

Abstract
We investigated mechanisms by which TLR9 signaling promoted the development of the protective response to C. neoformans in mice with cryptococcal pneumonia. The afferent (week1) and efferent (week3) phase immune parameters were analyzed in the infected wild type (TLR9+/+) and TLR-deficient (TLR9-/-) mice. TLR9 deletion diminished: 1) accumulation and activation of CD11b+ DCs and; 2) the induction of IFN-γ and CCR2 chemokines CCL7,CCL12 but not CCL2 at week 1; and 3) pulmonary accumulation and activation of the major effector cells: CD4+ and CD8+ T cells, CD11b+ lung dendritic cells (DCs) and exudate macrophages (ExMs) at week 3. The significance of CCL7 induction downstream of TLR9 signaling was investigated by determining whether CCL7 reconstitution would improve immunological parameters inC. neoformans infected TLR9-/- mice. Early reconstitution with CCL7: 1) improved accumulation and activation of CD11b+ DCs at week 1; 2) restored early IFN-γ production in the lungs; and 3) restored the accumulation of major effector cell subsets. CCL7 administration abolished the difference in lung fungal burdens between TLR9+/+ and TLR9-/- miceat week 3; however, significant reduction of fungal burdens between PBS and CCL7-treated mice has not been observed, suggesting that additional mechanism(s) apart from early CCL7 induction contribute to optimal fungal clearance in TLR9+/+ mice. Collectively, we show that TLR9 signaling during the afferent phase contributes to the development of protective immunity by promoting the early induction of CCL7 and IFN-γ and the subsequent early recruitment and activation of DCs and additional effector cells in mice with cryptococcal pneumonia.
Introduction
C. neoformans is a leading cause of fatal mycosis in HIV-positive individuals around the world (1) and is a common pathogen found in organ transplant recipients and patients with hematological malignancies (2) and other conditions characterized by impaired T cell function.Consistent with the clinical data, T cell-mediated immunity is required for clearance of C. neoformans in mouse models of cryptococcal infections (3-5). The protective anticryptococcal immune response in mice is marked by: 1) the recruitment of the effector T cells (CD4+ and CD8+) into the lungs (3-6); 2) Th1-type immune polarization and robust interferon (IFN)-γ production (7-11); and 3) the accumulation/activation of CD11b+ dendritic cells (DCs) and exudate macrophages (ExMs) in the lungs (12, 13). Thus, the interaction between the Th1 polarized effector T cells and the antigen presenting cellsin the lungs, primarily pulmonary DCs and macrophages, is needed for activation of the effector fungicidal mechanisms and clearance of C. neoformans(12, 14, 15).
Although the innate responses are insufficient to eliminate C. neoformans from the infected host, the innate immune system is the first line of defense against C. neoformans. Furthermore, the molecular signals elicited during the innate (afferent) phase of the immune response drive the development of the T cell mediated responses (14). During the innate phase, fungal organisms are sensed via innate pattern recognition receptors (PRRs), which results in a generation of “danger signals”, mainly cytokines and chemokines that trigger the development of the adaptive responses (16-19). Toll-like receptors (TLRs) represent the major class of PRRs known to recognize components of viral, bacterial and fungal pathogens and facilitate rapid innate responses to these organisms (18, 20-22). TLR2, TLR4 and TLR9 were demonstrated to recognize different components of C. neoformans(19, 23-26). However, only TLR9 signaling has been demonstrated to play a major role in generation of the protective immune response against cryptococcal lung infection in vivo(26, 27). Furthermore, the role of TLR9 signaling in response to various strains of C. neoformanshas been reported(25-28).
Our previous study shows that TLR9 deletion had profound effects on several aspects of the adaptive immune response against C. neoformans(26). Furthermore, complementary in vitro studies show that TLR9 signaling is crucial for activation of DCs during their encounter with this fungal pathogen(25). Together, these findings suggest that TLR9 activation is likely to be important early during the priming and initial development of adaptive anticryptococcal immunity which subsequently modulates the development of effector responses. However, the mechanism(s) by which TLR9 activation affects the development of protective anticryptococcal immunity in vivo are unknown. Experimental pulmonary infections with C. neoformans in mice have demonstrated that thechemokine and cytokine response during the first week post-infection (wpi) directs the subsequent development and polarization of adaptive responses in C. neoformans infected lungs (17, 29, 30). The major “early” cytokines driving Th1 development are IFN-γ , TNF-α and IL-12 (31-34), while CC chemokine receptor 2 (CCR2) ligands are critical for mononuclear leukocyte recruitment (12, 13, 15, 35, 36). This combination of an early cytokine/chemokine milieu is needed for optimal accumulation and maturation of DCs in the lungs, which in turn is required for the generation of protective Th1 immunity. Our previous studies demonstrated that CCR2 is required for the recruitment of CD11b+ lung DCs and ExMs(12, 13, 15)and Th1 polarization in C. neoformans-infected lungs (37). CCR2 ligands, chiefly CCL2/MCP-1 and CCL7/MCP-3 are induced early during the C. neoformans lung infection (13, 38). CCL2/MCP-1 was shown to contribute to the recruitment of CD4+ T cells and macrophages into the lung (38); however, studies by Traynor et al. suggested that CCR2 ligand(s) other than CCL2 may be more important for generation of a protective Th1 response (36).
Although the molecular mechanism by which TLR9 promotes the development of a protective response to C. neoformans remains unknown, a link between TLR9 signaling and the CCR2-axis has been established in a bacterial pneumonia model (39). TLR9 is required for generation of CCL2 and the accumulation/ maturation of DCs during innate immune response to Klebsiella(39). It remains to be determined, whether there is a mechanistic link between TLR9 signaling and the CCR2-axis during cryptococcal infection in the lungs.
In this manuscript, we provide insight into the mechanism whereby TLR9 signaling contributes to the development of adaptive immunity during pulmonary C. neoformans infection. TLR9 signaling facilitates CCL7 and CCL12 induction in the infected lungs during the innate phase of the immune response. Our findings show that early CCL7 induction contributes to the following important immune processes: 1) accumulation and activation of DCs, 2) IFN-γ production during the initial afferent phase of cryptococcal infection, and 3) the accumulation of effector cells during the efferent response to the organism. These data reveal the previously unknown role of CCL7 in the anticryptococcal host defenses.
Materials and Methods
Mice
Female wild type BALB/c mice were obtained from Jackson Laboratories (Bar Harbor, ME). TLR9-/- mice on a BALB/c background were bred at the University of Michigan/Ann Arbor VA Medical Center using micro-isolator cages covered with a filter top, with food/water provided ad libitum. All KO mice were back-bred to BALB/c background for at least 6 generations.Mice were aged to 8 to 10 weeks at the time of infection. At the time of data collection, mice were humanely euthanized by CO2 inhalation. All experiments were approved by the University Committee on the Use and Care of Animals and the VA IACUC.
C. neoformans
C. neoformans strain 52D (ATCC 24067) was recovered from 10% glycerol frozen stocks stored at -80 °C and grown to stationary phase at 37 °C in Sabouraud dextrose broth (1% neopeptone, 2% dextrose; Difco, Detroit, MI) on a shaker. The cultures were then washed in non-pyrogenic saline (Travenol, Deerfield, IL), counted on a hemocytometer and diluted to 3.3 ×105 yeast cells /ml in sterile non-pyrogenic saline.
Intratracheal inoculation of C. neoformans
Mice were anesthetized via intraperitoneal injection of Ketamine/Xylazine (Ketamine/Xylazine 100/6.8 mg/kg/BW) and were restrained on a foam plate. A small incision was made through the skin covering the trachea. The underlying salivary glands and muscles were separated. Infection was performed by intratracheal injection of 30 μl (104 CFU) via 30-gauge needle actuated from a 1ml tuberculin syringe with C. neoformans suspension (3.3 ×105/ml). After inoculation, the skin was closed with cyanoacrylate adhesive and the mice were monitored during recovery from the anesthesia.
CCL7 reconstitution
Following infection, TLR9+/+ and TLR9-/- mice were treated with either rCCL7 (100 ng/mouse,purified to contain less than 0.01ng of LPS per dose;.PeproTech, Rock Hill, NJ) or sterile phosphate-buffered saline (PBS) in a volume of 20 uLvia the intranasal route at days 3, 5 and 7.
Lung CFU assay
For determination of microbial burden in the lungs, small aliquots of dispersed lungs were collected following the digest procedure. Series of 10-fold dilutions of the lung samples were plated on Sabouraud dextrose agar plates in duplicates of 10μl aliquots and incubated at room temperature. C. neoformans colonies were counted 2 days later and the number of CFU was calculated on a per-organ basis.
Lung leukocyte isolation and culture
The lungs from each mouse were excised, washed in RPMI, minced with scissors, digested enzymatically at 37 °C for 30 minutes in 15 ml/mouse of digestion buffer [RPMI, 5% FBS, penicillin and streptomycin (Invitrogen, Grand Island, NY); 1 mg/ml Collagenase A (Roche Diagnostics, Indianapolis, IN); and 30 μg/ml DNase (Sigma, St. Louis, MO)] and processed as previously described(29, 30). The cell suspension and tissue fragments were further dispersed by repeated aspiration through the bore of a 10 ml syringe and were centrifuged. Erythrocytes in the cell pellets were lysed by addition of 3 ml of NH4Cl buffer (0.829% NH4Cl, 0.1% KHCO3, and 0.0372% Na2EDTA, pH 7.4) for 3 minutes followed by a ten-fold excess of RPMI. Cells were re-suspended and a second cycle of syringe dispersion and filtration through a sterile 100 μm nylon screen (Nitex, Kansas City, MO) was performed. The filtrate was centrifuged for 25 minutes at 1500 × g in the presence of 20% Percoll (Sigma) to separate leukocytes from cell debris and epithelial cells. Leukocyte pellets were re-suspended in 5 ml of complete RPMI media and enumerated on a hemocytometer following dilution in Trypan Blue (Sigma). Leukocytes were plated at 5×106 cells/ml in 24-well plates and supernatants were harvested at 24h for cytokine analysis.
Brochoalveolar lavage (BAL)
Euthanized mice were lavaged after cannulation of the trachea with polyethylene tubing (PE50), which was attached to a 23-gauge needle on a tuberculin syringe. The lungs were lavaged twice with 1ml of phosphate-bufferd saline containing 5×10-5 M 2-Mercaptoethanol (2-ME; Sigma, St. Louis, MO) and protein inhibitor cocktail (Roche Molecular Biochemicals, Indianapolis, Ind.). The recovered fluid (1.8-1.9 ml total) was spun at 1500 rpm, and the supernatant was removed and analyzed for cytokines.
Protein evaluation by ELISA
Cytokine and chemokine protein concentrations were measured by DuoSet (R&D Systems, Minneapolis, MN) and PromoKine(PromoCells GmbH, Heidelberg, Germany) kits following the manufacturer’s specifications. All plates were read on a Versamaxplate reader (Molecular Devices, Sunnyvale, CA).
qRT-PCR
Total RNA was prepared using RNeasyPlus Mini Kit (Qiagen, Valencia, CA, USA) and first-strand cDNA was synthesized using SuperScript™III (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Cytokine mRNA was quantified with SYBR Green-based detection using an MX 3000P system (Stratagene, La Jolla, CA) according to the manufacturer’s protocols. Forty cycles of PCR (94 °C for 15 s followed by 60 °C for 30 s and 72 °C for 30 s) were performed on a cDNA template. The mRNA levels were normalized to GAPDH mRNA levels and relative expression is shown as % of GAPDH.
Antibodies and flow cytometric analysis
For the flow cytometry experiment, the antibodies were purchased from Biolend, San Diego, CA, including rat anti-murine CD16/CD32 (Fc block), rat anti-murine CD45 conjugated to allophycocyanin (APC), hamster anti-murine CD11c or CD8 conjugated to pacific blue (PB), rat anti-murine CD11b or CD4 conjugated to allophycocyanin-Cy7 (APC-Cy7), rat anti-murine Ly6G conjugated to phycoerythrin-Cy7 (PE-Cy7), rat anti-murine CD3 conjugated to PerCP-Cy5.5, rat anti-murine CD19 conjugated to PerCP-Cy5.5, rat anti-murine MHC Class II (IA), CD40, CD80, CD86, or CD69 conjugated to phycoerythrin (PE), rat anti-murine Ly6C or CD45RB conjugated to Fluorescein isothiocyanate (FITC).
Staining was performed as previously described [15]. Data were collected on a FACS LSR2 flow cytometer using FACSDiva software (Becton Dickinson Immunocytometry Systems, Mountain View, CA). A minimum of 20,000 cells were evaluated from a predominantly leukocytic population identified by CD45+-stained cells per sample. The flow data was analyzed by FlowJo software (Tree Star Inc., San Carlos, CA). Total numbers of each cell population were calculated by multiplying the frequency of the population by the total number of leukocytes (the percentage of CD45+ cells multiplied by the original hemocytometer count of total cells).
Calculations and statistics
Statistical significance was calculated using Student’s t-test for individual paired comparisons or t-test with Bonferoni adjustment, whenever multiple groups were compared. Means with p values of <0.05 were considered significantly different. All values are reported as means ± standard errors (SEM).
Results
Impaired pulmonary clearance of C. neoformans in TLR9-deficient mice is associated with the diminished accumulation and activation of the major effector leukocyte subsets during the efferent phase of the immune response in the lungs
Our first objective in this study was to determine if the impaired clearance in TLR9-deficient mice was associated with changes in the status of major effector cells subsets during the efferent phase of the adaptive immune response. The TLR9+/+ and TLR9-/- mice were infected with C. neoformans and enzymatically dispersed lungs were evaluated for fungal burden and the number/activation status of T cells, DCs, and macrophages. Fungal clearance during the effector/efferent phase 3 weeks postinfection (wpi) of the immune response was significantly diminished in TLR9-/- mice (Figure 1A). This defect in clearance was associated with a diminished recruitment of specific effector cell subset populations evaluated by flow cytometric analysis. Specifically, both CD4+ and CD8+ cell numbers in the lungs were dramatically diminished, resulting in nearly 15 million fewer pulmonary CD3+ lymphocytes (T cells) in TLR9-/- mice compare to TLR9+/+ mice (Figure 1B). The activation phenotype of CD4+ T cells in the lung was then determined. We observed that the percentage of CD45RB on CD4+ CD45RB low-density (CD45RBlow) cells and CD69 on total CD4+ T cells in TLR9-/- mice was moderately diminished when compared with TLR9+/+ mice (Figure 1C).No difference was observed in the numbers of CD4+ and CD8+ T cells in the lungs of uninfected TLR9+/+ and TLR9-/- mice (2.15±0.71 versus 1.70±0.27 ×106/lung and 1.31±0.69 versus 0.73±0.14 ×106/lung, respectively).Collectively, it is likely that the profound decrease in total T cell numbers in conjunction with their diminished activation contributes to the defect in cryptococcal clearance observed in TLR9-deficient mice.
Figure 1. Impaired clearance of C. neoformans in TLR9-/- mice during the efferent phase of the immune response is associated with a diminished accumulation and activation of T cells.
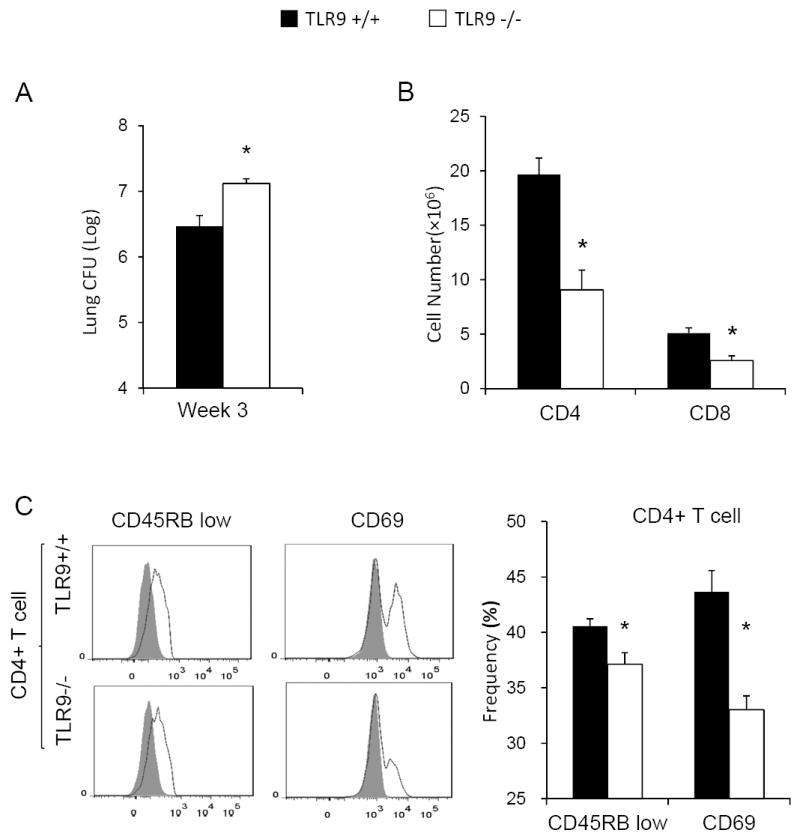
TLR9+/+ and TLR9-/- mice were inoculated intratracheally with 104C. neoformans 52D. The lungs were enzymatically dispersed at 3 weeks post-infection (wpi) for evaluation of fungal burdens (A) and pulmonary leukocyte analysis by flow cytometric analysis (B, C). Note that a 4-fold increase in fungal burden is accompanied by 50% decrease in total CD4+ and CD8+ T cell numbers (B) and diminished frequency of the activated effector CD4+ T-cell markers (C). Data represent mean ± SEM pooled from 2-3 separate matched experiments, N=6 and above for each of the analyzed parameters; * p< 0.05 in comparison between TLR9+/+ and TLR9-/- mice.
Execution of the effector functions by T cells depends on their interaction with antigen presenting cells (DCs and macrophages), which in turn results in activation of fungicidal mechanisms in these cells. We next sought to determine if TLR9 deletion affected the status of CD11b+ (conventional) DCs and major macrophage subsets in the lungs. DCs and macrophages were identified in the infected lungs using our established gating strategy (12).Briefly, autofluorescent (AF+) macrophages were distinguished from non-AF (AF-) DCs. Thereafter, relative expression of CD11b was used to separate CD11blo alveolar macrophages (AMs) from CD11bhiExMs and to identify CD11bhi DCs. (Figure 2A). Analysis of these three myeloid cell populations in the lungs at 3 wpi revealed a substantial defect in accumulation of CD11b+ DCs and ExMs in TLR9-/- mice, which contrasted with the robust recruitment of these subsets in TLR9+/+ mice at this time (Figure 2A). Interestingly, the number of AMs, was not affected by TLR9 deletion at 3 wpi (Figure 2A). Next we analyzed the expression of MHC Class II and two costimulatory molecules (CD40 and CD80) as these myeloid cell activation markers are crucial for their interactions with anti-fungal T cells. The CD11b+ DCs and ExMs from TLR9-deficient mice expressed less MHC Class II, CD40, and CD80 when compared with these populations in the lungs of TLR9+/+ mice (Figure 2B). In contrast, AM population obtained from infected TLR9+/+ and TLR9-/- mice showed no significant differences in the expression of these activation markers (Figure 2B), suggesting that TLR9 does not play a major role in activation of resident AM and further supporting the notion that AM are not an important effector cell during the efferent phase of pulmonary C. neoformans infection in mice (12, 40).No difference was observed in the numbers of AMs, ExMs and CD11b+ DCs in the lungs of uninfected TLR9+/+ and TLR9-/- mice (56.2±27.1 versus 56.3±11.0 ×104/lung; 8.5±3.6 versus 15.0±3.4 ×104/lung, and 36.7±11.5 versus 44.3±30.2 × 104/lung, respectively). Collectively, the analyses of CFU and pulmonary effector cell populations demonstrate that the impaired pulmonary clearance of C. neoformans in TLR9-deficient mice is linked to the impaired recruitment and activation of the major subsets of anticryptococcal effector cells.
Figure 2. Impaired clearance of C. neoformans in TLR9-/- mice is associated with a diminished accumulation and activation of CD11b+ dendritic cells (DCs), exudate macrophages (ExMs), but not alveolar macrophages (AMs).
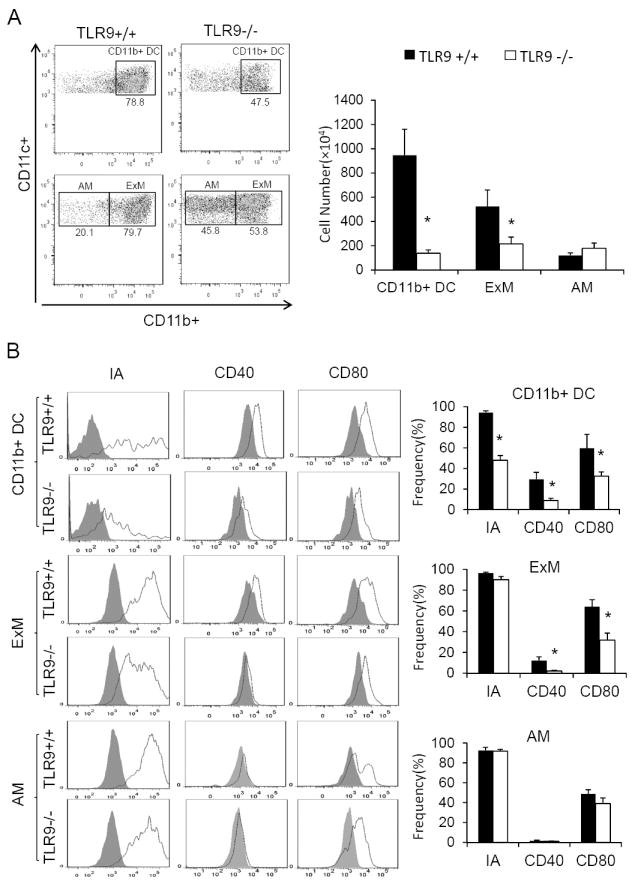
Lung leukocytes isolated from the infected lungs at 3 wpi were analyzed by flow cytometry. Myeloid cells were gated as described in Methods, with the final gating step of non-autofluorescent DCs (top scatter plots), and the auto-fluorescent AMs and ExMs (bottom scatter plots) displayed (A); The activation phenotype of mononuclear phagocytes was evaluated by the surface expression of MHCII (IA) and costimulatory molecules (CD40 and CD80). Stained samples are showed as solid line and isotype controls as shaded histograms. The bar graph presents mean frequencies of positive cells derived from these histograms(B). Note the dramatic deficiency in pulmonary DC and ExM numbers, diminished expression of activation markers on CD11b+ DCs and ExMs, in TLR9-/- mice and no effect of TLR9 deletion on these parameters in the AM subset. Data represent mean ± SEM pooled from 2-3 separate matched experiments, N=6 and above for each of the analyzed parameters; * p< 0.05 in comparison between TLR9+/+ and TLR9-/- mice.
TLR9 deletion results in impaired early accumulation and activation of pulmonary dendritic cells in C. neoformans infected lungs
Having determined that TLR9 signaling is required for the recruitment and activation of the major groups of the effector cells in C. neoformans infected lungs, we sought to identify possible upstream defects that could explain the defects in the efferent response found in TLR9-/- mice. We focused on recruitment/activation of pulmonary DCs at 1 wpi, a crucial step in the development of the adaptive immune response (32, 36). Although, an equivalent fungal burden has been observed in the lung of the TLR9-/- and TLR9+/+ mice at this time (Figure 3A), a significant decrease in total number of pulmonary CD11b+ DCs was observed in TLR9-/- mice compared to their TLR9+/+ counterparts (Figure 3B). Furthermore, a significantly lower percentage of TLR9-/- DCs expressed the activation markers MHC Class II or CD40 when compared with TLR9+/+ DCs (Figure 3C). Thus, TLR9 deletion profoundly impaired pulmonary DC accumulation and their activation during the afferent phase of the immune response. Interestingly, the numbers of macrophages were not yet affected by TLR9 deletion at this time (Figure 3B). Therefore, deficient TLR9 signaling impaired the recruitment and activation of pulmonary DCs which could explain the failure to develop a protective adaptive response against C. neoformans in TLR9-/- mice.
Figure 3. Afferent phase response in TLR9-/- mice is characterized by diminished recruitment and activation of pulmonary DCs at 1wpi.
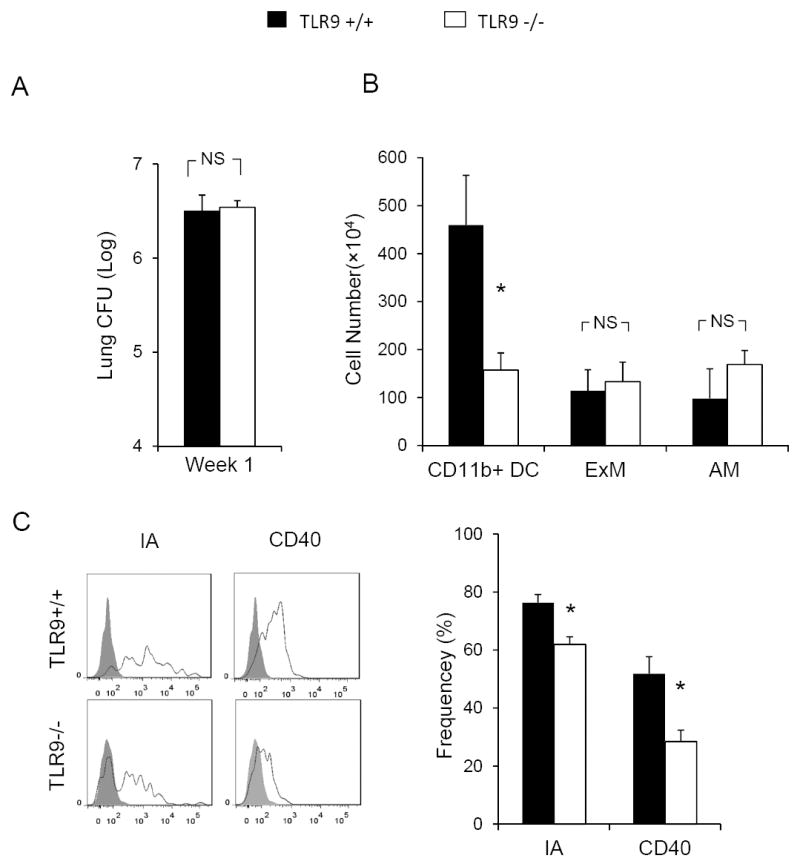
Fungal burden and flow cytometric analysis were performed on the dispersed lung samples from C. neoformans infected TLR9+/+ and TLR9-/- mice at 1 wpi. Pulmonary Leukocytes were isolated. CD11b+ DCs, AMs, and ExMs were gated as in Figure 2.In spite of equivalent fungal burden (A), a decreased number of CD11b+DCs was observed in TLR9-/-mice compared with TLR9+/+ mice (B). The expression of DC maturation surface markers MHC class II and CD40 was diminished in TLR9-/- mice compared to TLR9+/+ mice (C). Note that macrophage populations (B) are not significantly different between TLR9+/+ and TLR9-/- mice at 1 wpi. Data represent mean ± SEM pooled from 2-3 separate matched experiments, N=6 and above for each of the analyzed parameters; * p< 0.05 in comparison between TLR9+/+ and TLR9-/- mice.
Defect in Pulmonary DCs is associated with impaired early induction of CCR2 ligands and IFN-γ in C. neoformans infected lungs of TLR9-/- mice
Robust early expression of pulmonary inflammatory cytokines and chemokines is essential for the accumulation and activation of DCs and their subsequent “immune priming” effect during the development of a protective cellular response to C. neoformans (35, 36).In particular, CCR2 ligands promote DCs recruitment (13, 15, 41), while early induction of pro-inflammatory cytokines IL-12, TNF-α and IFN-γ correlates with the development of a protective Th1 response in cryptococcal infections (32-34). To determine if TLR9 deletion resulted in diminished expression of CCR2 ligands, RNA was isolated from lung leukocytes of both TLR9+/+ and TLR9-/-mice infected with C. neoformans at 1 wpi and evaluated by qRT-PCR. The relative expression data demonstrate that C. neoformans infection resulted in up-regulation of all known murine CCR2 ligands (CCL2/MCP-1; CCL7/MCP-3, and CCL12/MCP-5) in TLR9+/+ mice. CCL7 exhibited the highest expression level, followed by CCL2 and CCL12 (Figure 4A-C). By comparison, TLR9-/- mice showed a profound decrease in CCL7 and CCL12 induction whereas there was no change in the relative expression of CCL2. The dramatic defect in CCL7 production by lung leukocytes from TLR9-/- mice at 1 wpi has been confirmed at the protein level (Figure 4D). While TLR9+/+ mice demonstrate robust increase in CCL7 production at this time, TLR9-/- mice only show baseline (uninfected) level of CCL7 production. Additionally, the production of CCL2 protein was evaluated. Consistent with the outcomes of mRNA analysis, no difference between TLR9-/- and TLR9+/+ mice was observed (data not shown).These findings suggested that the deficiency in CCL7 (and possibly CCL12), rather than CCL2, contributed to decreased early accumulation of DCs in TLR9-/- mice (see Figure 3A).
Figure 4. TLR9 deletion alters early induction of CCR2 ligands: CCL7 and CCL12 during afferent phase of the immune response to C. neoformans in the infected lungs.
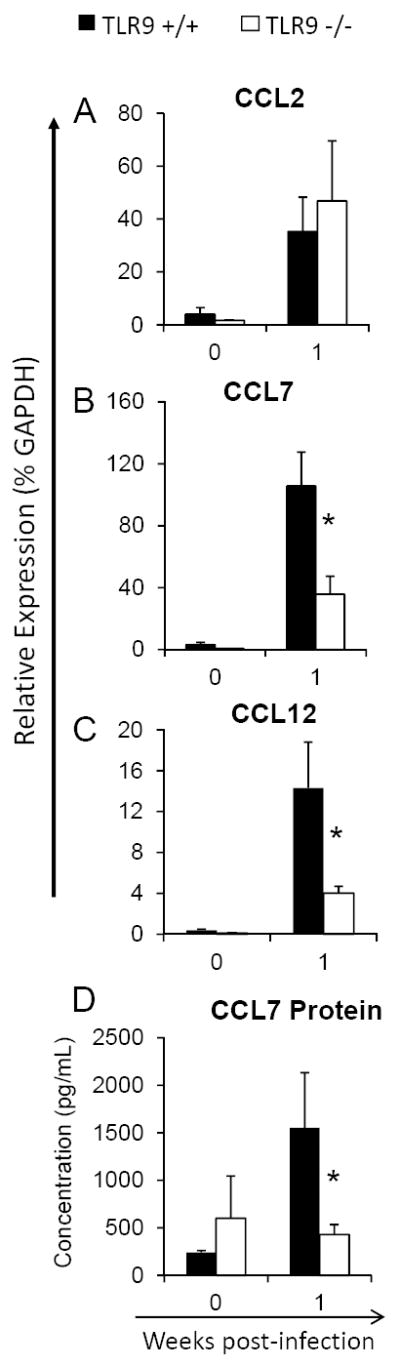
RNA was extracted from isolated lung leukocytes from C. neoformans infected TLR9+/+ and TLR9-/- mice, converted to cDNA and evaluated by qPCR for the relative expression of pulmonary chemokines. Chemokine protein levels were evaluated by ELISA in leukocyte culture supernatants. Baseline expression in uninfected mice 0 wpi and corresponding responses at 1 wpi are shown. Note a significant deficiency in the induction of CCR2 ligands CCL7/MCP3 (B and D) and CCL12/MCP5 (C) and equivalent expression of MCP1/CCL2 (A), compared to matched responses in TLR9+/+ mice. Data are expressed as mean relative expression ± SEM, pooled from 2 independent experiments, N=6 and above for each of the analyzed parameters; *p<0.05 in comparison with the matching TLR9+/+ mouse result.
We further explored if TLR9 deletion decreased the early induction of pro-inflammatory cytokines known to promote the development of protective immune response. We observed a significant decrease in IFN-γ expression (but not TNF-α or IL-12) in TLR9-/- mice at 1 wpi(Figure 5). Collectively, these data reveal a strong associationbetween TLR9, the expression of the CCR2 ligands CCL7 and CCL12, and the accumulation and activation of pulmonary DCs in mice which develop a protective immune response against cryptococcal lung infection.
Figure 5. TLR9 deletion alters early induction of Th1-type cytokine IFN-g during afferent phase of the immune response to C. neoformans in the infected lungs.
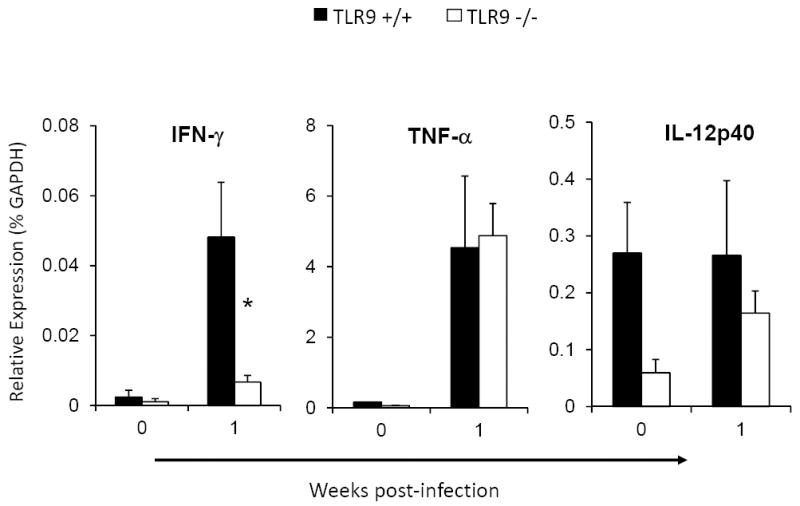
RNA was extracted from isolated lung leukocytes from C. neoformans infected TLR9+/+ and TLR9-/- mice, converted to cDNA and evaluated by qPCR for the relative expression of pulmonary cytokines/chemokines. Baseline expression in uninfected mice 0 wpi and corresponding responses at 1 wpi are shown. Note a significant deficiency in the induction of IFN-γ compared to matched responses in TLR9+/+ mice and equivalent expression of TNF-α and IL12p40 . Data are expressed as mean relative expression ± SEM (% GAPDH) (n=4-6). *p<0.05 in comparison with the matching TLR9+/+ mouse result.
CCL7 reconstitution improves the accumulation and activation of pulmonary dendritic cells and restores early IFN-γ production in TLR9 deficient mice during the afferent phase of the immune response to cryptococcal infection
TLR9 deletion resulted in a dramatic decrease in early CCL7 induction that occurred in concert with diminished early accumulation and activation of DCs and reduced IFN-γ induction in C. neoformans infected lungs. Such defects are highly associated with the development of a non-protective immune response against cryptococcal lung infection (32, 37, 42). To determine if decreased CCL7 was the mechanism leading to the immunological defects in TLR9-/- mice we studied the effects of the reconstitution of CCL7 on the outcomes of these immune parameters in C. neoformans-infected TLR9-/- mice. Following infection (day 0), we delivered CCL7 (100 ng/mouse in 20μl sterile PBS via the intranasal route) or sterile PBS (negative control) to TLR9+/+ and TLR9-/- mice at days 3, 5 and 7. At 1 wpi, CCL7 reconstitution in TLR9-/- mice resulted in a significant improvement (compared with TLR9-/- mice treated with PBS) in the accumulation of pulmonary DCs, to numbers comparable to those identified in TLR9+/+ mice treated with PBS(Figure 6A). Note that the numbers of DCs in CCL7-treated TLR9-/- mice remained lower than those identified in CCL7-treated TLR9+/+ mice suggesting that CCL7 could promote some additional DC recruitment in wild type animals. In addition to its effects on pulmonary DC number, CCL7 administration to TLR9-/- mice corrected the deficiencies in DC activation as the expression of both MHC class II and CD40 in CCL7-treated TLR9-/- mice were no longer significantly different from their CCL7-treated TLR9+/+ counterparts (Figure 6B). We also investigated whether CCL7 deficiency contributed to the diminished early induction of IFN-γ observed in TLR9-/- mice by assessing the level of IFN-γ in BAL fluid from CCL7 or PBS-treated TLR9+/+ and TLR9-/- mice at 1 wpi. ELISA analysis showed that CCL7 reconstitution restored pulmonary production of IFN-γ in CCL7-treated TLR9-/- mice when compared with CCL7-treated TLR9+/+ mice (Figure 6C). Thus, CCL7 reconstitution corrected the defect in early accumulation and activation of pulmonary DCs, and restored the early IFN-γ production in the lungs of TLR9 deficient mice.
Figure 6. CCL7 reconstitution improves the accumulation and activation of CD11b+ DCs and restores IFN-γ induction in TLR9-/- mice at week 1.
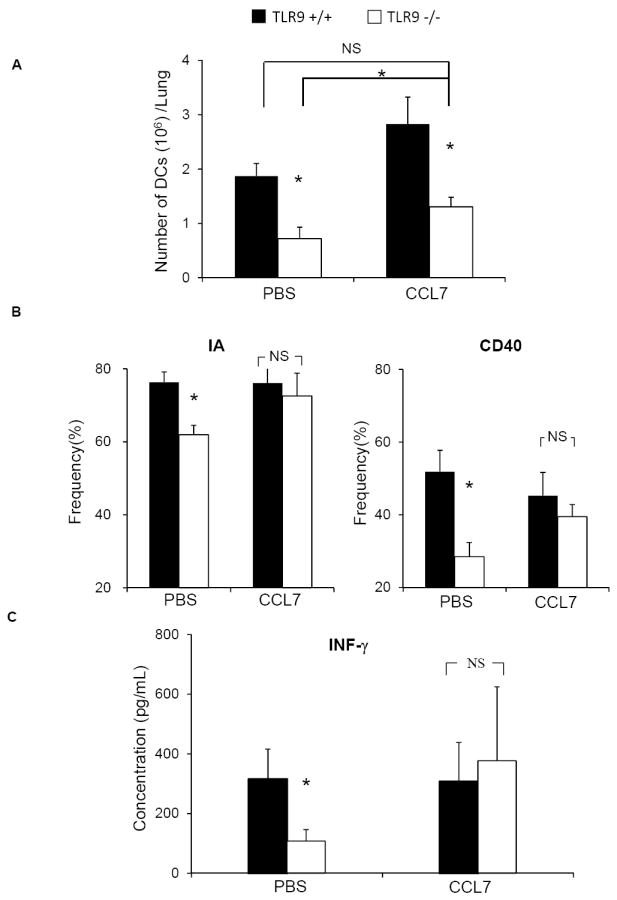
TLR9-/- and TLR9+/+ mice were inoculated intratracheally with 104C. neoformans and treated with either CCL7 (100 ng/mouse) or sterile PBS in a volume of 20 μL via intranasal inhalation on days 3, 5 and 7 post-infection. Pulmonary leukocytes were isolated at 1 wpi and evaluated by flow cytometric analysis (A-B). The BAL fluid was analyzed by ELISA to asses IFN-γ expression (C). Note that CCL7 reconstitution significantly improved the accumulation of CD11b+ DCs in the lungs of TLR9-/- mice (A) and corrected the defects in DC activation (B), and pulmonary production of IFN-γ (C). Data (mean ± SEM) represent 2 separate, matched experiments, N=6 and above for each of the analyzed parameters; * p< 0.05 in comparison between TLR9+/+ and TLR9-/- mice.
Reconstitution of CCL7 in TLR9 deficient mice corrects deficiencies in the recruitment of CD4+ and CD8 + T cells, CD11b+ DCs and exudate macrophages during the efferent phase of the immune response to C. neoformans
Early CCL7 reconstitution corrected the major defects identified in TLR9-/- mice during the afferent phase of the immune response; we next evaluated the effect of early CCL7 reconstitution on the effector cell subsets during the efferent phase of the response (3 wpi). Using the protocol described above, we delivered CCL7 or sterile PBS (negative control) to TLR9+/+ and TLR9-/- mice at days 3, 5 and 7. Numbers of lung leukocyte subsets were then assessed by flow cytometric analysis at 3 wpi. As expected, we observed a significant defect in total lymphocyte numbers including the specific number of both CD4+ and CD8+ T cell subsets in PBS-treated TLR9-/- mice compared with PBS-treated TLR9+/+ mice (Figure 7A). In contrast, results show that CCL7-treated TLR9-/- mice no longer displayed this impairment in T cell accumulation compared to CCL7-treated TLR9+/+ mice (Figure 7B). Next, the effects of CCL7 treatment on myeloid cell accumulation were evaluated. As anticipated, PBS-treated TLR9-/- mice had reduced numbers of CD11b+ DCs and ExMs relative to PBS-treated TLR9+/+ mice (Figure 8A). However, when TLR9-/- mice were reconstituted with CCL7 we observed equivalent accumulation of CD11b+ DCs and ExMs in the infected lungs in comparison with CCL7-treated TLR9+/+ mice (Figure 8B). Note that CCL7 administration did not affect the total number of AMs (Figure 8A and 8B), demonstrating that neither TLR9 signaling or subsequent CCL7 production affect AM accumulation in the lungs at 3 wpi. Thus CCL7 reconstitution during the first week of infection corrected the observed deficiency in the pulmonary accumulation of T cells, CD11b+ DCs and ExMs during the efferent phase of the immune response.
Figure 7. Early CCL7 reconstitution corrects accumulation of CD4+ and CD8+ T cells in TLR9-/-mice during efferent phase of the immune response (3 wpi).
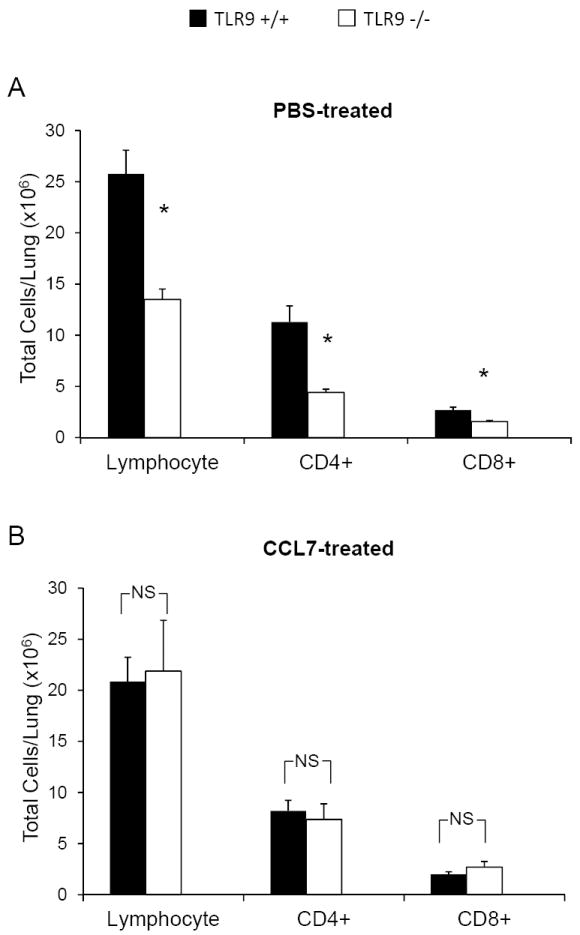
TLR9+/+ and TLR9-/- mice were inoculated with C. neoformans and treated with either CCL7 or sterile PBS and analyzed at 3wpi. Flow analysis demonstrated that in comparison with PBS treated group (A), CCL7 administration restored the pulmonary accumulation of total lymphocyte subset and both CD4+ and CD8+ T cell subsets in TLR9-/- mice (B). Data, (mean ± SEM) represent 2 separate, matched experiments, N=5 mice and above for each of the analyzed parameters; * p< 0.05 in comparison between TLR9+/+ and TLR9-/- mice; NS, no significant difference between TLR9-/- and TLR9+/+ groups.
Figure 8. Early CCL7 reconstitution corrects accumulation of pulmonary DCs and ExMs, but has no impact on AM population in TLR9-/- mice during the efferent phase of the immune response (3 wpi).
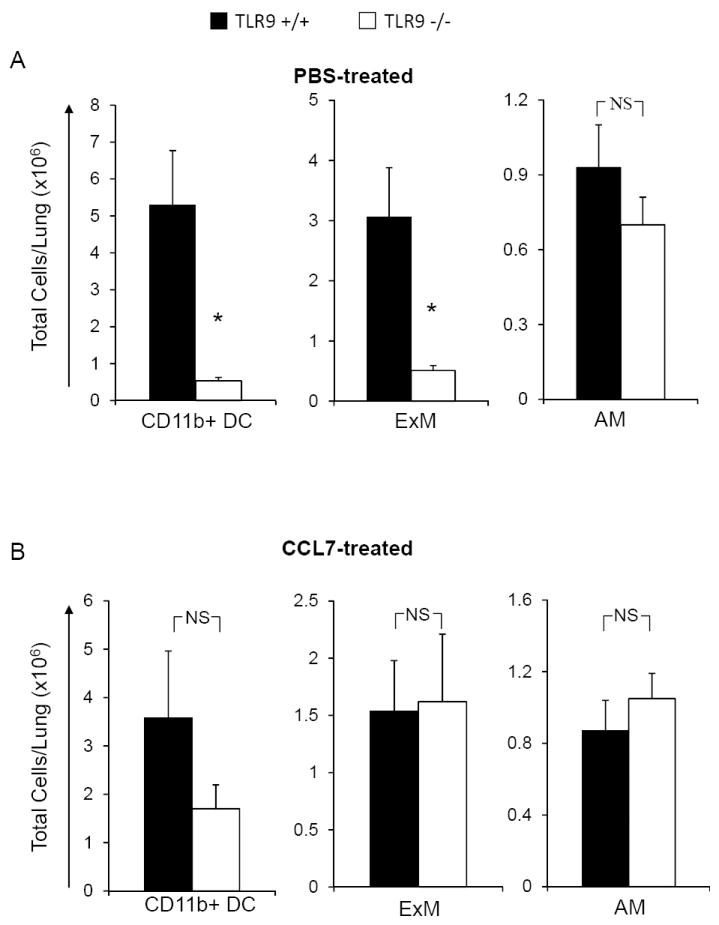
TLR9+/+ and TLR9-/- mice were inoculated with C. neoformans and treated with either CCL7 or sterile PBS and analyzed at 3wpi. Flow analysis demonstrated that in comparison with PBS treated group (A), CCL7 administration restored the pulmonary accumulation of CD11b+ DCs and ExMs (B). AM population displayed neither deficiency in TLR9-/- mice nor the effect of CCL7 administration (A) and (B). Data, (mean ± SEM) represent 2 separate, matched experiments, N=5 and above for each of the analyzed parameters; * p< 0.05 in comparison between TLR9+/+ and TLR9-/- mice; NS, no significant difference between TLR9-/- and TLR9+/+ groups.
CCL7 reconstitution mice abolishes the difference in pulmonary clearance of C. neoformans between TRL9+/+ and TLR9-/- mice but does not significantly improve fungal clearance
The final goal of this study was to determine if CCL7 reconstitution could abolish the effect of TLR9 deletion on C. neoformans clearance during the effector phase of the response (3 wpi). We analyzed the pulmonary fungal burden from infected TLR9+/+ and TLR9-/- mice lungs that had received either CCL7 or sterile PBS. As expected, our results showed that the fungal load of PBS-treated TLR9-/- mice was significantly higher than in PBS-treated TLR9+/+ mice (Figure 9). Results did show that the fungal burdens in CCL7-treated TLR9+/+ and TLR9-/- mice were no longer different from each other at 3 wpi (Figure 9); however, the fungal burdens in CCL7-treated TLR9-/- mice had not significantly improved when compared to PBS-treated TLR9-/- mice (Figure 9). These findings show that CCL7’s pronounced effect on cell recruitment is not the only mechanism whereby TLR9 contributes to the optimal clearance of the organism from the infected lungs.
Figure 9. Early CCL7 reconstitution abrogates the difference in pulmonary fungal burden between TLR9+/+ and TLR9-/- mice (3 wpi).
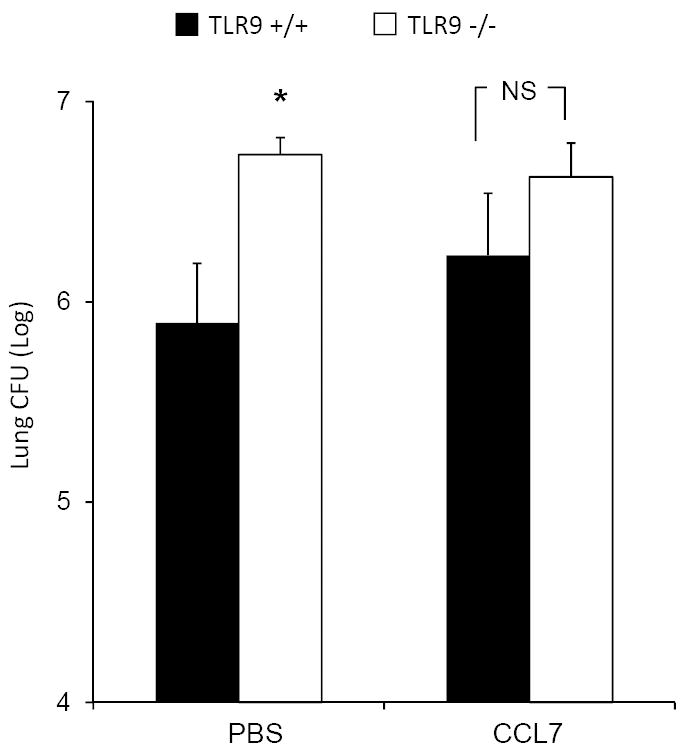
TLR9+/+ and TLR9-/- mice were inoculated with C. neoformans and treated with either CCL7 or sterile PBS as described in figure 5; the fungal burdens were compared at 3wpi. TLR9-/- mice treated with PBS alone show significant decrease in pulmonary clearance compared to TLR9+/+ mice (as shown in Figure 1), whereas TLR9-/- mice treated with CCL7 no longer display a significant increase in fungal burden compared to their wild type counterparts. However, no evident improvement in fungal clearance compared to the PBS treated group is observed. Data, (mean ± SEM) represent 2 separate, matched experiments, N=7 and above for each group; *p<0.05 in comparison between TLR9+/+ and TLR9-/- mice; NS, no significant difference between TLR9-/- and TLR9+/+ groups.
Discussion
The goal of this study was to evaluate the mechanism by which TLR9 promotes the development of protective immunity to C. neoformans. Our data demonstrate that C. neoformans infection results in a robust, early induction of CCR2 ligands, CCL7 and CCL12, which is a crucial downstream signal that aids to the development of protective immunity against cryptococcal infection in the lungs. This conclusion is supported by the following novel findings. First, deletion of TLR9 resulted in: 1) diminished induction of CCR2 ligands CCL7 and CCL12, and failure to induce INF-γ at the afferent phase of the immune response; 2) diminished early accumulation and activation of CD11b+ DCs; 3) impaired accumulation and activation of major effector leukocyte subsets such as T cells, CD11b+ DCs, ExMs; and 4) diminished fungal clearance during the efferent phase of the immune response in C. neoformans infected lungs. Second, delivery of CCL7 into the lungs of TLR9-/- mice during the first week of infection: a) corrected early induction of IFN-γ and the accumulation/activation of pulmonary CD11b+ DCs during the afferent phase of the immune response; and b) restored accumulation of CD4+ and CD8+ T cells, CD11b+ DCs, and ExMs, in the lungs of TLR9-/- mice during the efferent phase of the response c) alleviated the effect of TLR9 deletion on the fungal clearance. Collectively, these data reveal that early CCL7 induction downstream of TLR9 activation is a crucial step in the development of a protective immunity against cryptococcal lung infection, especially the recruitment of the essential effector cell subsets.
Our previous studies documented that TLR9 signaling was required for the development of the protective immune response in C. neoformans infected lungs (26), however, the mechanism by which TLR9 contributed to the development of the immune protection in vivo has been unknown. The finding that TLR9 affected clearance of C. neoformans from 3 wpi onwards suggested that TLR9 signaling was particularly important for proper execution of the adaptive (efferent) phase of the immune response. Consistent with this, we identified a group of major defects in the immunophenotype of TLR9-/- mice including diminished accumulation and activation of all major groups of cells engaged in execution of the effector phase of the immune response. We demonstrated that TLR9 signaling is required for pulmonary recruitment of CD4+ and CD8+ T cells and is required for optimal activation of CD4+ T cells (Figure 1C). Furthermore, we found that TLR9 signaling contributes to the robust accumulation and activation of CD11b+ DCs and ExMs, which are critical contributors to the protective immune response against cryptococcal lung. The CD11b+ DCs have a major role in initiation/priming of the adaptive immune response, however, recent data also support their role in activation of the effector T cells coming into the lungs and thus maintaining a Th1 “cytokine environment” during the effector phase of anticryptococcal response in the infected lungs (13, 43). Our recently published studies also demonstrate that ExM, and not AM, are the dominant macrophage subset expressing fungicidal molecules and are efficient killers of C. neoformansin vitro(12). Collectively, our data show that TLR9 signaling contributes to the recruitment and activation of T cells, CD11b+ DCs and ExMs, and the defect in accumulation and activation of these cells is associated with diminished adaptive clearance of C. neoformans (Figure 1A).
While the effects of TLR9 deletion were most profound during the efferent phase of the immune response, TLR9 is abundantly expressed by cells of the innate immune system and the importance of TLR9 signaling during the innate (afferent) phase of the immune response to C. neoformans has been demonstrated (25, 27). To understand the upstream mechanisms by which TLR9 signaling affected the development of the adaptive immunity, we analyzed the effects of TLR9 deletion on the afferent phase of the immune response. Consistent with our hypothesis, infected TLR9-/- mice displayed a profound defect in DC accumulation and activation (Figure 3). Our data suggests that impaired DC accumulation may be attributable to diminished production of CCR2 ligands, notably CCL7 and CCL12, in TLR9-/- mice, a finding compatible with the previously described role of CCR2 in mediating the recruitment of these cells to the lungs (13, 15, 36). Furthermore, TLR9 deletion affected expression of early IFN-γ, known to be important for activation of DCs (32-34). Interestingly, TLR9 deletion was not associated with decreased induction of IL-12, TNF-α or CCL2 (Figure 5), Collectively, these data provided a mechanistic explanation for the decreased accumulation and maturation of DCs in the infected lungs, which in turn was a likely cause of the downstream impairments observed during the efferent response in the lungs of TLR9-/- mice.
Since CCL7 is the most highly expressed chemokine in the infected lungs of the TLR9+/+ mice at 1wpi, and is significantly abrogated in the infected TLR9-/- mice, we tested if CCL7 reconstitution in TLR9-/- would restore the immunological defects resultant from TLR9 deletion. CCL7 reconstitution to TLR9-deficient mice improved the accumulation and activation of CD11b+ DCs and restored early induction of IFN-γ in the infected lungs. This suggests that the robust CCL7 induction that occurs downstream from TLR9 activation has an important role in the cascade of molecular events leading to the development of the adaptive immunity to C. neoformans. Furthermore, the immunological defects downstream of TLR9 deletion occurred independently of CCL2 (Figure 4A), which is consistent with a previous study that postulated the importance of CCL2-independent CCR2 pathway in anticryptococcal host defenses (36). Our data reveal that early CCL7 has an important role in optimal accumulation and activation of CD11b+ DCs in the lungs and induction of, Th1-priming, IFN-γ during the afferent phase of the immune response.
Another interesting finding, underscoring the pivotal role of early TLR9-mediated CCL7-induction, is the profound effect that CCL7-reconstitution had on the effector cell subsets during the efferent phase of the immune response. Although, the intranasal instillation of CCL7 into the lung does not perfectly mimic the indigenous production of this factor (which is most likely restricted to the infected areas of lungs and produced with continuous kinetics),CCL7 reconstitution in TLR9-/- mice during the first week of infection corrected the defect in accumulation of all major effector cell subsets present in the lungs at 3 wpi. Specifically, accumulation of CD4+ and CD8+ T cells, CD11b+ DCs, and ExMsin the lung of C. neoformans infected TLR9-/- mice was completely restored in CCL7 treated mice at 3 wpi (Figure 7-8). This result showed that the early TLR9-mediated CCL7 induction is required for proper accumulation of these effector cell subsets during cryptococcal lung infection.
Given that early CCL7 reconstitution corrected the defect of accumulation of the major effector cells at the efferent phase, we expected that CCL7 reconstitution would restore the pulmonary clearance of C. neoformans infected TLR9-/- mice. However, CCL7 reconstitution was not sufficient to completely correct the defect of pulmonary clearance in TLR9-/- mice. These results suggest the presence of another TLR9-mediated mechanism(s), in addition to its role in mediating effector cell recruitment, contributes to the development ofhostdefense against C. neoformans.
Apart from impaired CCL7 induction in TLR9-/- mice, we also observed a profound decrease in CCL12 induction at 1 wpi. This could be another potential mechanism that could contribute to the impaired clearance of C. neoformans. However, we believe that this hypothesis is rather unlikely. The relative expression of CCL12 is very low by comparison with other CCR2 ligands and is especially dwarfed by high expression of CCL7. Since, both of these ligands are thought to act mainly via CCR2 it is hard to expect that CCL12 would be needed apart from CCL7 to restore the clearance of C. neoformans in TLR9-/- mice. Future studies will be needed to determine whether this or other additional mechanism(s) contribute to the full development of the protective immunity against cryptococcal lung infection downstream from TLR9 signaling.
In summary, our findings substantially enhance our understanding of how TLR9 signaling promotes the development of the adaptive immune response during crytpotoccal infection in the lungs. The induction of CCL7, mediated by TLR9 signaling, is required for the recruitment and activation of CD11b+ DCs at the early time points post infection and subsequent accumulation of CD11b+ DCs, ExMs, CD4+ and CD8+ T cells during the efferent phase of the immune response. These findings define a novel role for CCL7 in the cryptococcal infection model that appears to be unique and non-redundant. Future studies utilizing CCL7KO mice and and/or neutralizing CCL7 will be needed to evaluate full impact of CCL7 in cryptococcal infection. Providing that these effects will prove to be equally important in humans, TLR9 and the CCL7/CCR2 axis could become targets for the development of novel biologic therapeuticsaimed at improving our ability to treat invasive fungal lung infections.
Acknowledgments
The authors wish to thank Dr. Xiumiao He for her help and acknowledge technical assistance of Ye Lu, Joanne Sonstein, Daniel Lyons, Daniel Meister, Michael Aljadah, and Zachary Hadd. We thank the Undergraduate Research Opportunity program at the University of Michigan for supporting the undergraduate students working in our laboratory.
Funding: This work was supported in part by Merit Review Grants (M.A.O. and G.B.T.) and Career Development Award (J.J.O.) from the Department of Veterans Affairs, NHLBI HL97456 and HL25243 (TJS), K08-HL094762 (U.B.).
References
- 1.Chuck SL, Sande MA. Infections with Cryptococcus neoformans in the acquired immunodeficiency syndrome. N Engl J Med. 1989;321:794–799. doi: 10.1056/NEJM198909213211205. [DOI] [PubMed] [Google Scholar]
- 2.Pappas PG, Perfect JR, Cloud GA, Larsen RA, Pankey GA, Lancaster DJ, Henderson H, Kauffman CA, Haas DW, Saccente M, Hamill RJ, Holloway MS, Warren RM, Dismukes WE. Cryptococcosis in human immunodeficiency virus-negative patients in the era of effective azole therapy. Clin Infect Dis. 2001;33:690–699. doi: 10.1086/322597. [DOI] [PubMed] [Google Scholar]
- 3.Huffnagle GB, Lipscomb MF, Lovchik JA, Hoag KA, Street NE. The role of CD4+ and CD8+ T cells in the protective inflammatory response to a pulmonary cryptococcal infection. J Leukoc Biol. 1994;55:35–42. doi: 10.1002/jlb.55.1.35. [DOI] [PubMed] [Google Scholar]
- 4.Huffnagle GB, Yates JL, Lipscomb MF. Immunity to a pulmonary Cryptococcus neoformans infection requires both CD4+ and CD8+ T cells. J Exp Med. 1991;173:793–800. doi: 10.1084/jem.173.4.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mody CH, Chen GH, Jackson C, Curtis JL, Toews GB. Depletion of murine CD8+ T cells in vivo decreases pulmonary clearance of a moderately virulent strain of Cryptococcus neoformans. J Lab Clin Med. 1993;121:765–773. [PubMed] [Google Scholar]
- 6.Huffnagle GB, Yates JL, Lipscomb MF. T cell-mediated immunity in the lung: a Cryptococcus neoformans pulmonary infection model using SCID and athymic nude mice. Infect Immun. 1991;59:1423–1433. doi: 10.1128/iai.59.4.1423-1433.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arora S, Hernandez Y, Erb-Downward JR, McDonald RA, Toews GB, Huffnagle GB. Role of IFN-gamma in regulating T2 immunity and the development of alternatively activated macrophages during allergic bronchopulmonary mycosis. J Immunol. 2005;174:6346–6356. doi: 10.4049/jimmunol.174.10.6346. [DOI] [PubMed] [Google Scholar]
- 8.Bava AJ, Afeltra J, Negroni R, Diez RA. Interferon gamma increases survival in murine experimental cryptococcosis. Rev Inst Med Trop Sao Paulo. 1995;37:391–396. doi: 10.1590/s0036-46651995000500003. [DOI] [PubMed] [Google Scholar]
- 9.Kawakami K, Tohyama M, Teruya K, Kudeken N, Xie Q, Saito A. Contribution of interferon-gamma in protecting mice during pulmonary and disseminated infection with Cryptococcus neoformans. FEMS Immunol Med Microbiol. 1996;13:123–130. doi: 10.1016/0928-8244(95)00093-3. [DOI] [PubMed] [Google Scholar]
- 10.Lovchik JA, Lyons CR, Lipscomb MF. A role for gamma interferon-induced nitric oxide in pulmonary clearance of Cryptococcus neoformans. Am J Respir Cell Mol Biol. 1995;13:116–124. doi: 10.1165/ajrcmb.13.1.7598935. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Y, Wang F, Tompkins KC, McNamara A, Jain AV, Moore BB, Toews GB, Huffnagle GB, Olszewski MA. Robust Th1 and Th17 immunity supports pulmonary clearance but cannot prevent systemic dissemination of highly virulent Cryptococcus neoformans H99. Am J Pathol. 2009;175:2489–2500. doi: 10.2353/ajpath.2009.090530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Osterholzer JJ, Chen GH, Olszewski MA, Zhang YM, Curtis JL, Huffnagle GB, Toews GB. Chemokine receptor 2-mediated accumulation of fungicidal exudate macrophages in mice that clear cryptococcal lung infection. Am J Pathol. 2011;178:198–211. doi: 10.1016/j.ajpath.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Osterholzer JJ, Curtis JL, Polak T, Ames T, Chen GH, McDonald R, Huffnagle GB, Toews GB. CCR2 mediates conventional dendritic cell recruitment and the formation of bronchovascular mononuclear cell infiltrates in the lungs of mice infected with Cryptococcus neoformans. J Immunol. 2008;181:610–620. doi: 10.4049/jimmunol.181.1.610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Olszewski MA, Zhang Y, Huffnagle GB. Mechanisms of cryptococcal virulence and persistence. Future Microbiol. 2010;5:1269–1288. doi: 10.2217/fmb.10.93. [DOI] [PubMed] [Google Scholar]
- 15.Osterholzer JJ, Chen GH, Olszewski MA, Curtis JL, Huffnagle GB, Toews GB. Accumulation of CD11b+ lung dendritic cells in response to fungal infection results from the CCR2-mediated recruitment and differentiation of Ly-6Chigh monocytes. J Immunol. 2009;183:8044–8053. doi: 10.4049/jimmunol.0902823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dan JM, Wang JP, Lee CK, Levitz SM. Cooperative stimulation of dendritic cells by Cryptococcus neoformans mannoproteins and CpG oligodeoxynucleotides. PLoS One. 2008;3:e2046. doi: 10.1371/journal.pone.0002046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huffnagle GB, Toews GB, Burdick MD, Boyd MB, McAllister KS, McDonald RA, Kunkel SL, Strieter RM. Afferent phase production of TNF-alpha is required for the development of protective T cell immunity to Cryptococcus neoformans. J Immunol. 1996;157:4529–4536. [PubMed] [Google Scholar]
- 18.Romani L. Immunity to fungal infections. Nat Rev Immunol. 2004;4:1–23. doi: 10.1038/nri1255. [DOI] [PubMed] [Google Scholar]
- 19.Yauch LE, Mansour MK, Shoham S, Rottman JB, Levitz SM. Involvement of CD14, toll-like receptors 2 and 4, and MyD88 in the host response to the fungal pathogen Cryptococcus neoformans in vivo. Infect Immun. 2004;72:5373–5382. doi: 10.1128/IAI.72.9.5373-5382.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 21.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 22.Kumagai Y, Takeuchi O, Akira S. Pathogen recognition by innate receptors. J Infect Chemother. 2008;14:86–92. doi: 10.1007/s10156-008-0596-1. [DOI] [PubMed] [Google Scholar]
- 23.Biondo C, Midiri A, Messina L, Tomasello F, Garufi G, Catania MR, Bombaci M, Beninati C, Teti G, Mancuso G. MyD88 and TLR2, but not TLR4, are required for host defense against Cryptococcus neoformans. Eur J Immunol. 2005;35:870–878. doi: 10.1002/eji.200425799. [DOI] [PubMed] [Google Scholar]
- 24.Nakamura K, Miyagi K, Koguchi Y, Kinjo Y, Uezu K, Kinjo T, Akamine M, Fujita J, Kawamura I, Mitsuyama M, Adachi Y, Ohno N, Takeda K, Akira S, Miyazato A, Kaku M, Kawakami K. Limited contribution of Toll-like receptor 2 and 4 to the host response to a fungal infectious pathogen, Cryptococcus neoformans. FEMS Immunol Med Microbiol. 2006;47:148–154. doi: 10.1111/j.1574-695X.2006.00078.x. [DOI] [PubMed] [Google Scholar]
- 25.Nakamura K, Miyazato A, Xiao G, Hatta M, Inden K, Aoyagi T, Shiratori K, Takeda K, Akira S, Saijo S, Iwakura Y, Adachi Y, Ohno N, Suzuki K, Fujita J, Kaku M, Kawakami K. Deoxynucleic acids from Cryptococcus neoformans activate myeloid dendritic cells via a TLR9-dependent pathway. J Immunol. 2008;180:4067–4074. doi: 10.4049/jimmunol.180.6.4067. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y, Wang F, Bhan U, Huffnagle GB, Toews GB, Standiford TJ, Olszewski MA. TLR9 signaling is required for generation of the adaptive immune protection in Cryptococcus neoformans-infected lungs. Am J Pathol. 2010;177:754–765. doi: 10.2353/ajpath.2010.091104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Edwards L, Williams AE, Krieg AM, Rae AJ, Snelgrove RJ, Hussell T. Stimulation via Toll-like receptor 9 reduces Cryptococcus neoformans-induced pulmonary inflammation in an IL-12-dependent manner. Eur J Immunol. 2005;35:273–281. doi: 10.1002/eji.200425640. [DOI] [PubMed] [Google Scholar]
- 28.Wang JP, Lee CK, Akalin A, Finberg RW, Levitz SM. Contributions of the MyD88-dependent receptors IL-18R, IL-1R, and TLR9 to host defenses following pulmonary challenge with Cryptococcus neoformans. PLoS One. 2011;6:e26232. doi: 10.1371/journal.pone.0026232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olszewski MA, Huffnagle GB, McDonald RA, Lindell DM, Moore BB, Cook DN, Toews GB. The role of macrophage inflammatory protein-1alpha/CCL3 in regulation of T cell-mediated immunity to Cryptococcus neoformans infection. J Immunol. 2000;165:6429–6436. doi: 10.4049/jimmunol.165.11.6429. [DOI] [PubMed] [Google Scholar]
- 30.Olszewski MA, Huffnagle GB, Traynor TR, McDonald RA, Cook DN, Toews GB. Regulatory effects of macrophage inflammatory protein 1alpha/CCL3 on the development of immunity to Cryptococcus neoformans depend on expression of early inflammatory cytokines. Infect Immun. 2001;69:6256–6263. doi: 10.1128/IAI.69.10.6256-6263.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bauman SK, Huffnagle GB, Murphy JW. Effects of tumor necrosis factor alpha on dendritic cell accumulation in lymph nodes draining the immunization site and the impact on the anticryptococcal cell-mediated immune response. Infect Immun. 2003;71:68–74. doi: 10.1128/IAI.71.1.68-74.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Herring AC, Falkowski NR, Chen GH, McDonald RA, Toews GB, Huffnagle GB. Transient neutralization of tumor necrosis factor alpha can produce a chronic fungal infection in an immunocompetent host: potential role of immature dendritic cells. Infect Immun. 2005;73:39–49. doi: 10.1128/IAI.73.1.39-49.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Herring AC, Lee J, McDonald RA, Toews GB, Huffnagle GB. Induction of interleukin-12 and gamma interferon requires tumor necrosis factor alpha for protective T1-cell-mediated immunity to pulmonary Cryptococcus neoformans infection. Infect Immun. 2002;70:2959–2964. doi: 10.1128/IAI.70.6.2959-2964.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoag KA, Lipscomb MF, Izzo AA, Street NE. IL-12 and IFN-gamma are required for initiating the protective Th1 response to pulmonary cryptococcosis in resistant C.B-17 mice. Am J Respir Cell Mol Biol. 1997;17:733–739. doi: 10.1165/ajrcmb.17.6.2879. [DOI] [PubMed] [Google Scholar]
- 35.Huffnagle GB, Traynor TR, McDonald RA, Olszewski MA, Lindell DM, Herring AC, Toews GB. Leukocyte recruitment during pulmonary Cryptococcus neoformans infection. Immunopharmacology. 2000;48:231–236. doi: 10.1016/s0162-3109(00)00222-8. [DOI] [PubMed] [Google Scholar]
- 36.Traynor TR, Herring AC, Dorf ME, Kuziel WA, Toews GB, Huffnagle GB. Differential roles of CC chemokine ligand 2/monocyte chemotactic protein-1 and CCR2 in the development of T1 immunity. J Immunol. 2002;168:4659–4666. doi: 10.4049/jimmunol.168.9.4659. [DOI] [PubMed] [Google Scholar]
- 37.Traynor TR, Kuziel WA, Toews GB, Huffnagle GB. CCR2 expression determines T1 versus T2 polarization during pulmonary Cryptococcus neoformans infection. J Immunol. 2000;164:2021–2027. doi: 10.4049/jimmunol.164.4.2021. [DOI] [PubMed] [Google Scholar]
- 38.Huffnagle GB, Strieter RM, Standiford TJ, McDonald RA, Burdick MD, Kunkel SL, Toews GB. The role of monocyte chemotactic protein-1 (MCP-1) in the recruitment of monocytes and CD4+ T cells during a pulmonary Cryptococcus neoformans infection. J Immunol. 1995;155:4790–4797. [PubMed] [Google Scholar]
- 39.Bhan U, Lukacs NW, Osterholzer JJ, Newstead MW, Zeng X, Moore TA, McMillan TR, Krieg AM, Akira S, Standiford TJ. TLR9 is required for protective innate immunity in Gram-negative bacterial pneumonia: role of dendritic cells. J Immunol. 2007;179:3937–3946. doi: 10.4049/jimmunol.179.6.3937. [DOI] [PubMed] [Google Scholar]
- 40.Shao XP, Mednick A, Alvarez M, van Rooijen N, Casadevall A, Goldman DL. An innate immune system cell is a major determinant of species-related susceptibility differences to fungal pneumonia. Journal of Immunology. 2005;175:3244–3251. doi: 10.4049/jimmunol.175.5.3244. [DOI] [PubMed] [Google Scholar]
- 41.Chiu BC, Freeman CM, Stolberg VR, Hu JS, Zeibecoglou K, Lu B, Gerard C, Charo IF, Lira SA, Chensue SW. Impaired lung dendritic cell activation in CCR2 knockout mice. Am J Pathol. 2004;165:1199–1209. doi: 10.1016/S0002-9440(10)63380-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huffnagle GB, Chen GH, Curtis JL, McDonald RA, Strieter RM, Toews GB. Down-regulation of the afferent phase of T cell-mediated pulmonary inflammation and immunity by a high melanin-producing strain of Cryptococcus neoformans. J Immunol. 1995;155:3507–3516. [PubMed] [Google Scholar]
- 43.Lindell DM, Ballinger MN, McDonald RA, Toews GB, Huffnagle GB. Diversity of the T-cell response to pulmonary Cryptococcus neoformans infection. Infect Immun. 2006;74:4538–4548. doi: 10.1128/IAI.00080-06. [DOI] [PMC free article] [PubMed] [Google Scholar]