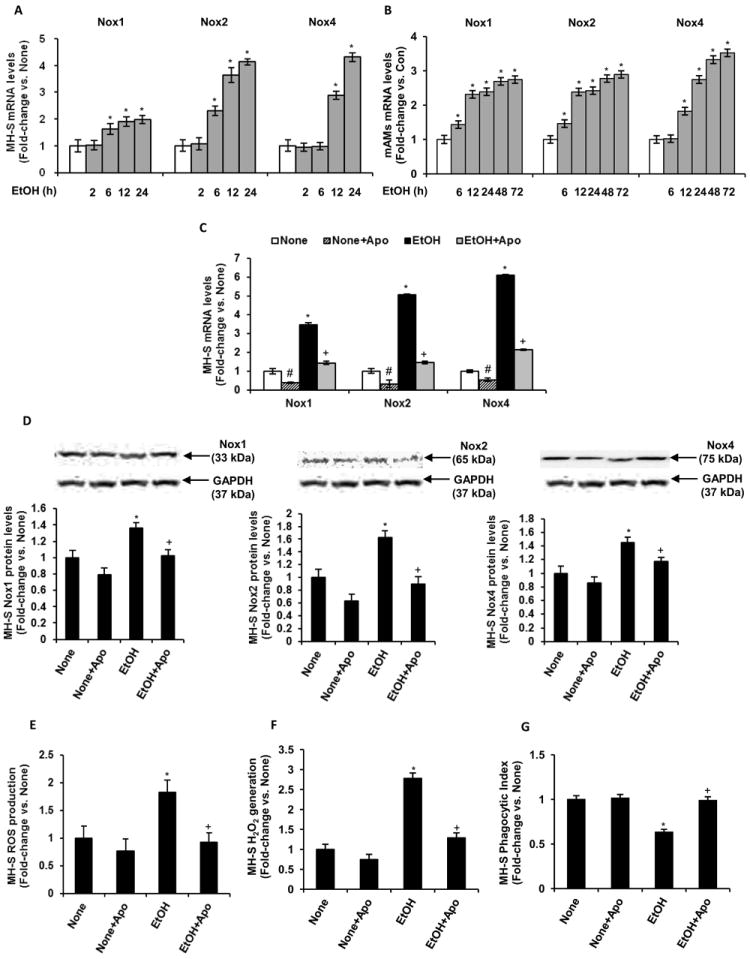
Ethanol mediated Nox4 expression and ROS generation in MH-S cells via regulation of Nox1 and Nox2. Cultured MH-S cells were either untreated (None) or ethanol-treated (EtOH, 0.08%) for 3 days or for the indicated durations. A, Time-course of Nox1, Nox2 and Nox4 mRNA expression in these MH-S cells treated with ethanol for 2, 6, 12 or 24 hours (n=3 independent experiments). *p<0.05, EtOH vs. None. B, Time-course Nox1, Nox2 and Nox4 mRNA expression in mAMs treated ex vivo without (Con) or with ethanol (EtOH, 0.08%) for 6, 12, 24, 48 or 72 hours (n=3 independent experiments). *p<0.05, EtOH vs. Con. Where indicated, cultured MH-S cells were treated with 300 μM apocynin (Apo) for 3 days, with or without ethanol exposure. Nox1, Nox2 and Nox4 mRNA (C) and protein (D) levels were measured in these MH-S cells (n=3-9 independent experiments). mRNA levels were measured by qRT-PCR analysis and normalized to 9s mRNA. #p<0.05, None+Apo vs. None; *p<0.05, EtOH vs. None; +p<0.05, EtOH+Apo vs. EtOH. Protein levels were measured by Western blotting analysis and normalized to GAPDH protein levels. E, Reactive oxygen species production was measured by DCFH-DA fluorescence assay (n=3 independent experiments, in duplicate). F, H2O2 generation was measured by Amplex Red assay (n=3 independent experiments, in duplicate). H2O2 values were normalized to cellular protein concentration. G, Phagocytic ability was assessed by phagocytosis assay (n=3 independent experiments, 10 fields per experimental condition). Phagocytic index was calculated from the percentage of phagocytic cells multiplied by the relative fluorescence units of S. aureus per cell. All values are expressed as mean ± SEM, relative to no treatment. *p<0.05, EtOH vs. None; +p<0.05, EtOH+Apo vs. EtOH.
To investigate potential interrelationships among Nox1, Nox2 and Nox4, MH-S cells were treated with and without 300 μM apocynin. In control MH-S cells, apocynin attenuated mRNA expression of Nox1, Nox2 and Nox4 (Fig. 3C). However, this did not result in decreased protein expression in Nox1, Nox2 or Nox4 during the 3 day culture period (Fig. 3D). In the ethanol ± apocynin group, apocynin inhibits Nox1 and Nox2 complex formation by impairing cytosolic p47phox translocation to the cell membrane (43). As shown in Fig. 3C, apocynin attenuated ethanol-induced mRNA expression of Nox1 by 82±8.6%, Nox2 by 89±5.6% and Nox4 by 78±4.9%. Apocynin treatment also abrogated ethanol-mediated increases in Nox1, Nox2 and Nox4 protein levels (Fig. 3D). Treatment with apocynin additionally attenuated ethanol-induced ROS production completely, as measured by DCFH-DA analysis (Fig. 3E), and reduced hydrogen peroxide generation by 84±1.4%, as measured by Amplex Red assay (Fig. 3F). Further, apocynin treatment completely reversed ethanol-mediated AM dysfunction (Fig. 3G) to rescue phagocytic ability.
Nox1 and Nox2 participate in ethanol-mediated Nox4 expression
Since apocynin treatment inhibits both Nox1 and Nox2 through regulation of their p47phox subunit, we used a siRNA silencing approach to determine which Nox protein may be specifically responsible for regulating Nox4 expression. MH-S cells transfected with Nox1 siRNA had reduced basal and ethanol-induced levels of Nox1 and Nox4 mRNA, whereas Nox2 mRNA expression was unaffected (Fig. 4A). Similarly, cells transfected with Nox2 siRNA exhibited diminished basal and ethanol-induced levels of Nox2 and Nox4 mRNA, whereas Nox1 mRNA expression was unaffected (Fig. 4B). Cells transfected with both Nox1 plus Nox2 siRNAs demonstrated reduced levels of Nox1, Nox2 and Nox4 mRNA expression (Fig. 4C). MH-S cells transfected with Nox4 siRNA had reduced basal and ethanol-induced Nox4 mRNA levels, whereas Nox1 and Nox2 levels were not altered (Fig. 4D).
Figure 4.
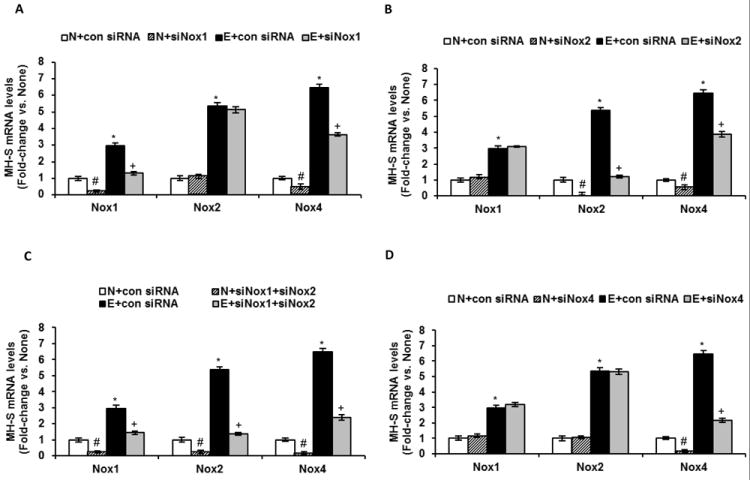
Ethanol mediated Nox4 mRNA expression in MH-S cells via upregulation of Nox1 and Nox2. Cultured MH-S cells were either untreated (N) or ethanol-treated (E, 0.08%) for 3 days. mRNA was isolated and Nox1, Nox2 and Nox4 mRNA levels were measured in Nox1 siRNA transfected cells (A), Nox2 siRNA transfected cells (B), Nox1+Nox2 siRNA co-transfected cells (C), and Nox4 siRNA transfected cells (D) by qRT-PCR and normalized to 9s mRNA (n=3-6 independent experiments). #p<0.05, N+siRNA vs. N; *p<0.05, E vs. N; +p<0.05, E+siRNA vs. E.
As shown in Figs. 5A-C, the effects of these siRNAs on ethanol-induced Nox mRNA expression were reflected in the protein levels of ethanol treated MH-S cells. Ethanol-induced Nox1 protein levels were completely attenuated with Nox1 siRNA (112±9.4%) and Nox1+Nox2 siRNAs (126±11.1%), but were unaffected by Nox2 and Nox4 siRNAs (Fig. 5A). Ethanol-mediated Nox2 protein levels were abrogated with Nox2 siRNA (84±10.5%) and Nox1+Nox2 siRNAs (123±11%), but were unchanged by Nox1 and Nox4 siRNAs (Fig. 5B). As shown in Fig. 5C, Nox4 protein levels augmented by ethanol were reduced with siRNAs for Nox1, Nox2, Nox1+Nox2, and Nox4. In vivo, a similar effect was seen in mAMs from commercially available (Jackson Laboratory) Nox2 knockout mice (Fig. 5D). Compared with control wild-type mice, control Nox2 knockout mice showed reduced Nox4 expression in their AMs. Further, AMs from ethanol-fed Nox2 knockout mice showed diminished Nox4 expression compared with ethanol-fed wild-type mice. Taken together, these results suggested that ethanol-induced Nox4 expression in mouse alveolar macrophages occurred through upregulation of Nox1 and Nox2 expression.
Figure 5.
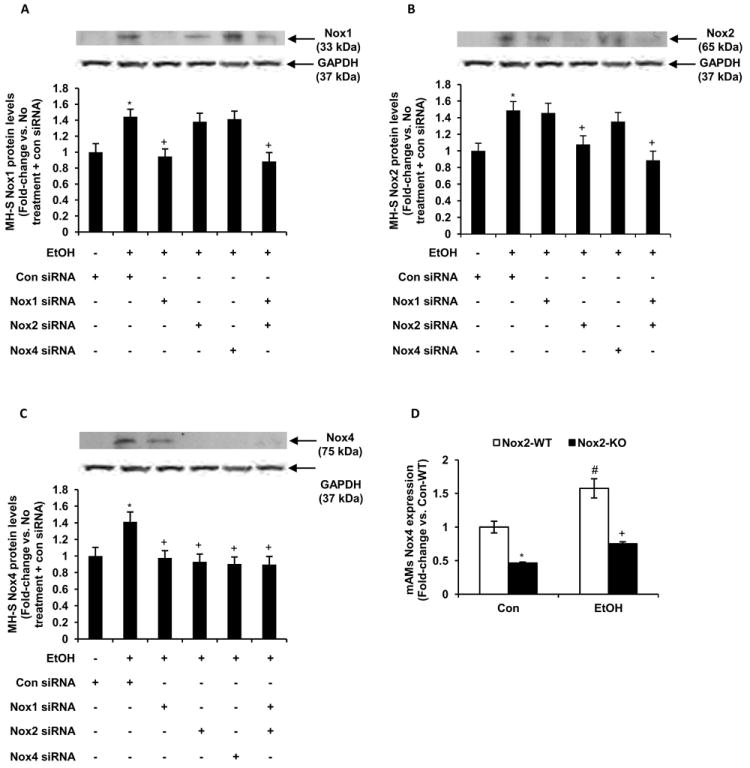
Ethanol induced Nox4 protein expression in vitro and in vivo via upregulation of Nox1 and Nox2. MH-S cells were transfected with control siRNA or siRNAs for Nox1, Nox2, Nox1+Nox2, or Nox4, and then cultured for 3 days with ethanol (EtOH, 0.08%) or without ethanol. Protein was isolated and protein levels of Nox1 (A), Nox2 (B) and Nox4 (C) were determined by Western blotting analysis and normalized to GAPDH protein levels (n=3 independent experiments). *p<0.05, EtOH vs. None; +p<0.05, EtOH+siRNA vs. EtOH. D, mAMs were collected from wildtype and Nox2 knockout mice that were either control-fed (Con) or ethanol-fed (EtOH). Expression of Nox4 was measured by computer analysis of confocal fluorescence microscopic images (n=5). Fluorescence values are expressed as mean relative fluorescent units (RFU) per cell ± SEM, relative to control. #p<0.05, Con-Nox2-KO vs. Con-Nox2-WT; *p<0.05, EtOH-Nox2-WT vs. Con-Nox2-WT; +p<0.05, EtOH+Nox2-KO vs. EtOH-Nox2-WT.
Nox1, Nox2 and Nox4 modulated ethanol-induced oxidative stress
Previous studies have shown that Nox1 and Nox2 produce superoxide anions (46, 47) and Nox4 generates hydrogen peroxide (48). As we have shown that ethanol increased Nox expression, we evaluated the contributions of Nox1, Nox2 and Nox4 to ethanol-mediated cellular oxidative stress. Using siRNAs for Nox1, Nox2, Nox1+Nox2, and Nox4 transfected MH-S cells, ethanol (0.08%) treatment for 3 days showed attenuation of ROS production, as measured by DCFH-DA analysis (Fig. 6A), and hydrogen peroxide generation (Fig. 6B), as measured by Amplex Red assay when compared with MH-S cells transfected with control siRNA. Further, mAMs isolated from wildtype, Nox1 knockout, or Nox2 knockout animals were treated with ethanol (0.08%) ex vivo for 3 days. Ethanol-induced ROS production (Fig. 6C), hydrogen peroxide generation (Fig. 6D), and impaired phagocytic ability (Fig. 6E) were attenuated in AMs from knockout compared to wild-type mice. Collectively, these results indicated that ethanol, by upregulating Nox1, Nox2 and Nox4, induced oxidative stress and dysfunction in mouse alveolar macrophages.
Figure 6.
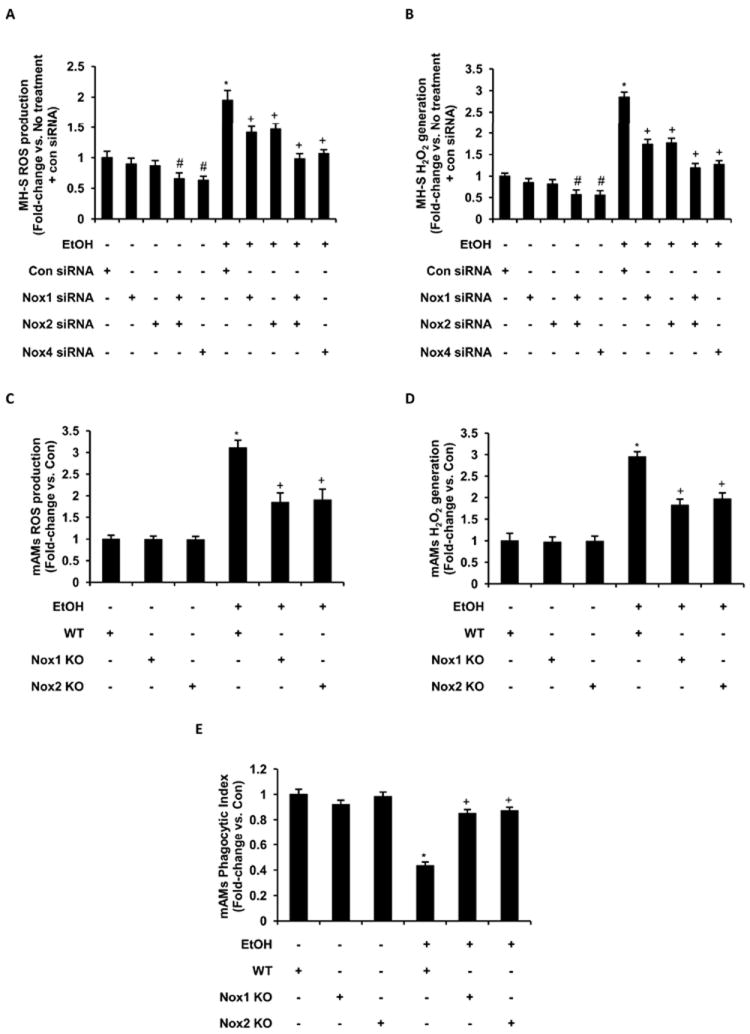
Oxidative stress in ethanol exposed MH-S and mAM cells was regulated by Nox1, Nox2 and Nox4 expression. MH-S cells were transfected with control siRNA (20 nM and 35 nM) or siRNAs for Nox1 (20 nM), Nox2 (20 nM), Nox1+Nox2, or Nox4 (35 nM), and then cultured for 3 days with ethanol (EtOH) or without ethanol. Control siRNA concentrations were adjusted accordingly for comparison, combined, and presented as a single con siRNA value. A, Reactive oxygen species production in transfected MH-S cells was measured by DCFH-DA fluorescence assay (n=3 independent experiments, in duplicate). B, H2O2 generation in transfected MH-S cells was measured by Amplex Red assay (n=3 independent experiments, in duplicate). H2O2 generation values were normalized to protein concentration. mAMs were isolated from wildtype (WT), Nox1 knockout (Nox1 KO), and Nox2 knockout (Nox2 KO) and cultured for 3 days with ethanol (EtOH) or without ethanol (Con). C, Reactive oxygen species production in these mAMs was measured by DCFH-DA fluorescence assay (n=3 independent experiments, in duplicate). D, H2O2 generation in these mAMs was measured by Amplex Red assay and normalized to protein concentration (n=3 independent experiments, in duplicate). E, Phagocytic ability was assessed in these mAMs by phagocytosis assay (n=3 independent experiments, 10 fields per experimental condition). Phagocytic index was calculated from the percentage of phagocytic cells multiplied by the relative fluorescence units of S. aureus per cell. All values are expressed as mean ± SEM, relative to no treatment. #p<0.05, No treatment+siRNA vs. No treatment+Con siRNA; *p<0.05, EtOH+Con siRNA vs. No treatment+Con siRNA or WT+EtOH vs. WT+Con; +p<0.05, EtOH+siRNA vs. EtOH+Con siRNA or Nox1/Nox2 KO+EtOH vs. WT+EtOH.
Chronic ethanol ingestion induced expression of Nox enzymes in human AMs
Since ethanol increased Nox enzyme expression in mAMs in vivo and in vitro, we determined the effects of ethanol on Nox expressions in hAMs. As shown in Fig. 7, compared to controls, AMs from chronic alcoholic patients show increased Nox1, Nox2 and Nox4 mRNA levels (Fig. 7A), as measured by qRT-PCR, and protein levels (Fig. 7B-C), as measured by computer analysis of confocal microscopic images. These data suggested that the mechanisms of ethanol-induced oxidative stress elucidated in mAMs may be similar in hAMs.
Figure 7.
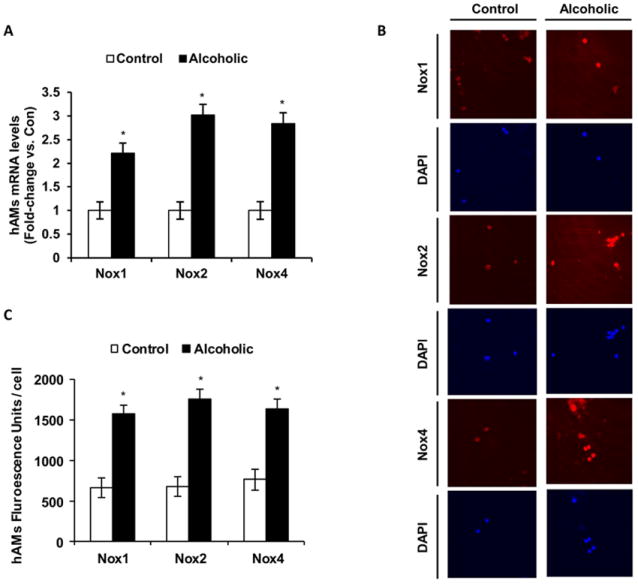
Ethanol enhanced Nox expression levels in hAMs. hAMs were collected from control (Control, n=5) and chronic alcoholic patients (Alcoholic, n=5). A, mRNA expression levels of Nox1, Nox2 and Nox4 were measured by qRT-PCR and normalized to 9s mRNA (n=10). B, Protein expression levels of Nox1, Nox2 and Nox4 were measured using computer analysis of confocal fluorescence microscopic images (n=10). C, Quantification of fluorescence images of Nox1, Nox2 and Nox4 protein levels. Fluorescence values are normalized to DAPI nuclear stain and are expressed as mean relative fluorescent units (RFU) per cell ± SEM. *p<0.05, Alcoholic vs. Control.
DISCUSSION
Chronic alcohol abuse is a comorbid variable associated with increased risk of ARDS and respiratory infections (3). AMs are critical to innate and acquired immunity (13) due to their ability to clear apoptotic cells and infectious particles from the lung by phagocytosis and respiratory burst (14). Chronic ethanol exposure impairs AM function (11) through mechanisms that remain to be defined and that may involve ethanol-induced oxidative stress (15). One potential mechanism for alcohol-induced oxidative stress is upregulation of the Nox family of proteins that comprise multi-component, membrane-associated NADPH oxidase enzymes that generate ROS (20). Previous studies showed that, in whole lung tissue, chronic ethanol ingestion increased the expression of Nox2, the classical phagocytic oxidase essential for ROS generation during respiratory burst (23). Although it is not involved in the respiratory burst, Nox4, a constitutively active isoform that generates ROS was similarly increased in the lung tissue of ethanol-fed mice. Under physiological conditions, the primary sources of ROS generation in AMs are the NADPH oxidases (18, 19). Nox1 (22), Nox2 (23) and Nox4 (21) are expressed in the lung. Since Nox proteins are central to the clearance of microbes through respiratory burst, it is important to understand how chronic ethanol ingestion alters the expression of Nox isoforms and their generation of ROS.
Our studies showed that chronic ethanol increased oxidative stress in mAMs in vivo and in vitro through upregulation of NADPH oxidase mRNA and protein expression. In our animal model, chronic ethanol ingestion also upregulated p22phox, a regulatory protein for Nox1, Nox2 and Nox4, as well as p47phox and p67phox, regulatory proteins for Nox1 and Nox2, in mAMs. Our data also demonstrated in MH-S cells and mAMs that ethanol increased Nox1, Nox2 and Nox4 mRNA expression in a time-dependent manner. While chronic ethanol exposure increased Nox expression in AMs, the mechanisms responsible for this increase remain unclear. Previous studies from our laboratories have found that chronic ethanol ingestion significantly attenuated antioxidant glutathione levels in lung tissue and BAL fluid (5-8) and augmented superoxide generation in lung tissue (23). Additionally, ethanol has been shown to enhance angiotensin II activity (49), which subsequently upregulates Nox expression (50). Recent studies suggest that chronic ethanol ingestion upregulates TGFβ1 expression (51), which has been implicated in the regulation of Nox4 (20). Further studies are warranted to elucidate the molecular mechanisms involved in ethanol-mediated Nox expression as well as its role in promoting AM oxidative stress.
Previous studies demonstrated that treating rats chronically fed ethanol with precursors to the antioxidant glutathione (procysteine or N-acetyl cysteine) normalized oxidative stress in the epithelial lining fluid and in AMs and restored phagocytic function (10-11), implicating alcohol-induced oxidative stress in AM dysfunction. Future studies will focus on the capacity of glutathione precursors or anti-oxidant treatments to attenuate alcohol-induced AM oxidative stress in MH-S cells. Recent ischemia-repurfusion studies suggest that oxidative stress can be involved in a positive feedback loop of further ROS generation (52). Hypoxia upregulates Nox1 and activates hypoxia-inducible factor-1 through increased ROS (22). The Nox1 promoter also contains an AP-1 binding site which is essential for its activity (53). LPS/interferon-γ induced ROS in monocytes upregulated the redox-sensitive transcription factor NF-κB to activate Nox2 expression (54). Studies show that ROS generated by thrombin-activated Nox4 induces NF-κB and hypoxia-inducible factor-1 (55). Further, the promoter of Nox4 contains response elements for several oxidative stress-related transcription factors, such as peroxisome proliferator-activated receptor, forkhead domain factor, hypoxia-inducible factor-1, nuclear respiratory factor-1 and NF-κB (56). Collectively, these studies suggest that alcohol-induced oxidative stress may stimulate that activity of redox-regulated transcription factors that promote increased expression of Nox subunits. While our studies show that chronic ethanol ingestion increased oxidative stress in mAMs in vivo and in vitro through upregulation of Nox enzymes, the specific mechanisms by which ROS generation upregulates Nox expression remain unknown.
Herein, our data demonstrated that apocynin attenuated ethanol-induced oxidative stress in addition to Nox1 and Nox2 expression; however, how apocynin inhibited ethanol-induced Nox1 and Nox2 mRNA expression in AMs is unclear. Apocynin is a commonly used pharmacological inhibitor of Nox1 and Nox2 complex formation through prevention of p47phox cytosolic translocation to the membrane (43). However, recent studies suggest that apocynin may act as an antioxidant in endothelial cells and smooth muscle cells, rather than specifically inhibiting NADPH oxidases (57). If apocynin can reduce ethanol-induced ROS in AMs, it may lead to subsequent down-regulation of Nox1 and Nox2 mRNA expression. In addition, apocynin may affect other regulators of Nox expression, such as p38 MAPK, Akt and ERK1/2 (57). Our studies show that apocynin treatment can reverse ethanol-mediated AM dysfunction, which may be an effect of apocynin’s ability to reduce ROS. Further studies are necessary to elucidate the mechanisms by which apocynin can attenuate ROS.
Our experiments using apocynin were confirmed by siRNAs for Nox1 and Nox2 to account for apocynin’s nonspecific functions. Experiments with apocynin and siRNAs for Nox1, Nox2 as well as Nox1 plus Nox2 reduced ethanol-induced Nox4 expression and oxidative stress in AMs. Our data showed that Nox1 or Nox2 induce Nox4, so silencing of Nox1, Nox2, or Nox4 have similar effects on hydrogen peroxide generation. Further, in AMs isolated from Nox1- or Nox2-knockout (KO) mice ROS production and AM dysfunction caused by ex vivo treatment with ethanol was attenuated. Taken together, these data suggested that the ethanol-mediated increase in AM Nox4 expression, oxidative stress, and dysfunction was secondary to increased Nox1 and Nox2. We also demonstrated that the expression levels of Nox1, Nox2 and Nox4 were increased in human AMs isolated from alcoholics compared to healthy controls. However, further studies are necessary to determine whether Nox1 or Nox2 expression drives Nox4 expression in the human AMs.
Nox1 and Nox2 play important roles in producing ROS (58). ROS increase TGFβ1 expression (59) which up-regulates Nox4 expression (20) through a Smad binding site in the Nox4 promoter. ROS also activate redox-sensitive transcription factors, such as NF-κB, that bind to and activate the Nox4 promoter to increase Nox4 expression (56). Based on these reports we speculate that ROS produced by Nox1 and Nox2 increase TGFβ1 and NF-κB signaling pathways to promote Nox4 expression. A more detailed examination of these pathways constitutes a focus of current studies in our labs. Regardless of the mechanisms involved, however, our study provides novel evidence of ethanol-induced Nox4 expression in AMs through upregulation of Nox1 and Nox2.
In summary, chronic ethanol ingestion increased AM oxidative stress through upregulation of Nox1, Nox2, and Nox4. Apocynin experiments showed that Nox1 and Nox2 complex formation, involving p47phox, is required for Nox4 expression. Silencing experiments utilizing siRNAs for Nox1 and Nox2 further demonstrated that Nox1 and Nox2 are required for Nox4 expression and ethanol-mediated oxidative stress. Nox1- and Nox2-KO experiments showed that either Nox1 or Nox2 are required for ethanol-induced AM oxidative stress and dysfunction. To our knowledge, this is the first report of ethanol’s ability to induce Nox1 and Nox2 expression in AMs, to increase Nox4 expression via increased expression of Nox1 or Nox2, and to subsequently promote oxidative stress. These studies suggest that strategies to reduce alcohol-mediated increases in AM Nox expression and activity may provide a novel therapeutic approach for attenuating ethanol-induced AM oxidative stress and dysfunction, resulting in reduced susceptibility to lung infection and injury.
Acknowledgments
We thank members of the clinical core of the Emory Alcohol and Lung Biology Center: Dr. Ashish Mehta, Dr. Annette Esper, and Amy Anderson for recruiting and enrolling patients for BAL collection in this study.
This work was supported by an NIAAA T32 training grant (5T32AA013528-08), the Emory Alcohol and Lung Biology Center (1P50AA135757) and Merit Review Funding from the Atlanta VA Medical Center.
References
- 1.Erickson SE, Martin GS, Davis JL, Matthay MA, Eisner MD. Recent trends in acute lung injury mortality: 1996-2005. Crit Care Med. 2009;37:1574–1579. doi: 10.1097/CCM.0b013e31819fefdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 3.Moss M, Parsons PE, Steinberg KP, Hudson LD, Guidot DM, Burnham EL, Eaton S, Cotsonis GA. Chronic alcohol abuse is associated with an increased incidence of acute respiratory distress syndrome and severity of multiple organ dysfunction in patients with septic shock. Crit Care Med. 2003;31:869–877. doi: 10.1097/01.CCM.0000055389.64497.11. [DOI] [PubMed] [Google Scholar]
- 4.Moss M, Bucher B, Moore FA, Moore EE, Parsons PE. The role of chronic alcohol abuse in the development of acute respiratory distress syndrome in adults. JAMA. 1996;275:50–54. [PubMed] [Google Scholar]
- 5.Brown LA, Harris FL, Bechara R, Guidot DM. Effect of chronic ethanol ingestion on alveolar type II cell: glutathione and inflammatory mediator-induced apoptosis. Alcohol Clin Exp Res. 2001;25:1078–1085. [PubMed] [Google Scholar]
- 6.Guidot DM, Modelska K, Lois M, Jain L, Moss IM, Pittet JF, Brown LA. Ethanol ingestion via glutathione depletion impairs alveolar epithelial barrier function in rats. Am J Physiol Lung Cell Mol Physiol. 2000;279:L127–135. doi: 10.1152/ajplung.2000.279.1.L127. [DOI] [PubMed] [Google Scholar]
- 7.Guidot DM, Brown LA. Mitochondrial glutathione replacement restores surfactant synthesis and secretion in alveolar epithelial cells of ethanol-fed rats. Alcohol Clin Exp Res. 2000;24:1070–1076. [PubMed] [Google Scholar]
- 8.Holguin F, Moss I, Brown LA, Guidot DM. Chronic ethanol ingestion impairs alveolar type II cell glutathione homeostasis and function and predisposes to endotoxin-mediated acute edematous lung injury in rats. J Clin Invest. 1998;101:761–768. doi: 10.1172/JCI1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moss M, Guidot DM, Wong-Lambertina M, Ten Hoor T, Perez RL, Brown LA. The effects of chronic alcohol abuse on pulmonary glutathione homeostasis. Am J Respir Crit Care Med. 2000;161:414–419. doi: 10.1164/ajrccm.161.2.9905002. [DOI] [PubMed] [Google Scholar]
- 10.Brown LA, Ping XD, Harris FL, Gauthier TW. Glutathione availability modulates alveolar macrophage function in the chronic ethanol-fed rat. Am J Physiol Lung Cell Mol Physiol. 2007;292:L824–832. doi: 10.1152/ajplung.00346.2006. [DOI] [PubMed] [Google Scholar]
- 11.Brown LA, Harris FL, Ping XD, Gauthier TW. Chronic ethanol ingestion and the risk of acute lung injury: a role for glutathione availability? Alcohol. 2004;33:191–197. doi: 10.1016/j.alcohol.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 12.Yeh MY, Burnham EL, Moss M, Brown LA. Chronic alcoholism alters systemic and pulmonary glutathione redox status. Am J Respir Crit Care Med. 2007;176:270–276. doi: 10.1164/rccm.200611-1722OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trapnell BC, Whitsett JA. Gm-CSF regulates pulmonary surfactant homeostasis and alveolar macrophage-mediated innate host defense. Annu Rev Physiol. 2002;64:775–802. doi: 10.1146/annurev.physiol.64.090601.113847. [DOI] [PubMed] [Google Scholar]
- 14.Lundahl J, Hallden G, Skold CM. Human blood monocytes, but not alveolar macrophages, reveal increased CD11b/CD18 expression and adhesion properties upon receptor-dependent activation. Eur Respir J. 1996;9:1188–1194. doi: 10.1183/09031936.96.09061188. [DOI] [PubMed] [Google Scholar]
- 15.Brown SD, Gauthier TW, Brown LA. Impaired terminal differentiation of pulmonary macrophages in a Guinea pig model of chronic ethanol ingestion. Alcohol Clin Exp Res. 2009;33:1782–1793. doi: 10.1111/j.1530-0277.2009.01017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1005–1028. doi: 10.1152/ajplung.2000.279.6.L1005. [DOI] [PubMed] [Google Scholar]
- 17.Fleury C, Mignotte B, Vayssiere JL. Mitochondrial reactive oxygen species in cell death signaling. Biochimie. 2002;84:131–141. doi: 10.1016/s0300-9084(02)01369-x. [DOI] [PubMed] [Google Scholar]
- 18.Forman HJ, Torres M. Reactive oxygen species and cell signaling: respiratory burst in macrophage signaling. Am J Respir Crit Care Med. 2002;166:S4–8. doi: 10.1164/rccm.2206007. [DOI] [PubMed] [Google Scholar]
- 19.Piotrowski WJ, Marczak J. Cellular sources of oxidants in the lung. Int J Occup Med Environ Health. 2000;13:369–385. [PubMed] [Google Scholar]
- 20.Brown DI, Griendling KK. Nox proteins in signal transduction. Free Radic Biol Med. 2009;47:1239–1253. doi: 10.1016/j.freeradbiomed.2009.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mittal M, Roth M, Konig P, Hofmann S, Dony E, Goyal P, Selbitz AC, Schermuly RT, Ghofrani HA, Kwapiszewska G, Kummer W, Klepetko W, Hoda MA, Fink L, Hanze J, Seeger W, Grimminger F, Schmidt HH, Weissmann N. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ Res. 2007;101:258–267. doi: 10.1161/CIRCRESAHA.107.148015. [DOI] [PubMed] [Google Scholar]
- 22.Goyal P, Weissmann N, Grimminger F, Hegel C, Bader L, Rose F, Fink L, Ghofrani HA, Schermuly RT, Schmidt HH, Seeger W, Hanze J. Upregulation of NAD(P)H oxidase 1 in hypoxia activates hypoxia-inducible factor 1 via increase in reactive oxygen species. Free Radic Biol Med. 2004;36:1279–1288. doi: 10.1016/j.freeradbiomed.2004.02.071. [DOI] [PubMed] [Google Scholar]
- 23.Polikandriotis JA, Rupnow HL, Elms SC, Clempus RE, Campbell DJ, Sutliff RL, Brown LA, Guidot DM, Hart CM. Chronic ethanol ingestion increases superoxide production and NADPH oxidase expression in the lung. Am J Respir Cell Mol Biol. 2006;34:314–319. doi: 10.1165/rcmb.2005-0320OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ambasta RK, Kumar P, Griendling KK, Schmidt HH, Busse R, Brandes RP. Direct interaction of the novel Nox proteins with p22phox is required for the formation of a functionally active NADPH oxidase. J Biol Chem. 2004;279:45935–45941. doi: 10.1074/jbc.M406486200. [DOI] [PubMed] [Google Scholar]
- 25.Hanna IR, Hilenski LL, Dikalova A, Taniyama Y, Dikalov S, Lyle A, Quinn MT, Lassegue B, Griendling KK. Functional association of nox1 with p22phox in vascular smooth muscle cells. Free Radic Biol Med. 2004;37:1542–1549. doi: 10.1016/j.freeradbiomed.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 26.Kawahara T, Ritsick D, Cheng G, Lambeth JD. Point mutations in the proline-rich region of p22phox are dominant inhibitors of Nox1- and Nox2-dependent reactive oxygen generation. J Biol Chem. 2005;280:31859–31869. doi: 10.1074/jbc.M501882200. [DOI] [PubMed] [Google Scholar]
- 27.Cheng G, Diebold BA, Hughes Y, Lambeth JD. Nox1-dependent reactive oxygen generation is regulated by Rac1. J Biol Chem. 2006;281:17718–17726. doi: 10.1074/jbc.M512751200. [DOI] [PubMed] [Google Scholar]
- 28.Sumimoto H, Miyano K, Takeya R. Molecular composition and regulation of the Nox family NAD(P)H oxidases. Biochem Biophys Res Commun. 2005;338:677–686. doi: 10.1016/j.bbrc.2005.08.210. [DOI] [PubMed] [Google Scholar]
- 29.Banfi B, Clark RA, Steger K, Krause KH. Two novel proteins activate superoxide generation by the NADPH oxidase NOX1. J Biol Chem. 2003;278:3510–3513. doi: 10.1074/jbc.C200613200. [DOI] [PubMed] [Google Scholar]
- 30.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 31.Ellmark SH, Dusting GJ, Fui MN, Guzzo-Pernell N, Drummond GR. The contribution of Nox4 to NADPH oxidase activity in mouse vascular smooth muscle. Cardiovasc Res. 2005;65:495–504. doi: 10.1016/j.cardiores.2004.10.026. [DOI] [PubMed] [Google Scholar]
- 32.Schilder YD, Heiss EH, Schachner D, Ziegler J, Reznicek G, Sorescu D, Dirsch VM. NADPH oxidases 1 and 4 mediate cellular senescence induced by resveratrol in human endothelial cells. Free Radic Biol Med. 2009;46:1598–1606. doi: 10.1016/j.freeradbiomed.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 33.Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S, Sorescu D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res. 2005;97:900–907. doi: 10.1161/01.RES.0000187457.24338.3D. [DOI] [PubMed] [Google Scholar]
- 34.Yang S, Zhang Y, Ries W, Key L. Expression of Nox4 in osteoclasts. J Cell Biochem. 2004;92:238–248. doi: 10.1002/jcb.20048. [DOI] [PubMed] [Google Scholar]
- 35.Li J, Stouffs M, Serrander L, Banfi B, Bettiol E, Charnay Y, Steger K, Krause KH, Jaconi ME. The NADPH oxidase NOX4 drives cardiac differentiation: Role in regulating cardiac transcription factors and MAP kinase activation. Mol Biol Cell. 2006;17:3978–3988. doi: 10.1091/mbc.E05-06-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clempus RE, Sorescu D, Dikalova AE, Pounkova L, Jo P, Sorescu GP, Schmidt HH, Lassegue B, Griendling KK. Nox4 is required for maintenance of the differentiated vascular smooth muscle cell phenotype. Arterioscler Thromb Vasc Biol. 2007;27:42–48. doi: 10.1161/01.ATV.0000251500.94478.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee YM, Kim BJ, Chun YS, So I, Choi H, Kim MS, Park JW. NOX4 as an oxygen sensor to regulate TASK-1 activity. Cell Signal. 2006;18:499–507. doi: 10.1016/j.cellsig.2005.05.025. [DOI] [PubMed] [Google Scholar]
- 38.Lyle AN, Deshpande NN, Taniyama Y, Seidel-Rogol B, Pounkova L, Du P, Papaharalambus C, Lassegue B, Griendling KK. Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ Res. 2009;105:249–259. doi: 10.1161/CIRCRESAHA.109.193722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dong J, Sulik KK, Chen SY. The role of NOX enzymes in ethanol-induced oxidative stress and apoptosis in mouse embryos. Toxicol Lett. 2010;193:94–100. doi: 10.1016/j.toxlet.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jerrells TR, Pavlik JA, DeVasure J, Vidlak D, Costello A, Strachota JM, Wyatt TA. Association of chronic alcohol consumption and increased susceptibility to and pathogenic effects of pulmonary infection with respiratory syncytial virus in mice. Alcohol. 2007;41:357–369. doi: 10.1016/j.alcohol.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Song K, Coleman RA, Zhu X, Alber C, Ballas ZK, Waldschmidt TJ, Cook RT. Chronic ethanol consumption by mice results in activated splenic T cells. J Leukoc Biol. 2002;72:1109–1116. [PubMed] [Google Scholar]
- 42.Pollock JD, Williams DA, Gifford MA, Li LL, Du X, Fisherman J, Orkin SH, Doerschuk CM, Dinauer MC. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet. 1995;9:202–209. doi: 10.1038/ng0295-202. [DOI] [PubMed] [Google Scholar]
- 43.Lemarie A, Bourdonnay E, Morzadec C, Fardel O, Vernhet L. Inorganic arsenic activates reduced NADPH oxidase in human primary macrophages through a Rho kinase/p38 kinase pathway. J Immunol. 2008;180:6010–6017. doi: 10.4049/jimmunol.180.9.6010. [DOI] [PubMed] [Google Scholar]
- 44.Patel N, Gonsalves CS, Yang M, Malik P, Kalra VK. Placenta growth factor induces 5-lipoxygenase-activating protein to increase leukotriene formation in sickle cell disease. Blood. 2009;113:1129–1138. doi: 10.1182/blood-2008-07-169821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perez RL, Kimani AP, King TE, Jr, Aguayo SM, Roman J. Bronchoalveolar lavage fluid D dimer levels are higher and more prevalent in black patients with pulmonary sarcoidosis. Respiration. 2007;74:297–303. doi: 10.1159/000091994. [DOI] [PubMed] [Google Scholar]
- 46.Kim YM, Guzik TJ, Zhang YH, Zhang MH, Kattach H, Ratnatunga C, Pillai R, Channon KM, Casadei B. A myocardial Nox2 containing NAD(P)H oxidase contributes to oxidative stress in human atrial fibrillation. Circ Res. 2005;97:629–636. doi: 10.1161/01.RES.0000183735.09871.61. [DOI] [PubMed] [Google Scholar]
- 47.Lassegue B, Sorescu D, Szocs K, Yin Q, Akers M, Zhang Y, Grant SL, Lambeth JD, Griendling KK. Novel gp91(phox) homologues in vascular smooth muscle cells : nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ Res. 2001;88:888–894. doi: 10.1161/hh0901.090299. [DOI] [PubMed] [Google Scholar]
- 48.Dikalov SI, Dikalova AE, Bikineyeva AT, Schmidt HH, Harrison DG, Griendling KK. Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free Radic Biol Med. 2008;45:1340–1351. doi: 10.1016/j.freeradbiomed.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bechara RI, Pelaez A, Palacio A, Joshi PC, Hart CM, Brown LA, Raynor R, Guidot DM. Angiotensin II mediates glutathione depletion, transforming growth factor-beta1 expression, and epithelial barrier dysfunction in the alcoholic rat lung. Am J Physiol Lung Cell Mol Physiol. 2005;289:L363–370. doi: 10.1152/ajplung.00141.2005. [DOI] [PubMed] [Google Scholar]
- 50.Seshiah PN, Weber DS, Rocic P, Valppu L, Taniyama Y, Griendling KK. Angiotensin II stimulation of NAD(P)H oxidase activity: upstream mediators. Circ Res. 2002;91:406–413. doi: 10.1161/01.res.0000033523.08033.16. [DOI] [PubMed] [Google Scholar]
- 51.Bechara RI, Brown LA, Roman J, Joshi PC, Guidot DM. Transforming growth factor beta1 expression and activation is increased in the alcoholic rat lung. Am J Respir Crit Care Med. 2004;170:188–194. doi: 10.1164/rccm.200304-478OC. [DOI] [PubMed] [Google Scholar]
- 52.Becker LB. New concepts in reactive oxygen species and cardiovascular reperfusion physiology. Cardiovasc Res. 2004;61:461–470. doi: 10.1016/j.cardiores.2003.10.025. [DOI] [PubMed] [Google Scholar]
- 53.Cevik MO, Katsuyama M, Kanda S, Kaneko T, Iwata K, Ibi M, Matsuno K, Kakehi T, Cui W, Sasaki M, Yabe-Nishimura C. The AP-1 site is essential for the promoter activity of NOX1/NADPH oxidase, a vascular superoxide-producing enzyme: Possible involvement of the ERK1/2-JunB pathway. Biochem Biophys Res Commun. 2008;374:351–355. doi: 10.1016/j.bbrc.2008.07.027. [DOI] [PubMed] [Google Scholar]
- 54.Anrather J, Racchumi G, Iadecola C. NF-kappaB regulates phagocytic NADPH oxidase by inducing the expression of gp91phox. J Biol Chem. 2006;281:5657–5667. doi: 10.1074/jbc.M506172200. [DOI] [PubMed] [Google Scholar]
- 55.Bonello S, Zahringer C, BelAiba RS, Djordjevic T, Hess J, Michiels C, Kietzmann T, Gorlach A. Reactive oxygen species activate the HIF-1alpha promoter via a functional NFkappaB site. Arterioscler Thromb Vasc Biol. 2007;27:755–761. doi: 10.1161/01.ATV.0000258979.92828.bc. [DOI] [PubMed] [Google Scholar]
- 56.Lu X, Murphy TC, Nanes MS, Hart CM. PPAR{gamma} regulates hypoxia-induced Nox4 expression in human pulmonary artery smooth muscle cells through NF-{kappa}B. Am J Physiol Lung Cell Mol Physiol. 2010;299:L559–566. doi: 10.1152/ajplung.00090.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Heumuller S, Wind S, Barbosa-Sicard E, Schmidt HH, Busse R, Schroder K, Brandes RP. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension. 2008;51:211–217. doi: 10.1161/HYPERTENSIONAHA.107.100214. [DOI] [PubMed] [Google Scholar]
- 58.Suh YA, Arnold RS, Lassegue B, Shi J, Xu X, Sorescu D, Chung AB, Griendling KK, Lambeth JD. Cell transformation by the superoxide-generating oxidase Mox1. Nature. 1999;401:79–82. doi: 10.1038/43459. [DOI] [PubMed] [Google Scholar]
- 59.Choi WS, Mitsumoto A, Kochevar IE. Involvement of reactive oxygen species in TGF-beta1-induced tropoelastin expression by human dermal fibroblasts. Photochem Photobiol. 2009;85:1425–1433. doi: 10.1111/j.1751-1097.2009.00611.x. [DOI] [PubMed] [Google Scholar]