

Abstract
The mechanisms that control toll-like receptor induced responses including endotoxin tolerance have been not well understood. The tuberous sclerosis complex 1 (TSC1) is a tumor suppressor that inhibits the mammalian target of rapamycin (mTOR). We show here that deficiency of TSC1 results in enhanced activation of not only mTOR complex 1 (mTORC1), but also JNK1/2, following lipopolysaccharide stimulation in macrophages. TSC1 deficient macrophages produce elevated proinflammatory cytokines and nitric oxide in response to multiple TLR ligands. Such enhanced TLR-induced responses can be inhibited by reducing mTORC1 and JNK1/2 activities with chemical inhibitors or small hairpin RNA, suggesting that TSC1 negatively controls TLR responses through both mTORC1 and JNK1/2. The impact of TSC1 deficiency appeared not limited to TLRs, as NOD- and -RIG-I/MDA-5 induced innate responses were also altered in TSC1 deficient macrophages. Furthermore, TSC1 deficiency appears to cause impaired induction of endotoxin tolerance in vitro and in vivo, which is correlated with increased JNK1/2 activation and can be reversed by JNK1/2 inhibition. Our results reveal a critical role of TSC1 in regulating innate immunity by negative control of mTORC1 and JNK1/2 activation.
Introduction
Toll-like receptors (TLR) recognize pathogen-associated molecular patterns and are critical for not only innate immunity but also adaptive immune responses against microbial infection (1-3). TLR-mediated responses are mediated by the Myd88-dependent and TRIF-dependent pathways. The MyD88-dependent pathway is utilized by all TLRs except TLR3 while the TRIF-dependent pathway is utilized by TLR3 and TLR4. These pathways eventually lead to the activation of IκB kinases (IKKs) and MAPKs JNK1/2, p38, and Erk1/2. Activated IKKs phosphorylate IκB, resulting in its degradation and the nuclear translocation of NFκB. These MAPKs activate multiple transcription factors such as AP1, which together with NFκB, promote transcription of cytokines and costimulatory molecules (4-9). While TLR-mediated responses are important for host defense against microbial infection, TLR-mediated responses have to be tightly regulated. Dysregulated TLR-induced inflammatory responses can contribute to the pathogenesis of diseases (10).
The mammalian target of rapamycin (mTOR) is a central regulator of cell growth and proliferation by integrating signals from nutrients, energy status, and growth factors. It has been implicated in numerous processes such as autophagy, transcription, protein synthesis, ribosome biogenesis, and cell survival (11-13). mTOR forms two signaling complexes, mTORC1 and mTORC2, with distinct signaling properties. mTORC1 phosphorylates pS6K1 and 4E-BP1 to promote cell growth and proliferation and is sensitive to rapamycin inhibition (14). mTORC2 phosphorylates Akt on serine 473 and PKCα to regulate cell survival and actin polymerization respectively (15, 16). Recently, mTORC2 has been found to be able to phosphorylate PKCθ to promote Th2 immune responses in T cells (17).
Mounting evidence indicates that mTOR-mediated signaling regulates both adaptive and innate immune cell development and functions (12, 18). In T cells, mTOR can be activated by the T cell receptor in a RasGRP1-dependent manner, and is negatively controlled by diacylglycerol kinases α and ζ (19). mTOR signaling regulates effector T cell differentiaton, regulatory T cell generation and function, memery T cell responses, and T cell trafficking (20-23). Similarly, LPS and several other TLR ligands have been shown to be able to induce mTORC1 and mTORC2 activation. Both positive and negative functions of mTOR in TLR-induced innate immune responses have been reported in various innate cell lineages using rapamycin as an mTOR inhibitor (24-31). While mTOR deficiency in innate immune cells has not been reported, defects of effector molecules downstream of mTOR have profound impacts on innate immunity. Deficiency of pS6K1/2 drastically decreases TLR- or virus-induced IFN-α production by plasmacytoid dendritic cell (pDCs) (32). In contrast, deficiency of 4E-BP1/2 results in enhanced IFNα and β production and resistance to viral infection in vitro and in vivo due to increased IRF-7 translation (33). Although these observations have suggested critical roles of mTOR signaling in innate immune response, the mechanisms and the importance of mTOR regulation in innate immunity are not well understood.
The tuberous sclerosis complex 1 (TSC1) associates with TSC2 to form a heterodimer. TSC1 stabilizes TSC2 and prevents its ubiquitin-mediated degradation (34). TSC1/2 complex negatively regulates mTORC1 through the GTPase activation property of TSC2 to RheB, a small GTPase protein that promotes to mTORC1 activation (35). Loss of function mutations in TSC1 or TSC2 result in tumorigensis correlated with elevated mTORC1 signaling (34). The role of TSC1 and the importance of mTOR regulation in the immune system have been poorly understood. Recent reports have demonstrated that TSC1 plays important roles in hematopoietic stem cells for the generation of multiple hematopoietic cell lineages (36, 37). In this report, we demonstrate that TSC1 deficiency results in increased expression of proinflammatory cytokines and nitric oxide (NO) in macrophages in response to TLR stimulation due to increased activation of mTORC1 and JNK1/2. Furthermore, TSC1 deficiency causes impairment of endotoxin tolerance in vitro and in vivo. Our data establish that TSC1 is an important negative regulator of TLR-induced innate immunity by inhibiting both mTORC1 and JNK1/2 activation.
Materials and methods
Mice and reagents
TSC1f/f mice and ERCre mice were described previously (38, 39). TSC1f/f-ERCre or TSC1f/f mice were intraperitoneally injected with 200 μl of 10 mg/ml Tamoxifen (Sigma, St. Louis, MO) on day 1, 2 and 5. Mice were used for experiment on day 8. All mice were generated and used in accordance with protocols approved by the Institutional Animal Care and Use Committee at Duke University. Lipopolysaccharide (LPS) from Escherichia coli O127:B8 was obtained from Sigma. Poly (I:C), Pam3CSK4, C12-iE-DAP, muramyl dipeptide (MDP), and LyoVec were purchased from Invivogen (San Diego, California). Rapamycin, SP600125, JNK inhibitor VIII, and SB203580 were purchased from EMD Biosciences (San Diego, CA).
Generation of BMMϕ
Bone marrow cells from femurs and tibias were flushed and plated into Petri dishes containing RPMI-10 (RPMI-1640 medium supplemented with 10% FBS, 100 U/ml penicillin, 1,000 U/ml streptomycin, and 20 mM L-glutamine) containing 15% L929 cell conditional medium as previously described (40). After 2–3 days of culture at 37°C in a CO2 incubator, nonadherent cells were transferred to new plates with fresh medium for another 3–5 days before they were used for experiments. More than 95% of cells were CD11b+ by flow cytometry analysis.
Phagocytosis
Listeria (L.) monocytogenes strain was grown overnight in brain heart infusion (BHI) broth at 37°C with shaking. Approximately 1 × 108 L. monocytogenes in 500 μl PBS were labeled with CFSE at 1 μg/ml for 15 min at room temperature with gentle shaking in the dark. Bacteria were washed twice with PBS and suspended in 1 ml RPMI 1640 medium. 1 × 106 BMMϕ in 1.0 ml medium were added to each well in a 12 well-plate. After overnight incubation, 1×107 CFSE-labeled L. monocytogenes were added to BMMϕ. The cells were incubated at 37°C for 0, 20, 40, and 60 min. After removal of culture medium, adherent BMMϕ were washed 2 times with 2.0 ml PBS and then fixed with PBS containing 1% paraformaldehyde for 5 min at room temperature. Cells were scraped off for flow cytometry analysis.
Stimulation of BMMϕ
Two hundred thousand BMMϕ from both TSC1f/f ERcre+ (TSC1KO) and TSC1f/f ERcre− (WT) were equally seeded in 24 well plates in 0.5 ml RPMI-10 in triplicates. After overnight incubation at 37°C at a CO2 incubator, BMMϕ were treated with 10 ng/ml LPS, Poly (I:C), Pam3CSK4, C12-iE-DAP (C12-iE), muramyl dipeptide (MDP), and Poly (I:C)/LyoVec (Poly(I:C)/LVec) for the indicated times. At each time points, supernatants were harvested for cytokine analyses and cells were used for making RNA using Trizol Reagent. For biochemical analysis, 0.5 × 106 BMMϕ were plated in 60 cm petri dishes and cultured at 37°C overnight. BMMϕ cells were then starved in serum-free medium for 2-3 hours and treated with 10 ng/ml LPS for 0, 15, 30, and 45 min. Cells were washed once with cold PBS, lysed in NP-40 or Triton X-100 lysis buffer with freshly added protease and phosphatase inhibitor cocktails (Sigma) and then subjected for western blot analysis. For induction of endotoxin tolerance, BMMϕ were treated with 1 ng/ml LPS prior to high dose of LPS restimulation (10 ng/ml). For the in vivo assay, tamoxifen treated TSC1f/f ERcre+ and TSC1f/f ERcre− mice were i.p. injected with 1 mg/kg and 2 mg/kg of LPS on day −3 and −2 and then with 40 mg/kg and 90 mg/kg of LPS on day 0 and day 2 in 200 μl PBS, respectively.
Cytokine and NO concentration
Cytokine levels in culture supernatant or in sera were determined using commercial ELISA kit for TNFα, IL12p40, IL6 (Biolegend, San Diego, CA) according to the manufacturer’s instruction. Nitric oxide was measured using Griess assay with NaNO2 as standard. Each value represents the mean of triplicate values.
RNA interference with small hairpin RNAs (shRNA)
pLKOpuro1-ShRaptor plasimd was purchased from Addgene (Cambridge, MA). The complementary oligonucleotides to generate the shRNA against mouse JNK1 were designed using BLOCK-iT™ RNAi Designer from Invitrogen (Carlsbad, CA). Oligo sequences that we used were as follows: ShJNK1-RNA 5′GGATGCAAATCTTTGCCAAGT 3′. Double stranded (ds) oligos were subcloned into pLKOpuro1. pLKOpuro1-ShLuc was used as a control. pLKOpuro1-ShLuc and pLKOpuro1 plasmids were generous gifts from Drs. Yang and Weinberg (Whitehead Institute for Biomedical Research, Cambridge, Massachusetts). pLKOpuro1-ShLuc and pLKOpuro1 targeted ShJNK1 were co-transfected with pCMV-VSVG and pHR’8.2ΔR (Dr. Weinberg) into 293T cells. After 48 hours, cultural media were collected and centrifuged for at 2000 rpm for 10 min at room temperature. Supernatants containing lentiviruses were used for transducing BMMϕ. To transduce BMMϕ, non-adherent cells on day 3 of in vitro BMMϕculture were transferred into in 6 well-plate (1 × 106 cells/well in 2 ml BMMϕ culture medium) followed by addition of 500 μl of viral supernatant together with polybrene at a final concentration of 1 μg/ml. The mixture of cells and viruses was spun at 2500 rpm for 90 min at room temperature. After overnight incubation at 37°C in a CO2 incubator, culture medium was replaced with 2 ml fresh medium containing 1 μg/ml puromycin. With two additional replacements of medium containing puromycin on day 2 and day 3 after transduction to remove dead cells, the selected cells were used for experiments on day 5 or day 6 after infection.
Real-time PCR
Total RNA was prepared by TRIzol® reagent (Invitrogen). Reverse transcription reaction was performed using random primers and SuperScript® III Reverse Transcriptase (Invitrogen). SYBR green real-time PCR was conducted using RealMasterMix on Realplex machine (Eppendorf) according to manufacturer’s protocol. The primer pairs used were as follows: β-Actin: forward 5′ TGTCCACCTTCCAGCAGATGT 3′ and reverse 5′ AGCTCAGTAACAGTCCGCCTAGA 3′; TNFα: forward 5′ CCCCAAAGGGATGAGAAGTT 3′ and reverse 5′CACTTGGTGGTTTGCTACGA 3′; IL12p40: forward 5′ TCTGAGCCACTCACATCTGC 3′ and reverse 5′ TTGGTGCTTCACACTTCAGG 3′; IL6: forward 5′TTCCATCCAGTTGCCTTCTTG 3′ and reverse 5′ TTGGGAGTGGTATCCTCTGTGA 3′; NOSII: forward 5′ CACCTTGGAGTTCACCCAGT 3′ and reverse 5′ACCACTCGTACTTGGGATGC3′. IFNβ: forword 5′ CCCTATGGAGATGACGGAGA 3′ and reverse 5′ CTGTCTGCTGGTGGAGTTCA 3′.
Western blot and antibodies
Protein concentrations in cell lysates were determined using Bio-Rad Protein Assay at OD595nm. Equivalent amounts of protein for each sample were subjected to SDS-polyacrylamide gel electrophoresis and transferred onto nitrocellulose membrane (BIO-RAD Laboratories). After blocking with 5% nonfat dry milk/PBS, the membranes were incubated with a primary antibody overnight at 4°C, washed with TBST (100mM Tris, pH 7.5, NaCl 0.9%, Tween 0.1%) three times, reacted with a secondary antibody for 45 min, and washed with TTBS three times. Protein bands were visualized by ECL (PerkinElmerLife Sciences). The blots were stripped and reprobed for loading control. Anti-phospho-Erk1/2, anti-phospho-p70S6K T421/S424, anti-phospho-4E-BP1 Thr37/Ser46, anti-phospho-Akt S473 (193H12), anti-phospho-IκBα, anti-phospho-p38, and anti-phospho-JNK1/2 (T183/Y185), anti-TSC1, anti-TSC2, anti-pErk1/2, anti-p70S6K, anti-4E-BP1, anti-AKT, anti-IκBα, anti-JNK1/2, and anti-p38 antibodies were purchased from Cell Signaling Technology. PE-conjugated anti-mouse F4/80, CD11b, and IgG control antibodies were obtained from Biolegend, Inc (San Diego, CA). Anti-NFκBp65 and anti-β-actin were purchased from Santa Cruz biotechnology and Sigma, respectively.
Immunofluorescence staining and confocal microscopy
WT and TSC1 KO BMMϕ were grown on chamber slides (BD Discovery Labware, Bedford, MA) in RPMI1640 supplemented with 15% L-cell conditional medium for 24 h at 37°C. The cells were treated with or without LPS at 10ng/mL for 30 and 60 hour. Immediately after LPS treatment, the cells were washed with ice-cold PBS twice, and then were fixed in 4% paraformaldehyde/PBS for 15 min, permeabilized with 0.2% Triton X-100/PBS for 20 min, and pre-blocked in 5% BSA/PBS overnight. The slides were then incubated with anti-mouse NFκBp65 antibody that was diluted at 1/100 in blocking solution for 2 hours, washed three times with PBS, and incubated with FITC-conjugated anti-mouse IgG antibody (Jackson ImmunoResearch Laboratories) for 1 h. After additional washes, the slides were incubated with DAPI for 3 min, and mounted with anti-fade solution. Cells were examined under the Zeiss LSM510 inverted confocal microscopy and images were acquired and analyzed with LSM image software.
Statistical analysis
For statistic analysis, the two-tail Student t-test was performed for all data except those from the in vivo endotoxin tolerance experiment. Endotoxin tolerance data were analyzed by the Fisher’s exact test. *, p<0.05; **, p<0.01.
Results
TSC1 and TSC2 expression in macrophages
To determine the role of TSC1 and the importance of tight control of mTOR signaling in innate immune responses, we first examined TSC1 and TSC2 expression in macrophages. Both TSC1 and TSC2 were detected in bone marrow derived macrophages (BMMϕ) and their expression was upregulated following LPS (TLR4 ligand) stimulation for 24 hours (Fig 1A). To investigate the role of TSC1 in innate immunity, we crossed TSC1 conditional knockout mice (TSC1f/f) mice (38) with the ERCre mice and generated TSC1f/f-ERCre mice to allow deletion of TSC1 following tamoxifen treatment. TSC1 was barely detectable in BMMϕ from tamoxifen treated TSC1f/f-ERCre (TSC1KO) mice (Fig 1B), confirming effective deletion of the gene. TSC2 protein was severely decreased in TSC1KO BMMϕ, suggesting that TSC1 is required for TSC2 stability in macrophages, which is consistent with the reported role of TSC1 in other cells (41). Almost all in vitro differentiated TSC1f/f (WT) and TSC1KO BMMϕ were CD11b and F4/80 positive, and TSC1KO BMMϕ expressed similar levels of CD11b and F4/80 to WT BMMϕ, suggesting that in vitro macrophage differentiation is not inhibited by TSC1 deficiency (Fig 1C). However, TSC1 deficiency obviously impacted on BMMϕ size. While similar to controls on day 3, TSC1KO BMMϕ progressively increased their size after seven days of in vitro differentiation (Fig 1D and 1E), indicating that TSC1 inhibits the growth of BMMϕ. Macrophages express MHC and costimulatory molecules that can be induced by LPS stimulation to regulate adaptive immune responses. TSC1KO BMMϕ expressed similar levels of MHC class II I-Ab, but elevated CD80 and CD86 co-stimulatory molecules before and after LPS stimulation compared to WT BMMϕ (Fig 1F). However, we could not rule out a potential contribution of enlarged sizes of TSC1KO BMMϕ to the increased detection of CD80 and CD86 levels in these cells.
Figure 1. Effects of TSC1 deficiency on macrophage morphology and expression of costimulatory molecules.

(A) Increased TSC1 and TSC2 proteins in BMMϕ following LPS stimulation. WT BMMϕ were left unstimulated or stimulated with LPS for 24 hours. TSC1 and TSC2 protein in cell lysates were detected by immunoblotting analysis with anti-TSC1 and TSC2 antibodies as well as an anti-β-actin antibody for loading control. (B) Diminished TSC2 proteins in TSC1KO BMMϕ. TSC1KO and WT BMMϕ lysates were subjected to immunoblotting analysis as in (A). (C) TSC1 deficiency does not inhibit BMMϕ differentiation in vitro. WT and TSC1KO BMMϕ were generated in vitro with M-CSF. CD11b and F4/80 expression on these BMMϕ on day 7 during in vitro differentiation were determined by flow cytometry. (D, E) Increased sizes of TSC1KO BMMϕ during in vitro differentiation. (D) Bar figures show BMMϕ sizes (mean ± SD) on indicated days from three experiments. *, P<0.05; **, P<0.01. (E) Typical microscopic morphology of WT and TSC1KO BMMϕ on day 8 during in vitro differentiation. (F) Increased costimulatory molecule expression in TSC1KO BMMϕ. WT and TSC1KO BMMϕ were left unstimulated or stimulated with LPS for 24 hours. MHC class II, CD80, and CD86 expression on CD11b+ BMMϕ was determined by flow cytometry. Data shown are representative of at least three experiments. G. Phagocytosis of L. monocytogenes by WT and TSC1KO BMMϕ. Macrophages were incubated with CFSE-labeled L. monocytogenes for 0, 20, 40 and 60 minutes. After washed and fixed with 1% paraformaldehyde, cells were subjected to flow-cytometry analysis to assess CFSE intensity. Data shown are representative of at least three (A-F) and two experiments (G).
To further determine if TSC1KO BMMϕ maintain macrophage properties, we examined their ability to phagocytize bacteria by incubating the cells with CFSE labeled L. monocytogenes for the indicated times followed by FACS analysis. As shown in Figure 1G, TSC1KO BMMϕ uptake similar amount of L. monocytogenes to WT BMMϕ, suggesting that TSC1 deficiency did not obviously affect BMMϕ phagocytosis. Together, these observations reveal that TSC1 and TSC2 expression is regulated by LPS stimulation, that TSC1 is critical for maintaining normal expression of TSC2, that TSC1 is not required for in vitro macrophage differentiation, and that TSC1 may inhibit costimulatory molecule expression in macrophages.
Altered proinflammatory cytokine production in the absence of TSC1
To investigate how the absence of TSC1 may affect TLR-mediated innate immune responses, we first measured TLR-induced cytokine production. Following LPS stimulation for different times, TSC1KO BMMϕ produced more proinflammatory cytokines TNFα, IL12p40, and IL6 at both the protein and mRNA levels than WT BMMϕ (Fig 2A-2B). Thus, TSC1 functions at least at the transcription level to inhibit the production of these cytokines. To test whether such enhanced proinflammatory responses is specific to TLR4, we stimulated WT and TSC1KO BMMϕ with Pam3CSK4 and Poly (I:C), ligands for TLR2 and TLR3, respectively. TSC1KO BMMϕ produced higher levels of TNFα than WT BMMϕ (Fig 2C), suggesting that TSC1 negatively controls cytokine production from multiple TLRs in macrophages.
Figure 2. Altered innate immune responses in TSC1KO BMMϕ.

(A, B) Increased proinflammatory cytokine production by TSC1KO BMMϕ. WT and TSC1KO BMMϕ were left unstimulated or stimulated with LPS at 10 ng/ml for the indicted times. Proteins (A) and mRNAs (B) of indicated cytokines in the supernatants and cells were measured by ELISA and quantitative real-time PCR, respectively. (C) WT and TSC1KO BMMϕ were left unstimulated (Unst) or stimulated with Poly(I:C) at 2 μg/mL and Pam3CSK4 at 1μg/mL overnight. TNFα protein production was determined by ELISA. (D) WT and TSC1KO BMMϕ were left unstimulated or stimulated with C12-iE-DAP (5 μg/ml) and MDP (10 μg/ml) overnight. TNFα mRNA Expression was determined by realtime-PCR. (E) WT and TSC1KO BMMϕ were left unstimulated or stimulated with Poly(I:C)/LyoVec (100 ng/ml) overnight. IFNβ mRNA expression was determined by realtime-PCR. (F) Elevated serum cytokine levels in TSC1KO mice following LPS injection. TSC1f/f and TSC1f/f-ERcre mice were injected three times with tamoxifen in five days. On day 7, mice were intraperitoneally injected with 10 mg/kg LPS. Serum cytokines after six hours of injection were measured by ELISA. (G) Increased NO production by TSC1KO BMMϕ following LPS stimulation. WT and TSC1KO BMMϕ were stimulated with 10 ng/ml LPS. NO in supernatant was measured 24 hours late (E). (H, I) Increased iNOS expression in TSC1KO BMMϕ. WT and TSC1KO BMMϕ were stimulated with 10 ng/ml LPS at the indicated times. iNOS protein (F) and mRNA (G) were determined by immunoblotting analysis and quantitative real-time RT-PCR, respectively. Data shown are means ± SD and are representative of at least three experiments. *, P<0.05; **, P<0.01.
NOD1, NOD2, RIG-I, and MDA-5 are important intracellular microbial pattern recognition molecules. To test whether TSC1 also plays a role in innate responses induced by these receptors, we stimulated WT and TSC1KO BMMϕ with C12-iE-DAP and MDP to activate NOD1 and NOD2 respectively. As shown in figure 2D, TNFα mRNA levels induced by these ligands were much lower in TSC1KO BMMϕ than in WT control. When stimulated with Poly(I:C)/LyoVec to activate RIG-I/MDA-5, TSC1KO BMMϕ produced higher levels of IFNβ than WT control (Fig 2E). Thus, the roles of TSC1 in innate immunity appear complex. It inhibits TLR and RIG-I/MDA-5 but promotes NOD1/2 induced innate immune responses.
To further assess the role of TSC1 in LPS-induced responses in vivo, we compared serum cytokine concentrations in tamoxifen treated TSC1f/f-ERCre and TSC1f/f control mice following LPS injection. As shown in figure 2F, higher serum concentrations of IL12p40, TNFα, and IL6 were detected in TSC1f/f-ERCre mice than in TSC1f/f mice. Together, these data demonstrate that TSC1 functions as a negative regulator of TLR-induced proinflammatory cytokine production in vitro and in vivo.
Increased nitric oxide production in TSC1 deficient macrophages
Nitric oxide (NO) is a highly reactive molecule involved in innate immunity that can be induced by TLR stimulation. Increased NO levels was detected in TSC1KO BMMϕ following LPS stimulation compared to WT BMMϕ (Fig 2G). In WT BMMϕ, NO synthase II (iNOS/NOS2, the enzyme responsible for NO production) can be induced 6 hours after LPS stimulation. In TSC1KO BMMϕ, LPS-induced iNOS protein expression was elevated (Fig 2H). iNOS mRNA could be detected 3 hours and further upregulated 6 and 24 hours after LPS stimulation. In TSC1KO BMMϕ, much higher levels of iNOS mRNA were detected during the course of LPS stimulation (Fig 2I), suggesting that TSC1 also inhibits iNOS production at least at the transcription level.
Enhanced JNK1/2 activation in TSC1 deficient macrophages
To explore the mechanisms by which TSC1 inhibits TLR induced innate immune responses, we examined how TSC1 deficiency may affect TLR signaling. A critical event in TLR signaling is the activation of IKKs. IKKs phosphorylate IκBα, leading to its degradation and subsequent NFκB nuclear translocation to activate gene transcription. IκBα phosphorylation and total IκBα protein level was not drastically altered in TSC1KO BMMϕ compared with WT BMMϕ (Fig 3A). Furthermore, NFκB translocation from the cytosol to the nucleus following LPS stimulation was similar between WT and TSC1KO BMMϕ (Fig 3B). Thus, absence of TSC1 does not obviously alter the activation of the IKK/NFκB pathway.
Figure 3. TSC1 inhibits JNK1/2 and mTORC1 but not IKK/NFκB activation in macrophage.

(A) Enhanced TLR4-induced JNK1/2 activation in the absence of TSC1. Cell lysates from unstimulated or LPS (10 ng/ml) stimulated WT (W) and TSC1KO (K) BMMϕ were subjected to immunoblotting analysis with the indicated antibodies. (B) Nuclear translocation of NFκB p65 in WT and TSC1KO BMMϕ following LPS stimulation. WT and TSC1KO BMMϕ, grown on chamber slides, were treated with or without LPS at 10ng/mL for 30 and 60 minutes. After fixation and permeablization, cells were stained with an anti-mouse p65 antibody followed by FITC-labeled anti-mouse secondary antibody. Nuclei were counter-stained with DAPI. Cells were examined under the Zeiss LSM510 inverted confocal microscopy. (C) Enhanced mTORC1 but decreased mTORC2 activation in the absence of TSC1. Cell lysates from unstimulated or LPS (10 ng/ml) stimulated WT and TSC1KO BMMϕ were subjected to immunoblotting analysis with the indicated antibodies. Data shown are representative of three experiments.
TLR-induced activation of MAPKs also contributes to inflammatory responses. LPS-induced Erk1/2 phosphorylation in TSC1KO BMMϕ was slightly higher than WT BMMϕ (Fig 3A). p38 phosphorylation in TSC1KO BMMϕ exhibited delayed kinetics compared to WT BMMϕ. However, JNK1/2 phosphorylation was significantly enhanced in TSC1KO BMMϕ following LPS stimulation, suggesting that TSC1 negatively controls TLR4-induced JNK1/2 activation in macrophages.
Enhanced mTORC1 but decreased mTORC2 activation in TSC1 deficient macrophages
We asked further how TSC1 deficiency may affect TLR-induced mTOR signaling in BMMϕ. As shown in figure 3C, TSC1KO BMMϕ displayed elevated phosphorylation of p70S6K1 and 4E-BP1 before and/or after LPS stimulation compared to WT BMMϕ, indicating that TSC1 inhibits TLR-induced mTORC1 activation. In contrast, Akt phosphorylation at S473, an mTORC2 dependent event, was decreased in TSC1KO BMMϕ compared to WT cells, suggesting that TSC1 is required for TLR-induced mTORC2 activation. TSC1/2 may promote mTORC2 signaling by directly participating in mTORC2 activation or by preventing mTORC1 mediated inhibitory mechanisms as demonstrated in other cell types. Together, these observations indicate that TSC1 inhibits mTORC1 activation but promotes mTORC2 activation following TLR4 stimulation in BMMϕ.
Enhanced mTORC1 activation in TSC1KO BMMϕ contributes elevated innate immune responses
To determine whether elevated mTORC1 and JNK1/2 activation contributes to the dysfunction of TSC1KO BMMϕ, we treated these cells with mTORC1 and JNK1/2 inhibitors. Treatment of TSC1KO BMMϕ with rapamycin (Rapa, R) reduced TSC1KO BMMϕ sizes (Fig 4A) and drastically decreased LPS-induced TNFα and NO production as well as iNOS expression in both WT and TSC1KO BMMϕ (Fig 4B-4E). To further confirm that the enhanced mTORC1 activation contributed to the elevated innate immune responses in TSC1KO BMMϕ, we transduced Raptor ShRNA (ShRapt) and a control ShRNA (ShLuc) into WT and TSC1KO BMMϕ to knock down Raptor expression. ShRapt reduced Raptor expression by about 70 % in both WT and TSC1KO BMMϕ (Fig 4F). Decreased Raptor expression resulted in reduced TNFα mRNA and protein expression in both WT and TSC1KO BMMϕ following LPS stimulation (Fig 4G-4H). Together, these data suggested that increased mTORC1 activity contributed to the enhanced TLR-induced responses in the absence of TSC1.
Figure 4. TSC1 inhibits mTORC1 to regulate macrophage function.
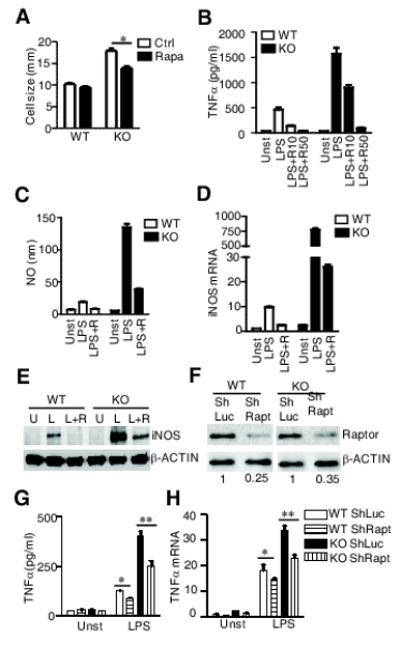
(A) Reduction of TSC1KO cell sizes by rapamycin. WT and TSC1KO BMMϕ were left untreated (Ctrl) or treated with 10 ng/ml rapymycin (rapa) for 24 hours and cell sizes were measure by CellometerTM Auto T4 (Nexcelom Bioscience). (B-E) Inhibition of TNFα and NO production by rapamycin. BMMϕ were pretreated with 0, 10 (R or R10), or 50 (R50) ng/ml of rapamycin for one hour, followed by stimulated with 0 (Unst or U) or 10 ng/ml LPS (L) overnight. TNFα (B), NO production (C), iNOS mRNA levels (D), and iNOS protein (E) were determined as described in Fig 2. (F-H) Decreased LPS-induced TNFα production by Raptor shRNA. Raptor protein levels in WT and TSC1KO BMMϕ transduced with either ShRaptor or ShLuc as control was detected by western blot (F). TNFα protein (G) and mRNA (H) in WT and TSC1KO BMMϕ transduced with the indicated shRNA following LPS (10 ng/ml) stimulation for 6 hours were determined by ELISA and realtime-qPCR, respectively.
Contribution of JNK1/2 activity to the elevated LPS induced innate immune responses in TSC1KO BMMϕ
Since JNK1/2 activation was elevated in TSC1KO BMMϕ, we further examined whether the elevated JNK1/2 activation might contribute to the enhanced LPS induced response in TSC1KO BMMϕ. Inhibition of JNK1/2 with SP600125 (SP) or JNK inhibitor VIII (VIII) resulted in diminished, but still elevated NO production in TSC1KO BMMϕ following LPS stimulation (Fig 5A). Simultaneous treatment with both rapamycin and JNK1/2 inhibitor further reduced NO production in TSC1KO BMMϕ (Fig 5B). The importance of JNK1/2 activity for LPS induced TNFα production in both WT and TSC1KO BMMϕ was further confirmed by knocking down JNK1 in these cells using ShRNA (Fig 5C-5E). Furthermore, while rapamycin treatment slightly reduced JNK1/2 phosphorylation in WT BMMϕ, it significantly reduced JNK1/2 phosphorylation in of TSC1KO BMMϕ (Fig 5F). Together, these data suggest that JNK1/2 activity is important for the elevated proinflammatory responses in TSC1KO BMMϕ and that increased mTORC1 activity partially contributes to the enhanced JNK1/2 activation in these cells.
Figure 5. Contribution of enhanced JNK1/2 activation to the elevated TLR-induced response in TSC1 deficient macrophages.

(A, B) Reduced LPS-mediated NO production by JNK inhibitors (A) or JNK inhibitor plus rapamycin (B). WT and TSC1KO BMMϕ were left unstimulated or stimulated with 10 ng/ml LPS in the presence or absence of JNK1/2 inhibitors (SP600125, SP; JNK inhibitor VIII, VIII), and/or rapamycin. NO in supernatant was measure 24 hours late. (C-E) Reduced LPS-induced TNFα production in TSC1KO BMMϕ by ShJNK1. TSC1KO BMMϕ were either transduced with ShJNK1 or ShLuc. JNK1 protein levels were determined by western blot. The numbers indicates relative JNK1 levels (C). TNFα protein (D) produced by and mRNA (E) in the transduced TSC1KO BMMϕ with (LPS) or without (Unst) 10 ng/ml LPS stimulation for 6 hours were determined by ELISA and realtime-qPCR, respectively. (F) Inhibition of LPS-induced JNK1/2 activation in TSC1KO BMMϕ by rapamycin. BMMϕ were pretreated with rapamycin for 45 minutes, stimulated with LPS for the indicated times, and lysed for immunoblotting analysis. Data shown are means ± SD and represent at least three experiments. *, P<0.05; **, P<0.01
Effects of TSC1 deficiency on the induction of endotoxin tolerance
TLR-induced tolerance is important in preventing uncontrolled proinflammatory responses detrimental to the host (42). The inhibitory role of TSC1 in TLR-mediated proinflammatory responses prompted us to examine how TSC1 deficiency may affect endotoxin tolerance. LPS-pretreated WT BMMϕ failed to produce TNFα and IL12p40 following secondary high dose LPS stimulation. However, LPS-pretreated TSC1KO BMMϕ still produced high levels of these cytokines following secondary LPS stimulation (Fig 6A-6B). Of note, LPS-pretreated TSC1KO BMMϕ produced less cytokines than the same cells without LPS-pretreatment following secondary LPS stimulation, suggesting that TSC1 deficiency only partially impaired the induction of LPS tolerance in vitro. To determine whether TSC1 deficiency affected the induction of LPS tolerance in vivo, we injected two low doses of LPS into tamoxifen pre-treated TSC1f/f and TSC1f/f-ERCre mice followed by two high doses of LPS challenge. All TSC1f/f mice survived high dose LPS challenges. In contrary, all TSC1f/f-ERcre mice succumbed to high dose LPS challenges, correlating with elevated serum IL6 and IL12p40 levels (Fig 6C-6D). Together, these observations suggest that TSC1 is important for the induction of endotoxin tolerance in vitro and in vivo, although we cannot rule out that dysregulated proinflammatory cytokine production caused by TSC1 deficiency may contribute to the impaired induction of endotoxin tolerance in TSC1 deficiency mice.
Figure 6. TSC1 deficiency results in impaired induction of LPS tolerance in vitro and in vivo.

(A, B) WT and TSC1KO BMMϕ with or without LPS (1 ng/ml, 24 hours) pretreatment were unstimulated or stimulated with 10 ng/ml LPS for various times. Proteins (A) and mRNA (B) of indicated cytokines were measured as in Fig 2. (C) Impaired endotoxin tolerance in TSC1 deficient mice. TSC1f/f (n=8) and TSC1f/f-ERcre (n=6) mice were injected with tamoxifen. Two days (day −3) after the last tamoxifen injection, mice were injected with 1 and 2 mg/kg of LPS on day −3 and −2 and then with 40 mg/kg and 90 mg/kg LPS on day 0 and day 2, respectively. Figure shown is a Kaplan-Meyer survival plot of TSC1 WT and KO mice. (D) Increased serum IL12p40 and IL6 levels in LPS-pretreated TSC1KO mice following secondary high dose LPS challenge. Mice were similarly treated as in (C) except serum cytokines were measured 6 hours after 40 mg/Kg LPS challenge. (E) Increased JNK1/2 activation in TSC1KO BMMϕ under tolerizing conditions. LPS-pretreated BMMϕ were subjected to secondary LPS stimulation followed immunoblotting analysis as in Fig 3A. (F) Effects of JNK1/2 inhibitor on LPS-induced NO and TNFα production by WT and TSC1KO BMMϕ pretreated with or without LPS. Data shown represent three (A, B, E, and F) or two (C, D) experiments.
Role of JNK1/2 activity for altered endotoxin tolerance in TSC1 deficient BMMϕ
To investigate the mechanisms by which TSC1 contribute to endotoxin tolerance, we analyzed LPS-induced signaling in macrophages that had already been pretreated with a low dose of LPS. We first examined mTORC1 signaling in LPS tolerated condition in WT BMMϕ. As shown in figure 6E, phosphorylation of S6K was not reduced and phosphorylation of 4E-BP1 was obviously increased in LPS-pretreated WT BMMϕ before and after secondary LPS stimulation. Since LPS stimulation for 24 hours upregulates TSC1/2 expression, this result suggests that mechanisms in addition to TSC1/2 maybe involved in regulating mTORC1 signaling in LPS tolerated macrophages. In TSC1KO BMMϕ, pretreatment with LPS did not obviously alter the hyper-phosphorylation status of S6K and 4E-BP1 in these cells. Phosphorylation of these proteins in LPS-pretreated TSC1KO BMMϕ remained high following secondary LPS stimulation.
Different from mTORC1 signaling, JNK1/2 phosphorylation was decreased in LPS-pretreated WT BMMϕ following secondary LPS stimulation (Fig 6E). However, JNK1/2 phosphorylation remained increased in LPS-pretreated TSC1KO BMMϕ following secondary LPS stimulation. Treatment of TSC1KO BMMϕ with SP600125 significantly reduced NO and TNFα production in the cells induced by secondary LPS stimulation (Fig 6F), suggesting that elevated JNK1/2 activation in TSC1KO BMMϕ may contribute to their resistance to endotoxin tolerance.
Discussion
The role of the TSC-mTOR pathway in TLR-induced innate immune responses has been controversial. Most published observations were based on in vitro and in vivo administration of rapamycin. Rapamycin has been found to be able to inhibit or increase TLR-induced proinflammatory cytokine production (24, 25, 27, 28, 30). Such differences could be attributed to the different cells examined. For example, rapamycin was found to be able to enhance IL-12 production in murine and human myeloid DCs by promoting NFκB activation, but inhibit IL-12 production in monocyte-derived DCs and bone marrow derived DCs (24, 27, 28). In addition, the length and timing of rapamycin treatment may have an impact its effects on TLR-induced innate immune responses. Our study has provided genetic evidence that TSC1 inhibits TLR-induced proinflammatory innate immune responses. Since enhanced proinflammatory responses in TSC1KO BMMϕ in response to LPS can be inhibited by rapamycin, our data support that mTORC1 positively regulates TLR-induced proinflammatory cytokine production. The PI3K/Akt pathway has been reported to be able to inhibit TLR induced cytokine production (43-45). At present, we cannot rule out that decreased mTORC2 signaling in TSC1 deficient macrophages may also contribute to the elevated cytokine production in these cells. In addition to regulating TLR-mediated innate immunity, we have also shown that TSC1 differentially controls NOD1/2 and RIG-I/MDA-5 induced innate immunity. TSC1 deficiency caused decreased TNFα production but increased IFNβ expression in BMMϕ after NOD1/2 and RIG-I stimulation respectively.
Our results are different from those observed in immortalized TSC2 deficient murine embryonic fibroblasts (MEFs) with compound p53 deficiency. In TSC2 deficient MEFs, TLR-induced mTORC1 signaling is enhanced, but proinflammatory responses are decreased as correlated with impaired IKK activation and NFκB translation to the nuclei (28). We have not observed obvious defect of IKK/NFκB activation in TSC1 deficient macrophages. LPS-induced IκB phosphorylation and degradation as well as NKκB p65 nuclear translocation are similar between WT and TSC1KO BMMϕ. The differences between these studies could be resulted from different cell types examined. Furthermore, the compound deficiency of both p53 and TSC2 in MEF cells could impact on the phenotype observed. Although the role of TSC1 in other cell lineages needs to be examined in the future, our data support that TSC1 is a negative regulator of TLR-induced response at least for BMMϕ and in vivo.
In addition to regulating mTOR signaling, our data reveal that TSC1 inhibits TLR-induced JNK1/2 activation and that enhanced JNK1/2 activation contributes to the enhanced proinflammatory response and resistance to endotoxin tolerance when TSC1 is deficient. Since enhanced JNK1/2 activation in TSC1 deficient BMMϕ can only be partially inhibited by acute rapamycin treatment, it suggests that mTORC1 functions as an upstream regulator for JNK1/2 activation during TLR signaling and that TSC1 may also control JNK1/2 activation through mTORC1-independent mechanisms. It has also been reported that TLR3 induced JNK1/2 activation in karotinocytes is inhibited by rapamycin (29). Thus, mTORC1 may function as a positive regulator of JNK1/2 activation for multiple TLRs.
In summary, we have demonstrated that TSC1 plays important roles in innate immune responses. TSC1 exerts differential roles for LPS induced mTORC1 and mTORC2 signaling by inhibiting mTORC1, but promoting mTORC2 activation in macrophages. In addition, TSC1 functions as a negative regulator for LPS-induced JNK1/2 activation and uncontrolled mTORC1 and JNK1/2 activation contributes to enhanced proinflammatory responses and impaired endotoxin tolerance caused by TSC1 deficiency.
Acknowledgement
We thank Dr. Weiguo Zhang and people in the Zhong lab for helpful discussions and for providing reagents. The authors declare no competing financial interests. H.P. was involved in the experimental design and execution, data analysis, and manuscript preparation. P.Z. participated in research. T.O. generated important reagents. X.P.Z. conceived the project and was involved in experimental design, data analysis, and manuscript preparation.
This study is supported by funding from the National Institute of Health (R01AI076357, R01AI079088, and R21AI079873), the American Cancer Society (RSG-08-186-01-LIB), and the American Heart Association to X-P.Z.
Abbreviations
- BMMϕ
bone marrow derived macrophage
- mTOR
mammalian target of rapamycin
- TSC
tuberous sclerosis complex
- KO
knockout
- muramyl dipeptide
MDP. 2
References
- 1.Takeuchi O, Akira S. Pattern Recognition Receptors and Inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 2.Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1:135–145. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- 3.Barton GM, Kagan JC. A cell biological view of Toll-like receptor function: regulation through compartmentalization. Nat Rev Immunol. 2009;9:535–542. doi: 10.1038/nri2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, Goode J, Lin P, Mann N, Mudd S, Crozat K, Sovath S, Han J, Beutler B. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature. 2003;424:743–748. doi: 10.1038/nature01889. [DOI] [PubMed] [Google Scholar]
- 5.Ermolaeva MA, Michallet MC, Papadopoulou N, Utermohlen O, Kranidioti K, Kollias G, Tschopp J, Pasparakis M. Function of TRADD in tumor necrosis factor receptor 1 signaling and in TRIF-dependent inflammatory responses. Nat Immunol. 2008;9:1037–1046. doi: 10.1038/ni.1638. [DOI] [PubMed] [Google Scholar]
- 6.Muzio M, Natoli G, Saccani S, Levrero M, Mantovani A. The human toll signaling pathway: divergence of nuclear factor kappaB and JNK/SAPK activation upstream of tumor necrosis factor receptor-associated factor 6 (TRAF6) J Exp Med. 1998;187:2097–2101. doi: 10.1084/jem.187.12.2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muzio M, Ni J, Feng P, Dixit VM. IRAK (Pelle) family member IRAK-2 and MyD88 as proximal mediators of IL-1 signaling. Science. 1997;278:1612–1615. doi: 10.1126/science.278.5343.1612. [DOI] [PubMed] [Google Scholar]
- 8.Pobezinskaya YL, Kim YS, Choksi S, Morgan MJ, Li T, Liu C, Liu Z. The function of TRADD in signaling through tumor necrosis factor receptor 1 and TRIF-dependent Toll-like receptors. Nat Immunol. 2008;9:1047–1054. doi: 10.1038/ni.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wesche H, Henzel WJ, Shillinglaw W, Li S, Cao Z. MyD88: an adapter that recruits IRAK to the IL-1 receptor complex. Immunity. 1997;7:837–847. doi: 10.1016/s1074-7613(00)80402-1. [DOI] [PubMed] [Google Scholar]
- 10.Liew FY, Xu D, Brint EK, O’Neill LA. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol. 2005;5:446–458. doi: 10.1038/nri1630. [DOI] [PubMed] [Google Scholar]
- 11.Sengupta S, Peterson TR, Sabatini DM. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol Cell. 2010;40:310–322. doi: 10.1016/j.molcel.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Powell JD, Delgoffe GM. The mammalian target of rapamycin: linking T cell differentiation, function, and metabolism. Immunity. 2010;33:301–311. doi: 10.1016/j.immuni.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhong XP, Shin J, Gorentla BK, O’Brien T, Srivatsan S, Xu L, Chen Y, Xie D, Pan H. Receptor signaling in immune cell development and function. Immunol Res. 2011;49:109–123. doi: 10.1007/s12026-010-8175-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, Tokunaga C, Avruch J, Yonezawa K. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002;110:177–189. doi: 10.1016/s0092-8674(02)00833-4. [DOI] [PubMed] [Google Scholar]
- 15.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 16.Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ, Sabatini DM. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell. 2006;11:859–871. doi: 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 17.Lee K, Gudapati P, Dragovic S, Spencer C, Joyce S, Killeen N, Magnuson MA, Boothby M. Mammalian target of rapamycin protein complex 2 regulates differentiation of Th1 and Th2 cell subsets via distinct signaling pathways. Immunity. 2010;32:743–753. doi: 10.1016/j.immuni.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thomson AW, Turnquist HR, Raimondi G. Immunoregulatory functions of mTOR inhibition. Nat Rev Immunol. 2009;9:324–337. doi: 10.1038/nri2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gorentla BK, Wan CK, Zhong XP. Negative regulation of mTOR activation by diacylglycerol kinases through inhibition of the diacylglycerol-RasGRP1-Ras-Mek1/2-Erk1/2 pathway. Blood. 2011;117:4022–31. doi: 10.1182/blood-2010-08-300731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, Ahmed R. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460:108–112. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gulen MF, Kang Z, Bulek K, Youzhong W, Kim TW, Chen Y, Altuntas CZ, Sass Bak-Jensen K, McGeachy MJ, Do JS, Xiao H, Delgoffe GM, Min B, Powell JD, Tuohy VK, Cua DJ, Li X. The receptor SIGIRR suppresses Th17 cell proliferation via inhibition of the interleukin-1 receptor pathway and mTOR kinase activation. Immunity. 2010;32:54–66. doi: 10.1016/j.immuni.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rao RR, Li Q, Odunsi K, Shrikant PA. The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity. 2010;32:67–78. doi: 10.1016/j.immuni.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, Worley PF, Kozma SC, Powell JD. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30:832–844. doi: 10.1016/j.immuni.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weichhart T, Saemann MD. The multiple facets of mTOR in immunity. Trends Immunol. 2009;30:218–226. doi: 10.1016/j.it.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 25.Hackstein H, Taner T, Zahorchak AF, Morelli AE, Logar AJ, Gessner A, Thomson AW. Rapamycin inhibits IL-4--induced dendritic cell maturation in vitro and dendritic cell mobilization and function in vivo. Blood. 2003;101:4457–4463. doi: 10.1182/blood-2002-11-3370. [DOI] [PubMed] [Google Scholar]
- 26.Turnquist HR, Cardinal J, Macedo C, Rosborough BR, Sumpter TL, Geller DA, Metes D, Thomson AW. mTOR and GSK-3 shape the CD4+ T-cell stimulatory and differentiation capacity of myeloid DCs after exposure to LPS. Blood. 2010;115:4758–4769. doi: 10.1182/blood-2009-10-251488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haidinger M, Poglitsch M, Geyeregger R, Kasturi S, Zeyda M, Zlabinger GJ, Pulendran B, Horl WH, Saemann MD, Weichhart T. A versatile role of mammalian target of rapamycin in human dendritic cell function and differentiation. J Immunol. 2010;185:3919–3931. doi: 10.4049/jimmunol.1000296. [DOI] [PubMed] [Google Scholar]
- 28.Weichhart T, Costantino G, Poglitsch M, Rosner M, Zeyda M, Stuhlmeier KM, Kolbe T, Stulnig TM, Horl WH, Hengstschlager M, Muller M, Saemann MD. The TSC-mTOR signaling pathway regulates the innate inflammatory response. Immunity. 2008;29:565–577. doi: 10.1016/j.immuni.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 29.Zhao J, Benakanakere M, Hosur K, Galicia J, Martin M, Kinane D. Mammalian target of rapamycin (mTOR) regulates TLR3 induced cytokines in human oral keratinocytes. Mol Immunol. 2010 doi: 10.1016/j.molimm.2010.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmitz F, Heit A, Dreher S, Tittel K, Mages J, Haas T, Krug A, Janssen K, Kirschning CV, Wagner H. Mammalian target of rapamycin (mTOR) orchestrates the defense program of innate immune cells. Wiener Klinische Wochenschrift. 2008;120:36–36. doi: 10.1002/eji.200838761. [DOI] [PubMed] [Google Scholar]
- 31.Ohtani M, Nagai S, Kondo S, Mizuno S, Nakamura K, Tanabe M, Takeuchi T, Matsuda S, Koyasu S. Mammalian target of rapamycin and glycogen synthase kinase 3 differentially regulate lipopolysaccharide-induced interleukin-12 production in dendritic cells. Blood. 2008;112:635–643. doi: 10.1182/blood-2008-02-137430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cao WP, Manicassamy S, Tang H, Kasturi SP, Pirani A, Murthy N, Pulendran B. Toll-like receptor-mediated induction of type I interferon in plasmacytoid dendritic cells requires the rapamycin-sensitive PI(3) K-mTOR-p70S6K pathway. Nat immunol. 2008;9:1157–1164. doi: 10.1038/ni.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Colina R, Costa-Mattioli M, Dowling RJ, Jaramillo M, Tai LH, Breitbach CJ, Martineau Y, Larsson O, Rong L, Svitkin YV, Makrigiannis AP, Bell JC, Sonenberg N. Translational control of the innate immune response through IRF-7. Nature. 2008;452:323–328. doi: 10.1038/nature06730. [DOI] [PubMed] [Google Scholar]
- 34.Huang J, Manning BD. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J. 2008;412:179–190. doi: 10.1042/BJ20080281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17:1829–1834. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gan B, Sahin E, Jiang S, Sanchez-Aguilera A, Scott KL, Chin L, Williams DA, Kwiatkowski DJ, DePinho RA. mTORC1-dependent and -independent regulation of stem cell renewal, differentiation, and mobilization. Proc Natl Acad Sci U S A. 2008;105:19384–19389. doi: 10.1073/pnas.0810584105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen C, Liu Y, Liu R, Ikenoue T, Guan KL, Zheng P. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J Exp Med. 2008;205:2397–2408. doi: 10.1084/jem.20081297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kwiatkowski DJ, Zhang HB, Bandura JL, Heiberger KM, Glogauer M, el-Hashemite N, Onda H. A mouse model of TSC1 reveals sex-dependent lethality from liver hemangiomas, and up-regulation of p70S6 kinase activity in Tsc1 null cells. Human Mol Genet. 2002;11:525–534. doi: 10.1093/hmg/11.5.525. [DOI] [PubMed] [Google Scholar]
- 39.Shapiro-Shelef M, Lin KI, Savitsky D, Liao J, Calame K. Blimp-1 is required for maintenance of long-lived plasma cells in the bone marrow. J Exp Med. 2005;202:1471–1476. doi: 10.1084/jem.20051611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu CH, Machado FS, Guo R, Nichols KE, Burks AW, Aliberti JC, Zhong XP. Diacylglycerol kinase zeta regulates microbial recognition and host resistance to Toxoplasma gondii. J Exp Med. 2007;204:781–792. doi: 10.1084/jem.20061856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chong-Kopera H, Inoki K, Li Y, Zhu T, Garcia-Gonzalo FR, Rosa JL, Guan KL. TSC1 stabilizes TSC2 by inhibiting the interaction between TSC2 and the HERC1 ubiquitin ligase. J Biol Chem. 2006;281:8313–8316. doi: 10.1074/jbc.C500451200. [DOI] [PubMed] [Google Scholar]
- 42.Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. 2009;30:475–487. doi: 10.1016/j.it.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 43.Fukao T, Koyasu S. PI3K and negative regulation of TLR signaling. Trends Immunol. 2003;24:358–363. doi: 10.1016/s1471-4906(03)00139-x. [DOI] [PubMed] [Google Scholar]
- 44.Luyendyk JP, Schabbauer GA, Tencati M, Holscher T, Pawlinski R, Mackman N. Genetic analysis of the role of the PI3K-Akt pathway in lipopolysaccharide-induced cytokine and tissue factor gene expression in monocytes/macrophages. J Immunol. 2008;180:4218–4226. doi: 10.4049/jimmunol.180.6.4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol. 2005;6:777–784. doi: 10.1038/ni1221. [DOI] [PMC free article] [PubMed] [Google Scholar]