

The intramolecular haloamination of alkenes is a direct method for the synthesis of halogen-functionalized nitrogen heterocycles.[1] Vicinal amino halides are highly versatile synthetic intermediates that are especially useful in drug discovery and combinatorial chemistry, and are also of interest in their own right as chemotherapeutic agents.[2] The synthesis of chiral nitrogen heterocycles using this method has been dominated by substrate-controlled reactions where the starting amine is already chiral.[3] Alternatively, some chiral 2-halomethyl nitrogen heterocycles are also available through multistep functional-group conversion from a limited number of naturally occuring chiral nitrogen heterocycles.[4] An alkene haloamination/cyclization in which the enantioselectivity was contolled by a chiral catalyst would be a powerful method for obtaining chiral products from achiral substrates and would expand the diversity of the product structure and the flexibility of the synthetic sequence. Herein is reported the first metal-catalyzed enantioselective alkene aminohalogenation/cyclization.
The enantioselective halogenation of alkenes has been a subject of vigorous investigations in recent years.[5–8] Organocatalytic methods have been especially fruitful, thus enabling the development of an enantioselective, electrophile-initiated, intramolecular alkene halolactonization,[5] with the first enantioselective bromoaminocylization of alkenes being accomplished by organocatalysis in 2011.[7] The mechanistically distinct nucleophile-initiated organocatalytic intermolecular enantioselective aminohalogenation of enones has also been reported.[8] These organocatalytic methods are generally specialized for a specific halogen and for a specific alkene substitution pattern. Transition-metal-catalyzed asymmetric aminohalogenation methods have the potential to extend the range of alkene substrates and scope of halogen atoms, but although a number of racemic alkene aminohalogenation/cyclization reactions catalyzed by Pd and to a lesser extent Cu salts have been reported,[9] these methods have not been extended to enantioselective versions. The development of an enantioselective Pd- or Cu-catalyzed aminohalogenation is a challenge because of: a) competing background electrophilic halogenation that does not require the agency of the chiral metal catalyst; b) different modes of aminometallation, for example, cis aminometallation and trans aminometallation, can be competitive, thus making a specific enantiodetermining aminometallation transition-state geometry difficult to achieve; and; c) the halide could act as a ligand and coordinate to the metal, thus disrupting the enantiodetermining step. Herein, we report a different strategy for the catalytic enantioselective alkene aminohalogenation/cyclization that relies on radical-based atom transfer to install the carbon–halogen bond, thereby minimizing the possibility for unselective electrophilic background aminohalogenation and complications that derive from nucleophilic or electrophilic halogen sources (Scheme 1).
Scheme 1.
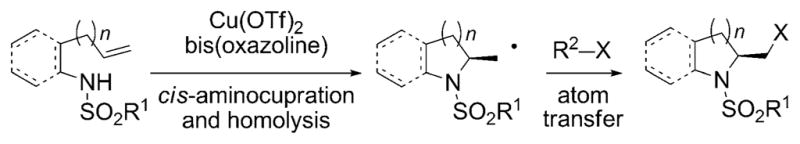
Aminohalogenation by atom transfer. Tf = trifluoromethyl-sulfonyl.
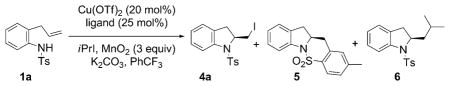
In recent years, our group has developed a family of Cu-catalyzed enantioselective alkene amination reactions, including carboamination,[10] aminooxygenation,[11] and diamination.[12] These reactions occur through a cis aminocupration followed by homolysis of the C–CuII bond, thereby generating a primary carbon radical.[10c] The fate of the carbon radical is influenced by the substrate structure and reaction components, and both determine which difunctionalized product is formed.[13] We hypothesized that if we could identify a halogen atom donor with a greater propensity for atom transfer than for electrophilic halogenation under our Cu-catalyzed reaction conditions, the enantioselective aminocupration step would be favored over non-catalyzed (racemic) background processes. An initial screen of several halogen sources (NBS, CBr4, NIS, I2, and 2,4,4,6-tetrabromocyclohexa-2,5-dienone) revealed that significant background aminohalogenation occurred under the reaction conditions in the absence of the [Cu(R,R)-Ph-box](OTf)2 catalyst. Fortunately, however, we did find that 2-iodopropane[14] did not produce any aminoiodination product (4a) under these reaction conditions in the absence of catalyst (Table 1, entry 1). When the reaction was carried out in the presence of the chiral catalyst (20 mol%) under these reaction conditions 2-iodomethylindoline 4a was obtained in 84% yield and with 70% enantiomeric excess (Table 1, entry 2). As well as the desired aminoiodination product two carboamination products 5 and 6 (< 10% each) were obtained, where 5 is the product of intramolecular addition onto the tosyl ring and 6 is the product of intermolecular addition to the isopropyl radical. We believe that most of the isopropyl radical is oxidized to propene under these reaction conditions in analogy to similar secondary radical intermediates under similar reaction conditions.[10c]
Table 1.
Optimization of the enantioselective aminoiodination.[a]
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Ligand | RI (equiv) | t[h] | T [°C] | Yield[b]4a [%] | ee [%][c] |
| 1[d] | none | 6 | 24 | 120 | – | – |
| 2 | 2 | 6 | 24 | 120 | 84 | 70 |
| 3[e] | 2 | 6 | 24 | 120 | 74 | 80 |
| 4[e] | 2 | 6 | 6 | 105 | 85 | 89 |
| 5[e] | 2 | 4 | 6 | 105 | 62 | n.d.[f] |
| 6[e] | 2 | 6 | 6 | 95 | 52 | 85 |
| 7[e] | 3 | 6 | 6 | 105 | 84 | 91 |
| 8[e,g] | 2 | 6 | 6 | 105 | 56 | 74 |
| 9[e,h] | 2 | 6 | 6 | 105 | 70 | 85 |
| 10[e,h] | 2 | 9 | 6 | 105 | 80 | 89 |
| 11[e,i] | 2 | 9 | 6 | 105 | 70 | 83 |
| 12[e,i] | 3 | 9 | 6 | 105 | 66 | 84 |
| 13[e,i,j] | 3 | 12 | 24 | 105 | 74 | 88 (>98)[k] |
Reactions run with 50.0 mg (0.174 mmol) of 1a at 0.1M concentration with 300 mol% of MnO2 and RI = iPrI unless otherwise noted.
Yield of product isolated by flash chromatography on SiO2.
Enantioselectivity determined by HPLC on a chiral stationary phase.
Reaction run without Cu(OTf)2. N-iPr-N-tosyl-2-allylaniline was the major product.
Activated molecular sieves (4 Å; 20 mgmL−1) were used.
Not determined.
Reaction run with 15 mol% of Cu(OTf)2 and 18 mol% of (R,R)-Ph-box.
Reaction run with 250 mg (0.870 mmol) of 1a.
Reaction run with 500 mg (1.74 mmol) of 1a.
400 mol% of MnO2 was used.
After one recrystallization. Reactions run 1–3 times each. Ts = p-toluenesulfonyl.
![[k]](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/1359/3324620/57b4defa7ee9/nihms362194u2.jpg)
A variety of reaction conditions (temperature, time, catalyst loading, ligand structure, amount of oxidant and 2-iodopropane, iodoalkane structure, and the use of 4 Å molecular sieves) were examined to identify optimal conditions for enantioselective aminoiodination. We found that the use of activated molecular sieves (4 Å) generally increased the yield and selectivity and rendered the results more reproducible (Table 1, entry 3). The temperature could be lowered to 105°C and the reaction time could be reduced to 6 hours without reducing the yield of 2a (Table 1, entry 4). Changing from 2-iodopropane to 2-iodobutane and iodocyclohexane did not increase the yield of the desired product (not shown). Both the (R,R)-Ph-box ligand 2 and the (4R,5S)-di-Ph-box ligand 3 performed well in the reaction (Table 1, entries 4 and 7). A reduction of the amount of iPrI to 4 equivalents resulted in diminished yield (Table 1, entry 5), as did reduction in temperature to 95°C (Table 1, entry 6). When the catalyst loading was decreased to 15 mol% a decrease in yield and enantioselectivity was observed (Table 1, entry 8). Both yield and enantioselectivity are diminished in the presence of water and we hypothesize that at low catalyst loading adventitious water becomes a significant issue. When the reaction using the commercially available (R,R)-Ph-box ligand 2 was scaled up from a 0.174 mmol scale to a 0.870 mmol scale, with respect to 1a, the enantioselectivity was consistent although the yield was reduced, but for the larger scale reaction an increase in the amount of iPrI to 9 equivalents resulted in an increase in the yield (Table 1, compare entries 4, 9, and 10). The more soluble (4R,5S)-diphenylbis(oxazoline) ligand 3 provided better reproducibility than ligand 2 on a 1.74 mmol (500 mg) scale of 1a (Table 1, entry 12).[11] A further increase in yield and selectivity was obtainedon this scale when the amount of MnO2 was increased to 400 mol%, the iPrI was increased to 12 equivalents, and the reaction time was increased to 24 hours (Table 1, entry 13).[15] Recrystallization provided 4a in >98% ee.
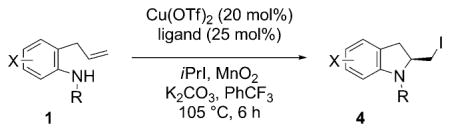
A variety of substituted 2-iodomethyl indolines 4 were synthesized (Table 2) using the optimized reaction conditions (Table 1, entry 4). Good yields and enantioselectivities were observed with a range of para-substituted N-tosyl-2-allylanilines 1 (X = Me, CN, F, Cl, MeO; Table 2, entries 1–5). The meta-methoxy substrate 1g also performed well (Table 2, entry 6) but the ortho-methoxy substrate 1h reacted with significant reduction in enantioselectivity (Table 2, entry 7). A variety of substituents on the nitrogen atom were also examined. The benzenesulfonyl substrate 1i gave similar yield and selectivity to the tosyl substrate 1a (Table 2, entry 8 and Table 1, entry 4), but the nosyl substrate 1j was less reactive and required higher catalyst loading (40 mol% of Cu(OTf)2 and 50 mol% of 3) for efficient conversion. The N-mesyl and N-trimethylsilylethylsulfonyl substrates 1k and 1l underwent aminoiodination with slightly diminished enantio-selectivity and better results were achieved when ligand 3 was used. The highest yielding reaction was that of N-3,5-di-tert-butylbenzenesulfonyl substrate 1m to give the aminoiodination adduct 4m; the yield of the intramolecular carboamination side-product analogous to 5 was significantly diminished for this reaction.
Table 2.
Scope of indoline synthesis.[a]
 | ||||
|---|---|---|---|---|
| Entry | Substrate | Ligand | Yield [%][b] | ee [%][c] |
| 1 | 1b, X =4-Me, R =Ts | 2 | 85 | 90 |
| 2 | 1c, X =4-CN, R =Ts | 2 | 85 | 84 |
| 3 | 1d, X =4-F, R =Ts | 2 | 80 | 89 |
| 4 | 1e, X =4-Cl, R =Ts | 2 | 83 | 87 |
| 5 | 1f, X =4-OMe, R =Ts | 2 | 72 | 87 |
| 6 | 1g, X =3-OMe, R =Ts | 2 | 71 | 88 |
| 7 | 1h, X =2-OMe, R =Ts | 2 | 80 | 15 |
| 8 | 1i, X =H, R =Bs | 2 | 77 | 88 |
| 9[d] | 1j, X =H, R =Ns | 3 | 72 | 80 |
| 10 | 1k, X =H, R =Ms | 3 | 85 | 81 |
| 11 | 1l, X =H, R =SES | 3 | 85 | 82 |
| 12 | 1m, X =H, R =3,5-di-tBu-C6H3SO2 | 2 | 90 | n.d.[e] |
Reaction conditions from Table 1, entry 4 were used. The indicated ligand, 2 or 3, was used in each reaction.
Yield of product isolated by flash chromatography on SiO2.
Enantioselectivity determined by HPLC on a chiral stationary phase.
40 mol% of Cu(OTf)2 and 50 mol% of 3 were used.
Not determined. The enantiomers could not be separated by HPLC on a chiral stationary phase. Bs = benzenesulfonyl, Ms = methanesulfonyl, Ns = 4-nitrophenylsulfonyl, SES = 2-tri-methylsilylethanesulfonyl.
The less-reactive 4-pentenylsulfonamides 7 required the use of the (4R,5S)-di-Ph-box ligand 3 and longer reaction times (11h) for optimal yield and enantioselectivity (Table 3). Under these reaction conditions, up to 93% ee was observed. Substitution on the carbon chain (R1 ≠ H) was beneficial to the reactivity and enantioselectivity. Substrate 7e, which has no carbon chain substituents (R1 = H), reacted with lower enantioselectivity (Table 3, entry 5) but substrate 7f, which also has no carbon chain substituents but has a 3,5-di-tert-butylbenzenesulfonyl substituent on the nitrogen atom, reacted with increased yield and enantioselectivity (Table 3, entry 6). An increase in the number of substituents on the olefin led to formation of a tertiary amine-bearing stereo-center but with significantly diminished enantioselectivity (Table 3, entry 9). It is interesting to note that 1,1-disubstituted[7a] and internal[7b] alkenes perform best in organocatalytic aminobrominations while monosubstituted terminal alkenes are superior substrates in our reaction. A singular example of a tetrahydroisoquinoline synthesis was examined (Table 3, entry 10), but this reaction was also poorly selective. In general, the enantioselectivity is higher with more-reactive substrates and it is possible that with less-reactive substrates a Cu-catalyzed background reaction can occur if the enantioselective cis-aminocupration step is relatively higher in energy. The absolute configurations of indoline 4a and pyrrolidine 8g were assigned by X-ray crystallography and all other products were assigned by analogy.[16]
Table 3.
Scope of pyrrolidine synthesis.[a]
| Entry | Substrate | Product | Yield [%][b] | ee [%][c] |
|---|---|---|---|---|
|
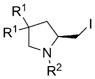
|
|||
| 1 | 7a, R1 =Me, R2 =Ts | 8a | 81 | 88 |
| 2 | 7b, R1 =Me, R2 =Ms | 8b | 78 | 43 |
| 3 | 7c, R1 =Me, R2 =Ns | 8c | 80 | 60 |
| 4 | 7d, R1 =Ph, R2 =Ts | 8d | 85 | 93 |
| 5 | 7e, R1 =H, R2 =Ts | 8e | 77 | 73 |
| 6 | 7f, R1 =H, R2 =3,5-di-tBu-C6H3SO2 | 8f | 85 | 88 |
| 7 |
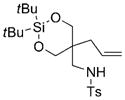 7g |
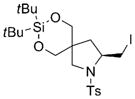 8g |
78 | 92 |
| 8 |
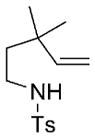 9 |
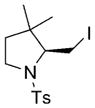 10 |
78 | 83 |
| 9 |
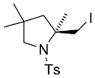
11 |
 12 |
74 | 16 |
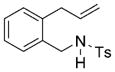
| 10[d] |
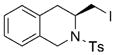
 13 |
 14 |
73 | 27 |
Reactions run as described in Table 1, entry 7 except reaction time was extended to 11h.
Yield of product isolated by flash chromatography on SiO2.
Enantioselectivity determined by HPLC on a chiral stationary phase.
Ligand 2 was used.
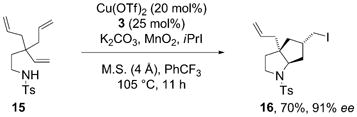
In an example of a radical cascade reaction, 4,4-diallyl-4-pentenylsulfonamide 15 underwent a double cyclization that terminated in atom transfer iodination. Bicyclic diastereomer 16 was formed exclusively in 70% yield and 91% ee [Eq. (1)].
 |
(1) |
The utility of the new vicinal aminoiodides as chiral synthetic intermediates is illustrated in Scheme 2. p-Fluoro-2-iodomethylindoline 2d (obtained on a larger scale in 75% yield and 92% ee using reaction conditions analogous to Table 1, entry 12) underwent an SN2 reaction with NaN3 to provide the chiral diamine 17 in 98% yield and an SN2 reaction with thiophenol to provide the chiral phenylthiosulfonamide 18 in 98% yield. This same indoline underwent Fe-catalyzed[17a,b] C–C bond formation with phenylmagnesium bromide, thus generating adduct 19 in 80% yield. The optical purity of the substrate was retained in all three reactions.
Scheme 2.

Substitution reactions of the chiral alkyliodide. acac = acetoacetonate, DMF = N,N′-dimethylformamide, HMTA = hexamethylene-tetramine, TMEDA= N,N,N′,N′-tetramethylethylenediamine.
Extension of the atom transfer step to other halogen atom donors was next explored (Scheme 3). Chlorine atom transfer was explored using 1,1-dichloroethylene, tetrachloroethylene, 2-chloropropane, tert-butylchloride, and tetrachlorocyclopropene as potential atom transfer reagents. Of these reagents, only 1,1-dichloroethene and tert-butylchloride provided the aminochloride 4n, and only 1,1-dichloroethylene (9 equiv) provided a highly enantioselective reaction. Bromine atom transfer was explored using 2-bromopropane, tert-butylbromide, and (2,2-dibromo-1-methylcyclopropyl)benzene. Of these reagents only (2,2-dibromo-1-methylcyclopropyl)benzene[18] (6 equiv) provided the aminobromide 4o in moderate yield and in good enantiomeric excess. These data indicate that the identification of an atom transfer reagent that can provide both good yield and enantioselectivity in such a transformation can be challenging.
Scheme 3.

Enantioselective aminochlorination and aminobromination.
In summary, we have disclosed the first metal-catalyzed enantioselective alkene aminohalogenation reaction. Our reaction provides a range of chiral 2-iodomethyl indolines and pyrrolidines in good to excellent yield and enantioselectivity. We have also identified reagents that will perform efficient enantioselective alkene aminobromination and aminochlorination. Such halogen generality in enantioselective alkene halofunctionalization is unusual as most processes are optimal for delivery of only one halogen.[5–7] The haloamine products are highly crystalline and thus amenable to chiral amplification by recrystallization (Table 1, entry 13). As demonstrated in Scheme 2, this method can be coupled with substitution reactions to provide a variety of functionalized chiral heterocycles, an application which should be very useful to organic synthesis and medicinal chemistry. Finally, this aminohalogenation is the first example of an efficient atom transfer process occurring in this class of Cu-catalyzed alkene amination reactions.[13]
Supplementary Material
Acknowledgments
Dedicated to William R. Roush on the occasion of his 60th birthday.
Footnotes
We are grateful to the National Institute of General Medical Sciences, National Institutes of Health (grant no. GM078383) for support of this research. We thank Prof. Nancy Totah for helpful discussions and Tim Liwosz for preliminary aminochlorination experiments. William W. Brennessel and the X-ray crystallographic facility at the University of Rochester are gratefully acknowledged for the X-ray structures of 4a and 8g.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201109044.
References
- 1.Selected haloamination/cyclization reactions: Horning DE, Muchowski JM. Can J Chem. 1974;52:1321–1330.Knapp S, Levorse AT. J Org Chem. 1988;53:4006–4014.Friesen RW, Giroux A, Cook KL. Tetrahedron Lett. 1993;34:5983–5986.Kobayashi K, Miyamoto K, Morikawa O, Konoshi H. Bull Chem Soc Jpn. 2005;78:886–889.Kobayashi K, Kondo S, Hashimoto K, Fukamachi S, Morikawa O, Konoshi H. Heterocycles. 2007;71:1827–1835.Klein JEMN, Muller-Bunz H, Evans P. Org Biomol Chem. 2009;7:986–995. doi: 10.1039/b819610a.
- 2.Selected bioactive vicinal amino halides: Qiu J, Silverman RB. J Med Chem. 2000;43:706–720. doi: 10.1021/jm9904755.Pors K, Shnyder SD, Teesdale-Spittle PH, Hartley JA, Zloh M, Searcey M, Patterson LH. J Med Chem. 2006;49:7013–7023. doi: 10.1021/jm0608154.Kuang Y, Balikrishnan K, Gandhi V, Peng X. J Am Chem Soc. 2011;133:19278–19281. doi: 10.1021/ja2073824.
- 3.Selected diastereoselective aminohalogenation/cyclizations: Tamaru Y, Kawamura S, Bando T, Tanaka K, Hojo M, Yoshida Z. J Org Chem. 1988;53:5491–5501.Williams DR, Osterhout MH, McGill JM. Tetrahedron Lett. 1989;30:1327–1330.Davies SG, Nicholson RL, Price PD, Roberts PM, Russell AJ, Savory ED, Smith AD, Thomson JE. Tetrahedron: Asymmetry. 2009;20:758–772.Yeung YY, Hong S, Corey EJ. J Am Chem Soc. 2006;128:6310–6311. doi: 10.1021/ja0616433.Beshore DC, Smith AB. J Am Chem Soc. 2007;129:4148–4149. doi: 10.1021/ja070336+.Cakmak M, Mayer P, Trauner D. Nat Chem. 2011;3:543–545. doi: 10.1038/nchem.1072.Moriyama K, Izumisawa Y, Togo H. J Org Chem. 2011;76:7249–7255. doi: 10.1021/jo201113r.Su S, Rodriguez RA, Baran PS. J Am Chem Soc. 2011;133:13922–13925. doi: 10.1021/ja206191g.Xu X, Kotti SRSS, Liu JY, Cannon JF, Headley AD, Li GG. Org Lett. 2004;6:4881–4884. doi: 10.1021/ol048045i.
- 4.Park SH, Kang HJ, Ko S, Park S, Chang S. Tetrahedron: Asymmetry. 2001;12:2621–2624.Zhu S, Zhang Q, Gudise C, Wei L, Smith E, Zeng Y. Bioorg Med Chem. 2009;17:4496–4502. doi: 10.1016/j.bmc.2009.05.011.
- 5.Catalytic enantioselective halolactonizations and haloetherifications: Kang SH, Lee SB, Park CM. J Am Chem Soc. 2003;125:15748–15749. doi: 10.1021/ja0369921.Zhou L, Tan CK, Jiang X, Chen F, Yeung YY. J Am Chem Soc. 2010;132:15474–15476. doi: 10.1021/ja1048972.Chen G, Ma S. Angew Chem. 2010;122:8484–8486.Angew Chem Int Ed. 2010;49:8306–8308. doi: 10.1002/anie.201003114.Veitch GE, Jacobsen EN. Angew Chem. 2010;122:7490–7493. doi: 10.1002/anie.201003681.Murai K, Matsushita T, Nakamura A, Fukushima S, Shimura M, Fujioka H. Angew Chem. 2010;122:9360–9363. doi: 10.1002/anie.201005409.Angew Chem Int Ed. 2010;49:9174–9177. doi: 10.1002/anie.201005409.Whitehead DC, Yousefi R, Jaganathan A, Borhan B. J Am Chem Soc. 2010;132:3298–3299. doi: 10.1021/ja100502f.Yousefi R, Whitehead DC, Mueller JM, Staples RJ, Borhan B. Org Lett. 2011;13:608–611. doi: 10.1021/ol102850m.Hennecke U, Muller CH, Frohlich R. Org Lett. 2011;13:860–863. doi: 10.1021/ol1028805.Denmark SE, Burk MT. Org Lett. 2012;14:256–259. doi: 10.1021/ol203033k.
- 6.Enantioselective halonium-induced polyene cyclizations and dihalogenations: Sakakura A, Ukai A, Ishihara K. Nature. 2007;445:900–903. doi: 10.1038/nature05553.Snyder SA, Treitler DS, Schall A. Tetrahedron. 2010;66:4796–4804.Monaco MR, Bella M. Angew Chem. 2011;123:11238–11240.Angew Chem Int Ed. 2011;50:11044–11046. doi: 10.1002/anie.201104843.and references therein.
- 7.a) Zhou L, Chen J, Tan CK, Yeung YY. J Am Chem Soc. 2011;133:9164–9167. doi: 10.1021/ja201627h. [DOI] [PubMed] [Google Scholar]; b) Huang D, Wang H, Xue F, Guan H, Li L, Peng X, Shi Y. Org Lett. 2011;13:6350–6353. doi: 10.1021/ol202527g. [DOI] [PubMed] [Google Scholar]
- 8.Intermolecular catalytic enantioselective aminohalogenations: Appayee C, Brenner-Moyer SE. Org Lett. 2010;12:3356–3359. doi: 10.1021/ol101167z.Cai Y, Liu X, Hui Y, Jiang J, Wang W, Chen W, Lin LS, Feng X. Angew Chem. 2010;122:6296–6300.Angew Chem Int Ed. 2010;49:6160–6164. doi: 10.1002/anie.201002355.
- 9.Pd- and Cu-catalyzed aminohalogenation/cyclizations: Manzoni MR, Zabawa TP, Kasi D, Chemler SR. Organometallics. 2004;23:5618–5621.Lei A, Lu X, Liu G. Tetrahedron Lett. 2004;45:1785–1788.Michael FE, Sibbald PA, Cochran BM. Org Lett. 2008;10:793–796. doi: 10.1021/ol702922c.Helaja J, Gottlich R. Chem Commun. 2002:720–721. doi: 10.1039/b201209j.Wu T, Yin G, Liu G. J Am Chem Soc. 2009;131:16354–16355. doi: 10.1021/ja9076588.Yin G, Wu T, Liu G. Chem Eur J. 2012;18:451–455. doi: 10.1002/chem.201102776.
- 10.a) Zeng W, Chemler SR. J Am Chem Soc. 2007;129:12948–12949. doi: 10.1021/ja0762240. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Miao L, Haque I, Manzoni MR, Tham WS, Chemler SR. Org Lett. 2010;12:4739–4741. doi: 10.1021/ol102233g. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sherman ES, Fuller PH, Kasi D, Chemler SR. J Org Chem. 2007;72:3896–3905. doi: 10.1021/jo070321u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Fuller PH, Kim JW, Chemler SR. J Am Chem Soc. 2008;130:17638–17639. doi: 10.1021/ja806585m. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Paderes MC, Chemler SR. Eur J Org Chem. 2011:3679–3684. doi: 10.1002/ejoc.201100444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sequeira FC, Turnpenny BW, Chemler SR. Angew Chem. 2010;122:6509–6512. doi: 10.1002/anie.201003499. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2010;49:6365–6368. doi: 10.1002/anie.201003499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chemler SR. J Organomet Chem. 2011;696:150–158. doi: 10.1016/j.jorganchem.2010.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blank O, Wetzel A, Ullrich D, Heinrich MR. Eur J Org Chem. 2008:3179–3189. [Google Scholar]
- 15.Reaction scale sensitivity may be due to surface area heating and reaction mixing differences. Commercially available <5 micron MnO2 (85%) is used in these reactions.
- 16.CCDC 858586 (4a), 858585 (8g) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- 17.Fe-catalyzed coupling of alkylhalides with Grignard reagents: Cahiez G, Duplais C, Moyeux A. Org Lett. 2007;9:3253–3254. doi: 10.1021/ol7016092.Cahiez G, Habiak V, Duplais C, Moyeux A. Angew Chem. 2007;119:4442–4444. doi: 10.1002/anie.200700742.Angew Chem Int Ed. 2007;46:4364–4366. doi: 10.1002/anie.200700742.Hsu SF, Ko CW, Wu YT. Adv Synth Catal. 2011;353:1756–1762.Zhou J, Fu GC. J Am Chem Soc. 2004;126:1340–1341. doi: 10.1021/ja039889k.Molander GA, Argintaru OA, Aron I, Dreher SD. Org Lett. 2010;12:5783–5785. doi: 10.1021/ol102717x.
- 18.a) Nishii Y, Nagano T, Gotoh H, Nagase R, Motoyoshiya J, Aoyama H, Tanabe Y. Org Lett. 2007;9:563–566. doi: 10.1021/ol062673d. [DOI] [PubMed] [Google Scholar]; b) Fedorynski M. Chem Rev. 2003;103:1099–1132. doi: 10.1021/cr0100087. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.