

Abstract
The efficacy of alloSCT is limited by graft-versus-host disease (GVHD). Host hematopoietic antigen presenting cells (APCs) are important initiators of GVHD making them logical targets for GVHD prevention. Conventional dendritic cells (cDCs) are key APCs for T cell responses in other models of T cell immunity and they are sufficient for GVHD induction. However, we report here in two polyclonal GVHD models in which host hematopoietic APCs are essential, that GVHD was not decreased when recipient cDCs were inducibly or constitutively deleted. Additional profound depletion of plasmacytoid DCs and B cells, with or without partial depletion of CD11b+ cells, also did not ameliorate GVHD. These data indicate that, in contrast to pathogen models, there is a surprising redundancy as to which host cells can initiate GVHD. Alternatively, very low numbers of targeted APCs were sufficient. We hypothesize the difference in APC requirements in pathogen and GVHD models relates to the availability of target antigens. In anti-pathogen responses specialized APCs are uniquely equipped to acquire and present exogenous antigens whereas in GVHD all host cells directly present alloantigens. These studies make it unlikely that reagent-based host APC depletion will prevent GVHD in the clinic.
Introduction
Allogeneic hematopoietic stem cell transplantation (alloSCT) can be a life-saving therapy for hematologic malignancies and acquired or inherited nonmalignant disorders of blood cells. Mature donor T cells in allografts play important roles in alloSCT. They contribute to T cell reconstitution in the recipient, promote donor hematopoietic engraftment and mediate an anti-tumor effect called graft-vs.-leukemia (GVL). Unfortunately, donor T cells can broadly target host tissues causing GVHD(1). Because of GVHD, all patients receive some type of prophylactic immunosuppression, either by depleting T cells from the allograft, or more commonly, with pharmacologic agents that inhibit T cell function. However, even with pharmacologic immunosuppression, GVHD remains a major cause of morbidity and mortality. Novel approaches are clearly needed.
GVHD is initiated by antigen presenting cells (APCs) that prime alloreactive donor T cells (1-6). Recipient APCs that survive conditioning are essential for GVHD in MHC-mismatched transplants and in CD8-mediated GVHD across only minor histocompatibility antigens (miHAs) (1-4, 7, 8). They also have an important and nonredundant role in CD4-mediated GVHD across miHAs (6). Thus, recipient APCs would be a logical target for suppressing GVHD.
APCs, which include dendritic cells (DCs), B cells, macrophages and basophils, are diverse cells that have in common the ability to prime T cells. Among APCs, DCs are perhaps the most efficacious in priming naïve T cells (9), which are potent inducers of GVHD (1). Consistent with this, in an MHC-mismatched alloBMT model, infusing wild type recipient conventional DCs (cDCs) or plasmacytoid DCs (pDCs) can partially restore GVHD in mice with MHC-deficient hematopoietic cells (3, 4).
The importance of cDCs in adaptive T cell responses has been highlighted by data from experiments studying immune responses in mice in which cDCs can be inducibly depleted or mice with a constitutive absence of cDCs. Both anti-viral and anti-bacterial T cell responses were greatly diminished after induced depletion of CD11c+ cells (10-17). cDC depletion also blunted T cell responses in allergic asthma (18) and in a model of anti-tumor immunity (19). Mice that constitutively lack cDCs with a variable depletion of pDCs have impaired anti-viral clearance (20) and diminished T cell responses in a mouse lupus model (21).
In sum, these data made DCs attractive cells to target to prevent GVHD. We therefore studied the role of host DCs in two models of GVHD in which host hematopoietic APCs are absolutely required for GVHD induction (2, 8, 22). Contrary to expectations, neither induced nor constitutive profound depletion of CD11c+ cDCs mitigated clinical or histopathologic GVHD. The addition of antibody-based depletion of pDCs and B cells and partial depletion of additional CD11b+ cells also did not prevent GVHD. These data highlight the unique aspects of T cell activation in GVHD and suggest that there are redundant populations of APCs capable of priming allogeneic T cells and/or that very few residual host APCs are sufficient. These results make it unlikely that reagent based host DC depletion will be effective in preventing GVHD in the clinic.
Methods
Mice
C57BL/6 (B6) CD45.1 mice were purchased from Taconic/NCI Frederick (Frederick, MD). C3H.SW and B6bm12 mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and bred at Yale. C3H.SW β2M-/- mice (5) were bred at Yale. CD11c-DTR (10) and CD11b-DTR mice (23) were purchased from The Jackson Laboratory and bred at Yale. These mice were crossed to create mice heterozygous for both transgenes and typed by PCR as described (10, 23). CD11c-cre mice were a gift of B. Reizis (24)(Columbia University, NY). Rosa26-eGFP-DT C57BL/6 mice were provided by J.P. Martinez-Barbera (25) (University College, London). The Rosa26-eGFP-DT locus contains the gene for the diphtheria toxin α chain (DTA) that is preceded by a loxP-flanked STOP cassette so that expression of the toxin is restricted to those cells expressing Cre recombinase. These mice were intercrossed to obtain constitutively DC-deficient CD11c-cre +Rosa26-eGFP-DT+ mice (CD11c:DTA).
Cell purifications
CD8 cells were purified via depletion from lymph node (LN) using biotin-conjugated antibodies, streptavidin microbeads and an AutoMACs cell separator (Miltenyi Biotec, Auburn, CA) as described (5). CD8 cells were >90% pure with CD4+ T cell contamination of less than 3%. All donor bone marrow (BM) was depleted of T cells using anti-Thy1.2 microbeads and the AutoMACs (Miltenyi Biotec).
Bone marrow transplantation
All transplants were performed according to protocols approved by the Yale University Institutional Animal Care and Use Committee. To create BM chimeras, mice were irradiated (500cGy × 2) and reconstituted with 107 T cell-depleted donor BM cells. To induce GVHD, chimeras were reirradiated (450cGy × 2) and reconstituted with T cell-depleted donor BM with or without T cells. In the C3H.SW→B6 system, GVHD was induced by 2×106 CD8 cells. In the B6bm12→B6 model, GVHD was induced by splenocytes containing 106 CD4 cells. Mice were weighed 2-3x/week; weights from mice that died, were sacrificed due to predefined measures of morbidity or were censored when experiments of different lengths were combined were included in averages for subsequent time points at the last value recorded. Deaths prior to day 11 in the C3H.SW→B6 model were scored as not being GVHD related based on our extensive experience in this model, and these mice were censored (indicated by ticks on survival curves). Skin disease was scored when mice were weighed; minimal criteria for clinical skin disease was fur loss in an area >1cm2.
Assessment of APC engraftment in BM chimeras
Spleens and LNs were digested with collagenase D as described (8). To distinguish residual recipient (CD45.1+) and donor-derived (CD45.2+) DCs in initial BM chimeras, cells were stained with antibodies against CD45.1 or CD45.2 (clones A20 and 104; BD Pharmingen), CD11c (APC; clone HL3; BD Pharmingen), a cocktail of biotin-conjugated antibodies against Gr-1 (RB6-8C5; BD Pharmingen), CD19 (1D3; BD Pharmingen) or CD45RB (B220), Ly76 (TER119; BD Pharmingen), and Thy1.2 (30H12; lab-prepared). Cells were stained with streptavidin-PerCP (BD Pharmingen). DCs were identified as CD11c+lineage-. A cohort of chimeras were sacrificed for analysis prior to each second GVHD-inducing transplant. CD11c+ DCs were at least >96% donor-derived and in most mice >98% donor-derived.
Measurement of serum cytokines
Serum cytokines were measured using the Bioplex kit Th1/Th2 panel (Bio-Rad; Hercules, CA) and a Luminex 100 system (Luminex; Austin, TX) as previously described (26).
DT treatment and B cell and pDC depletion
DT (Sigma) was resuspended in sterile water. Females, which were less than 25gms, were treated with 100ng of DT whereas males which were mostly >25gms were weighed and treated with 4ng/gm. The BST2 hybridoma(27) was a gift from Marco Colonna (Washington University School of Medicine, MO) and was lab-prepared. Anti-mouse CD20 (an IgG2aκ derivative of clone 18B12(28)) was provided by Marilyn Kehry (Biogen/IDEC, San Diego, CA).
Scoring of pathology
In the C3H.SW→B6 strain pairing some CD8 recipients reached endpoints for extent of skin disease and morbidity that required earlier sacrifice and pathology was harvested at that time. Otherwise pathology was harvested at the conclusion of each experiment. Skin and gastrointestinal pathology were scored by J.M. and A.J.D., respectively, as previously described (5, 29).
Statistics
Significance for differences in weight loss was calculated by an unpaired t test. P values for differences in survival and the incidence of skin disease were calculated by a log-rank test. P values for histology scores and serum cytokine levels were calculated by Mann-Whitney. Error bars for weights represent standard error measurements (GraphPad Prism).
Results
DT treatment depletes CD11c+ cells in CD11c-DTR→B6 radiation chimeras
To deplete cDCs we exploited mice transgenic for a construct that targets the simian diphtheria toxin receptor (DTR) to CD11c+ cells (CD11c-DTR). Diphtheria toxin (DT) treatment of these mice rapidly and profoundly depletes cDCs (10, 30). Because repeated DT treatment is lethal due to promiscuous transgene expression (30), we used as hosts in allogeneic bone marrow transplant (alloBMT) experiments B6 CD11c-DTR (H-2b; CD45.2)→B6 (CD45.1) BM chimeras in which >96% of LN and splenic CD11c+ cells were donor-derived ((29) and not shown). To confirm that DT-treatment depletes splenic and lymph node (LN) DCs, CD11c-DTR (CD45.2; heterozygous for the CD11c-DTR transgene)→B6 CD45.1 radiation chimeras were treated with a single dose of DT and cohorts were sacrificed daily for 4 days to quantitate the number of CD45.2+CD11c+ cells. Consistent with prior reports, within 24 hours of DT treatment there was a profound depletion of CD11c-DTR CD11c+ cells, which gradually recovered over the following 72 hours (Figure 1A, B). We next determined whether DT treatment augmented DC depletion beyond what is induced by radiation. CD11c-DTR→B6 (CD45.1) and control B6→B6 (CD45.1) chimeras received DT followed several hours later by split-dose radiation. Splenic DCs were quantitated 24 and 48 hours later. DT treatment further diminished splenic DC numbers both 24 and 48 hours after radiation (Figure 1C, D). In a separate experiment, B6 CD11c-DTR mice were irradiated and reconstituted with C3H.SW bone marrow (BM) and 2×106 CD8 cells. On days - 1 and +1, mice were treated with 4ng/gm (100ng/mouse) of DT or PBS. Cohorts were sacrificed on days +3 and +6 to quantitate host DCs. In transplanted mice, at day +3 there was a nearly 200-fold reduction in splenic DC numbers and greater than 100-fold reduction in LN DCs in DT-treated mice (Supplemental Figure 1A, representative flow cytometry; Supplemental Figure 1B, quantitation). The absolute DC numbers were quite small in DT-treated mice—less than 200/spleen and 50 in pooled cervical, subscapular, mesenteric, axillary and inguinal LNs. At day +6 we could detect no host DCs in DT-treated mice and <200/spleen in non-DT treated mice (Supplemental Figure 1B). We could detect no host DCs in LNs of untreated or DT-treated mice at day +6 (Supplemental Figure 1B).
Figure 1. DT depletes cDCs in CD11c-DTR→B6 BM chimeras.

(A, B) CD11c-DTR+/−→B6 CD45.1 and WT→B6 CD45.1 BM chimeras were treated a single 100ng dose of DT i.p and cohorts were sacrificed daily for 4 days to quantitate spleen and LN live lineage-CD45.2+CD11c+ cells. (A) Representative flow cytometry, gating on live/lineage- cells. (B) Numbers of live lineage-CD11c+ cells per spleen (left panel) and in cervical, axillary, inguinal and mesenteric LNs combined (right panel) in DT-treated CD11c-DTR+/− →B6 chimeras. To determine whether DT-treatment depleted cDCs beyond what is achieved with radiation, CD11c-DTR+/−→B6 CD45.1 BM chimeras were injected with DT or PBS followed by 450cGy × 2 several hours later. Splenic CD11c+ cells were enumerated 1 and 2 days after treatment. (C) Representative flow cytometry, gating on live/lineage- cells. (D) Numbers of splenic live lineage-CD11c+ cells. P<0.0120 comparing number of splenic cDCs with or without DT treatment at day +1 and day +2.
Host cDC depletion does not prevent GVHD in the C3H.SW→B6 GVHD model
In an initial experiment we DT-treated CD11c-DTR→B6 (CD45.1) chimeras on day -1 prior to transplantation with C3H.SW (H-2b) BM with no T cells or purified C3H.SW CD8 cells. Despite profound cDC depletion, neither clinical nor histopathologic GVHD was reduced as compared to non-DT-treated CD11c-DTR→B6 chimeras or DT-treated wt→wt chimeras (not shown). We next considered the possibility that prolonged DT treatment would be required as there could be radiation-resistant CD11c-host cDC progenitors that are not killed by a single DT treatment but that later mature into functional cDCs (31). We therefore repeated the prior experiment (in the C3H.SW→B6 model) except DT-treatment was on days -1, 0, +1, +3, +5, and +7. Again neither clinical nor histopathologic GVHD was reduced (Figure 2).
Figure 2. Depletion of host CD11c+ cells does not diminish CD8-mediated GVHD in the C3H.SW→B6 strain pairing.

CD11c-DTR→B6 (CD45.1) chimeras (“CD11c+”) or control wt→wt chimeras (“CD11c-”) mice were injected with DT (100ng/mouse i.p.) on days -1, 0, +1, +3, +5 and +7. On day 0 chimeras were reirradiated (450cGy × 2) and reconstituted with 107 T cell-depleted C3H.SW BM cells, with or without 2×106 C3H.SW CD8 cells. An additional group of CD11c+ chimeras were transplanted but not injected with DT. (A) Weight loss. P<0.02 comparing any CD8 recipient group with its BM alone control beginning on day +25; P>0.13 comparing any CD8 recipient group to any other CD8 recipient group beginning on day +11. (B) Incidence of skin disease. P<0.04 comparing any CD8 recipient group with its BM alone control; P>0.93 comparing any CD8 recipient group to any other CD8 recipient group. (C) Organ pathology scores. * P≤0.05 compared to its BM alone control; P>0.29 comparing any CD8 recipient group to any other CD8 recipient group. Data are representative of 3 independent experiments.
Although we previously found that C3H.SW→B6 and β2M-/-→B6 BM chimeras are resistant to GVHD when retransplanted with C3H.SW BM and CD8 cells (8), we considered the possibility that with DT treatment, donor APCs were sufficient. We therefore repeated the experiment described in Figure 2, except we used C3H.SW β2M-/- BM donors, thereby preventing antigen presentation by donor APCs (5). All recipients were treated with anti-NK1.1 to prevent rejection of donor β2M-/- cells. DT-treated CD11c-DTR→B6 CD8 chimeric recipients developed similar weight loss and histopathologic GVHD of liver as did the control CD8 recipient groups (Figure 3). In the colon, there was reduced colon histopathology in DT-treated Tg+ CD8-recipients as compared to DT-treated Tg- CD8-recipients, but not as compared to non-DT-treated Tg+ CD8-recipients (Figure 3). No CD8 recipient group developed significant clinical or histopathologic skin GVHD, as we previously found when donor BM was β2M-/- in this model (5) (not shown). These data indicate that the failure of host cDC depletion to prevent GVHD was not due to antigen presentation by donor APCs.
Figure 3. Failure of cDC depletion to decrease GVHD is not due to donor T cell activation by donor APCs.
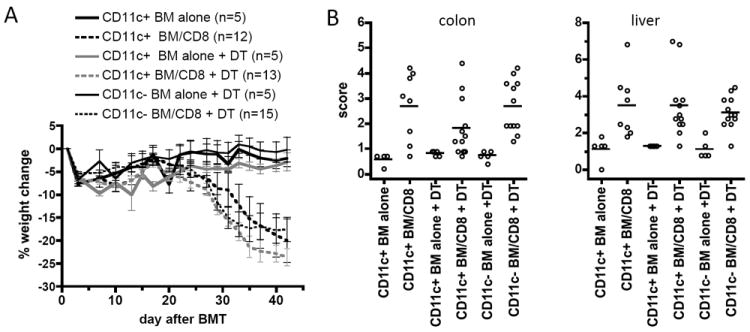
Mice were transplanted as in Figure 2 except donor BM was C3H.SW β2M-/-. All mice were treated with 0.2mg anti-NK1.1 (PK136) on days -2, -1 and +7 as described (5, 8). (A) Weight loss. P<0.03 comparing CD11c+ and CD11c- DT-treated CD8 recipient groups with their BM alone controls beginning on day +27; P<0.044 comparing the CD11c+ no DT group with its BM alone control beginning on day +35. P>0.135 comparing any CD8 recipient group to any other CD8 recipient group beginning on day +15. (B) Organ pathology scores. Skin and ear GVHD were not significant, consistent with the reduced cutaneous GVHD we previously observed when donor BM is β2M-/- (5). P<0.013 comparing any CD8 recipient group with its BM alone control; P>0.23 comparing any CD8 recipient group to any other CD8 recipient group except for P=0.035 comparing colon scores in CD11c+ and CD11c- DT-treated CD8 recipients. Data are from 1 experiment.
cDC depletion does not prevent GVHD in the B6bm12→(B6→B6bm12) GVHD model
We next considered the possibility that residual non-transgenic cDCs in the CD11c-DTR →B6 chimeras were initiating GVHD. We thought this was unlikely given the quality of donor engraftment and the aforementioned resistance of C3H.SW→B6 and β2M-/-→B6 chimeras to GVHD induction (8) and GVL (32). To formally exclude priming by residual host non-transgenic APCs we used the MHCII-mismatched B6bm12→B6 model in which allogeneic host hematopoietic cells are sufficient for GVHD (2, 29). To prevent allogeneic T cell priming by residual host nontransgenic APCs we made B6 CD11c-DTR→B6bm12 chimeras and retransplanted them with B6bm12 BM and spleen cells. In such chimeras any residual B6bm12 hematopoietic APCs and all nonhematopoietic cells are syngeneic to donor B6bm12 GVHD-inducing CD4 cells. Further, because B6 and B6bm12 mice only differ by a few amino acids in I-A (33), there are essentially no miHAs to be presented by donor APCs. Therefore allogeneic stimulation of donor B6bm12 CD4 cells in these chimeras is exclusively by B6 background hematopoietic APCs. Host DT-treatment reduced neither weight loss nor serum IFN-γ levels (characteristic of GVHD in this model (22, 34) as compared to non-DT treated CD11c-DTR→B6bm12 and DT-treated transgene-negative→B6bm12 CD4 recipients (Figure 4A, B). Liver pathology was present in all CD4 recipient groups in one of two repetitions, and it was modestly reduced in DT-treated CD11c+ CD4 recipients (Figure 4C). Therefore when antigen presentation by residual host type APCs is excluded and the only functional APCs are derived from CD11c-DTR transgene-positive BM, DT-mediated depletion of cDCs was insufficient to prevent GVHD, though in one experiment there was reduced liver histopathology in cDC-depleted CD4-recipients.
Figure 4. Depletion of host CD11c+ cells does not diminish CD4-mediated GVHD in the B6bm12→(B6→B6bm12) model.
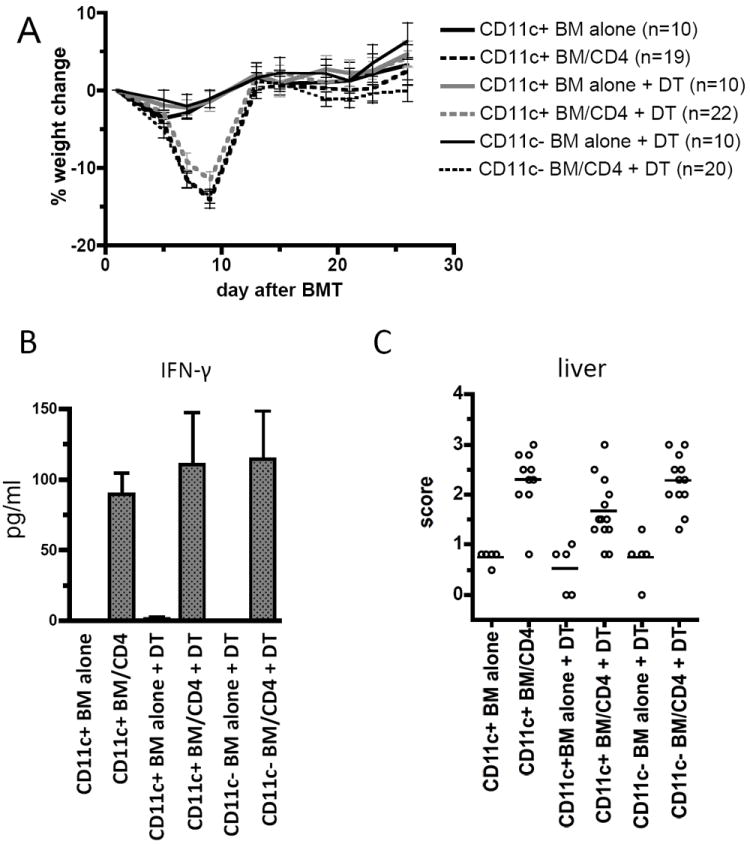
CD11c-DTR→B6bm12 chimeras (CD11c+) or control wt→B6bm12 chimeras (CD11c-) mice were injected with DT (4ng/gm i.p.) on days -1, 0, +1, +3, +5 and +7. On day 0, chimeras were reirradiated (450cGy × 2) and reconstituted with T cell-depleted B6bm12 BM with or without B6bm12 splenocytes containing 106 CD4 cells. An additional CD11c+ group was transplanted without DT injection. (A) Weight loss. P≤0.0003 comparing any CD4 recipient group with its BM alone control on days +7 and +9; P>0.06 comparing any CD4 recipient group to any other CD4 recipient group at all days except day +26. (B) Serum IFN-γ levels measured by Luminex at day +7 (3 BM alone and 7 spleen cell recipients for each group). P=0.0083 comparing any CD4 recipient group to its BM alone control; P>0.7 comparing any CD4 recipient group to any other CD4 recipient group. (C) Liver pathology scores from one of two experiments. P≤0.0029 comparing any CD4 recipient group to its BM alone control; P<0.027 comparing DT-treated CD11c+ and CD11c- CD4-recipients and DT-treated and untreated CD11c+ CD4 recipients.
Additional depletion of pDCs, B cells and partial depletion of CD11b+ cells does not mitigate GVHD
To broaden the scope of APC depletion we added antibody-mediated pDC (27) and B cell depletion (28). B cells were depleted as we previously reported (28). pDCs were depleted by injecting the anti-BM stromal derived antigen antibody BST-2 on days -2 and -1 prior to BMT (Supplemental Figure 2A). In the C3H.SW→(CD11cDTR→B6) model, additional depletion of B cells and pDCs did not affect clinical or histologic GVHD (not shown). We next crossed the CD11c-DTR mice to CD11b-DTR mice (23) (CD11c/b-DTR). DT treatment of CD11b-DTR mice depletes tissue macrophages and induces a modest reduction in subsets of CD11b+ cells in spleen and LN ((23, 35-41) and Supplemental Figure 2). We created CD11c/b-DTR→B6 (CD45.1; referred to as Tg+) and wt→B6 (CD45.1; WT) BM chimeras and used them as recipients in the C3H.SW→B6 model. All chimeras, Tg+ or WT, were DT-treated every other day from day -5 to day +5. All Tg+ chimeras received anti-CD20 2 weeks prior to transplant. We further added pDC depletion to one group of Tg+ chimeras. DT-treated and B cell-depleted Tg+ chimeras developed similar clinical GVHD as did WT chimeras as measured by weight loss and incidence of clinical skin disease, whether or not pDCs were additionally depleted (Figure 5A, B). Survival was worse in DT-treated Tg+ DT recipients (Figure 5C) as compared to DT-treated Tg- CD8 recipients. Histopathologic GVHD of the skin and ear were similar in all CD8 recipient groups (Figure 5D). Colon and liver GVHD were reduced in DT-treated Tg+ CD8 recipients (not shown). However this difference is difficult to interpret as some of the most severely affected Tg+ CD8 recipients died prior to tissue harvest or had their tissues collected at earlier times when sacrificed secondary to the extent of skin disease.
Figure 5. DT treatment of CD11c/b-DTR→B6 (Tg+) chimeras with B cell depletion, with or without pDC depletion, does not decrease GVHD.

All BM chimeric hosts (Tg+ and Tg-) were DT-treated (4ng/gm i.p.) every other day from day -5 to day +5. All Tg+ chimeras received 0.25mg of anti-CD20 two weeks prior to transplantation as previously described (28). One group of Tg+ chimeras also received 0.5mg BST2 (27) i.p on days -2 and -1 to deplete pDCs (Supplemental Figure 2A). Mice were otherwise transplanted as in Figure 3. DT treatment and B cell and pDC depletion did not diminish weight loss (A) or incidence of clinical skin disease (B). P≤0.036 comparing weight change in any CD8 recipient to its BM alone control, beginning on day +20. P≥0.28 comparing weights of Tg+ + BST2 vs Tg- CD8 recipients beginning on day +13. P=0.042 and P=0.024 comparing incidence of skin disease in Tg+ CD8 recipients with or without BST2 treatment to the combined BM alone controls. P=0.09 comparing Tg- CD8 recipients and BM alone controls. Survival was inferior in Tg+ CD8 recipients, with or without pDC depletion, as compared to Tg- CD8 recipients (C; P<0.027). (D) Histopathology scores. P<0.05 comparing skin or ear scores in any CD8 recipient group versus its BM alone control. P=0.014 and P=0.083 comparing skin and ear scores (respectively) of Tg+ + BST2 to Tg- CD8 recipients. Data combined from 2 experiments, terminated on days +29 and +39; the Tg+ BM alone group was only present in the experiment which ended on day +29.
We also tested this approach in the B6bm12 model. We created CD11c/b-DTR→B6bm12 (Tg+) and WT→B6bm12 chimeras. All mice were DT-treated every other day from day-5 to day +5 and Tg+ chimeras additionally were B cell- and pDC-depleted. These chimeras were retransplanted with B6bm12 BM and spleen cells. Weight loss and serum IFN-γ levels were similar in DT-treated and B cell/pDC-depleted Tg+ chimeras and control DT-treated WT chimeras (Figure 6A, B). Liver GVHD, which developed in one of two repetitions, was significant in both spleen cell recipient groups, and scores were lower in Tg+ spleen cell recipients (Figure 6C). However, scores in the Tg- BM alone group were higher than in the Tg+ BM alone group and thus the difference in spleen cell recipient groups could have been due to inflammation unrelated to GVHD.
Figure 6. DT treatment of CD11c/b-DTR→B6bm12 (Tg+) chimeras with B cell depletion and pDC depletion does not decrease GVHD.

CD11c/b-DTR→B6bm12 chimeras (Tg+) or control wt→B6bm12 chimeras (Tg-) mice were injected with DT (4ng/gm i.p.) every other day from day -5 to day +5. All Tg+ mice received 0.25mg of anti-CD20 on day -14 and with 0.5mg of BST2 i.p. on days -2 and -1 to deplete B cells and pDCs, respectively. On day 0, chimeras were reirradiated (450cGy × 2) and reconstituted with T cell-depleted B6bm12 BM with or without B6bm12 splenocytes containing 106 CD4 cells. (A) Weight change. P<0.03 comparing weights of Tg+ or WT→B6bm12 (WT) spleen cell recipients versus their BM alone controls from days +5 to +27. P>0.05 comparing Tg+ and WT spleen recipients on days +5, +7, +12 and +14. P<0.0001 comparing serum IFN-γ levels (B) at day +7 in Tg+ or Tg- spleen cell recipients versus BM alone controls (4 Tg+ and WT BM alone controls; 10 Tg+ and WT spleen cell recipients). (C) Liver histopathology scores. P=0.353 comparing spleen cell recipient groups. P=0.14 and 0.04 comparing Tg+ and WT spleen cell recipients to their BM alone controls, respectively. P=0.03 comparing Tg+ and WT spleen cell recipients. P=0.095 comparing Tg+ and WT BM alone groups. Data combined from 2 experiments.
Lastly we used as hosts in the C3H.SW→B6 system CD11c-cre transgenic mice (24) crossed to mice which have a floxed STOP cassette flanking the DT-α chain gene, inserted into the ROSA26 locus (25) (CD11c:DTA). CD11c:DTA mice have a constitutive absence of CD11c+ cDCs, Langerhans cells (LCs) and most pDCs ((20, 21); Figure 7A). We further added antibody-mediated B cell depletion and pDC depletion. Both CD8 recipient groups lost weight relative to their BM alone controls (Figure 7B) and there were more deaths in CD11c:DTA CD8 recipients (Figure 7C). Histopathologic skin, ear, and colon GVHD were similar in both CD8 groups (Figure 7D).
Figure 7. GVHD is not reduced in CD11c:DTA recipients.

(A) Shown are staining of representative Tg- and CD11c:DTA splenocytes for CD11c+MHCII+ (left panel) and BST2+B220+ pDCs (right panel). (B-D) CD11c:DTA or control CD11c-cre or ROSA26stop/flox/stop DT mice were irradiated (475cGy × 2) and reconstituted with 107 T cell-depleted C3H.SW BM with no T cells or with 2×106 C3H.SW CD8 cells. CD11c:DTA mice were also depleted of B cells and pDCs as described in Figure 5. (B) Weight change. P<0.006 comparing CD11c:DTA or control CD8 recipients with their BM alone controls from day +22 onward. (C) Survival. P<0.0001 comparing CD8 recipient groups. (D) Histopathology scores. P≤0.008 comparing skin, ear and colon scores in CD11c:DTA CD8 recipients as compared to the CD11c:DTA BM alone controls. P<0.0003 comparing skin and colon scores and P=0.053 comparing ear scores in control CD8 recipients and BM alone controls. P≥0.15 comparing all histopathology scores in CD11c:DTA and control CD8 recipients. Liver GVHD could not be evaluated due to a high baseline level of inflammation in the CD11:DT BM alone group (not shown). Data combined from two experiments with similar results.
Discussion
Contrary to expectations, depletion of host cDCs failed to significantly mitigate GVHD in two models in which host hematopoietic APCs are necessary and sufficient for GVHD induction. Importantly, we excluded priming by donor or residual Tg- host APCs. We cannot completely exclude that GVHD was induced by residual Tg+ cDCs not killed by DT treatment or by expression of DTA. However, cDC ablation was profound in DT-treated CD11c-DTR and CD11c/b-DTR BM chimeras and in CD11c:DTA mice, and this degree of cDC ablation was sufficient to block T cell responses in many other model systems (20, 30, 42), (21). From these data we conclude that host cDCs are not required for GVHD induction or that a very small number of cDCs, insufficient to initiate substantial T cell response in other systems, are adequate in GVHD. Either way, targeting host cDCs will be unlikely to prevent GVHD in the clinic. Additional partial depletion of host CD11b+ cells along with B cells and pDCs did not meaningfully mitigate GVHD in either model system or reduce serum IFN-γ levels in the B6bm12 model.
Although overall cDC depletion did not reduce GVHD, liver GVHD was reduced in DT-treated mice in one experiment in the B6bm12 model (Figure 4) and colon GVHD relative to DT-treated Tg- CD8-recipients in one experiment in the C3H.SW→B6 system (but not relative to PBS-treated Tg+ CD8-recipients; Figure 3). It is possible that these reductions were a true consequence of cDC depletion.
Our results raise the question of what other APCs could be priming donor T cells. We can exclude Langerhans cells (LCs) as they are neither sufficient nor required for GVHD in the C3H.SW→B6 model (8, 43) and are depleted in CD11c:DTA mice (21). In B6→B6bm12 chimeras, LCs remained B6bm12 in origin (43) and therefore would not prime syngeneic B6bm12 donor CD4 cells. Although we used state of the art methods to deplete APCs, all host CD11b+ cells were not eliminated in DT-treated CD11b-DTR mice, and we were unable to employ higher doses of DT which may have improve depletion due to lethality in Tg+ and Tg- mice (not shown). Future studies testing the importance of these cells will require the development of less toxic depletion methods.
Could nonhematopoietic host cells be priming donor T cells? This is unlikely in the C3H.SW→B6 system wherein others and we have previously shown host hematopoietic cells to be essential APCs (8, 32). Host hematopoietic cells were also required for lethal GVHD in the BALB/c (H-2d)→B6 (H-2b) MHC-mismatched strain pairing (7). In the B6bm12→ (B6→B6bm12) model, nonhematopoietic cells were B6bm12 and would not have primed syngeneic B6bm12 CD4 cells.
As to why APC requirements differ in GVHD than in other models of T cell immunity, we hypothesize this relates to differences in the availability and distribution of target antigens. In most models of T cell immunity wherein CD11c+ APCs have been shown to be critical, APCs are directly infected or more commonly must acquire, process and present exogenous antigens. cDCs and other specialized APCs have receptors and antigen processing mechanisms that facilitate this (44, 45) (46, 47). Further, because in infections only a minority of APCs acquire pathogen-derived antigens, such specialized properties of APCs and their activation by pattern recognition receptors may be pivotal for the efficient attraction, priming and programming of rare antigen-reactive T cells (48, 49). In contrast, in GVHD all host cells express and directly present alloantigens, a situation akin to a viral infection of every cell. Therefore the specialized properties of “professional APCs” that evolved to respond to pathogens may not be critical for alloreactive T cell activation. Consistent with this, GVHD induction in the same models presented here do not require that APCs be stimulated through TLRs or the IL-1R in response to IL-1 generated by inflammasome activation (29). In sum these studies demonstrate that the APC requirements for activation of T cells in alloBMT are unique and clearly distinct from those in other models. Host APC-directed approaches for preventing GVHD in the clinic cannot focus only on cDCs, pDCs and B cells and will need to additionally target other APCs classically considered to be less potent or alternatively interfere with antigen processing mechanisms found on all cells.
Supplementary Material
Acknowledgments
We thank the technicians of the Yale animal facilities for excellent care.
This work was supported by HL083072 and AI064343. WDS was a Clinical Scholar of the Leukemia and Lymphoma Society.
Footnotes
Author contributions. HL designed and performed experiments, analyzed data and wrote the paper. CM and HST performed experiments. AJD and JM scored histopathology. DMR and FGL assisted in experimental design and data analysis. WDS designed experiments, analyzed data and wrote the paper.
References
- 1.Shlomchik WD. Graft-versus-host disease. Nat Rev Immunol. 2007;7:340–352. doi: 10.1038/nri2000. [DOI] [PubMed] [Google Scholar]
- 2.Teshima T, Ordemann R, Reddy P, Gagin S, Liu C, Cooke KR, Ferrara JL. Acute graft-versus-host disease does not require alloantigen expression on host epithelium. Nat Med. 2002;8:575–581. doi: 10.1038/nm0602-575. [DOI] [PubMed] [Google Scholar]
- 3.Duffner UA, Maeda Y, Cooke KR, Reddy P, Ordemann R, Liu C, Ferrara JL, Teshima T. Host dendritic cells alone are sufficient to initiate acute graft-versus-host disease. J Immunol. 2004;172:7393–7398. doi: 10.4049/jimmunol.172.12.7393. [DOI] [PubMed] [Google Scholar]
- 4.Koyama M, Hashimoto D, Aoyama K, Matsuoka K, Karube K, Niiro H, Harada M, Tanimoto M, Akashi K, Teshima T. Plasmacytoid dendritic cells prime alloreactive T cells to mediate graft-versus-host disease as antigen-presenting cells. Blood. 2009;113:2088–2095. doi: 10.1182/blood-2008-07-168609. [DOI] [PubMed] [Google Scholar]
- 5.Matte CC, Liu J, Cormier J, Anderson BE, Athanasiadis I, Jain D, McNiff J, Shlomchik WD. Donor APCs are required for maximal GVHD but not for GVL. Nat Med. 2004;10:987–992. doi: 10.1038/nm1089. [DOI] [PubMed] [Google Scholar]
- 6.Anderson BE, McNiff JM, Jain D, Blazar BR, Shlomchik WD, Shlomchik MJ. Distinct roles for donor- and host-derived antigen-presenting cells and costimulatory molecules in murine chronic graft-versus-host disease: requirements depend on target organ. Blood. 2005;105:2227–2234. doi: 10.1182/blood-2004-08-3032. [DOI] [PubMed] [Google Scholar]
- 7.Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, Posati S, Rogaia D, Frassoni F, Aversa F, Martelli MF, Velardi A. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. 2002;295:2097–2100. doi: 10.1126/science.1068440. [DOI] [PubMed] [Google Scholar]
- 8.Shlomchik WD, Couzens MS, Tang CB, McNiff J, Robert ME, Liu J, Shlomchik MJ, Emerson SG. Prevention of graft versus host disease by inactivation of host antigen-presenting cells. Science. 1999;285:412–415. doi: 10.1126/science.285.5426.412. [DOI] [PubMed] [Google Scholar]
- 9.Shortman K, Liu YJ. Mouse and human dendritic cell subtypes. Nat Rev Immunol. 2002;2:151–161. doi: 10.1038/nri746. [DOI] [PubMed] [Google Scholar]
- 10.Jung S, Unutmaz D, Wong P, Sano G, De los Santos K, Sparwasser T, Wu S, Vuthoori S, Ko K, Zavala F, Pamer EG, Littman DR, Lang RA. In vivo depletion of CD11c(+) dendritic cells abrogates priming of CD8(+) T cells by exogenous cell-associated antigens. Immunity. 2002;17:211–220. doi: 10.1016/s1074-7613(02)00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Probst HC, van den Broek M. Priming of CTLs by lymphocytic choriomeningitis virus depends on dendritic cells. Journal of Immunology. 2005;174:3920–3924. doi: 10.4049/jimmunol.174.7.3920. [DOI] [PubMed] [Google Scholar]
- 12.Tian T, Woodworth J, Sköld M, Behar SM. In vivo depletion of CD11c+ cells delays the CD4+ T cell response to Mycobacterium tuberculosis and exacerbates the outcome of infection. Journal of Immunology. 2005;175:3268–3272. doi: 10.4049/jimmunol.175.5.3268. [DOI] [PubMed] [Google Scholar]
- 13.Ciavarra RP, Stephens A, Nagy S, Sekellick M, Steel C. Evaluation of immunological paradigms in a virus model: Are dendritic cells critical for antiviral immunity and viral clearance? Journal of Immunology. 2006;177:492–500. doi: 10.4049/jimmunol.177.1.492. [DOI] [PubMed] [Google Scholar]
- 14.Kassim SH, Rajasagi NK, Zhao X, Chervenak R, Jennings SR. In vivo ablation of CD11c-positive dendritic cells increases susceptibility to herpes simplex virus type 1 infection and diminishes NK and T-cell responses. J Virol. 2006;80:3985–3993. doi: 10.1128/JVI.80.8.3985-3993.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu CH, Fan YT, Dias A, Esper L, Corn RA, Bafica A, Machado FS, Aliberti J. Cutting edge: Dendritic cells are essential for in vivo IL-12 production and development of resistance against Toxoplasma gondii infection in mice. Journal of Immunology. 2006;177:31–35. doi: 10.4049/jimmunol.177.1.31. [DOI] [PubMed] [Google Scholar]
- 16.Tait ED, Jordan KA, Dupont CD, Harris TH, Gregg B, Wilson EH, Pepper M, Dzierszinski F, Roos DS, Hunter CA. Virulence of Toxoplasma gondii Is Associated with Distinct Dendritic Cell Responses and Reduced Numbers of Activated CD8(+) T Cells. Journal of Immunology. 2010;185:1502–1512. doi: 10.4049/jimmunol.0903450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steel CD, Hahto SM, Ciavarra RP. Peripheral dendritic cells are essential for both the innate and adaptive antiviral immune responses in the central nervous system. Virology. 2009;387:117–126. doi: 10.1016/j.virol.2009.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Rijt LS, Jung S, Kleinjan A, Vos N, Willart M, Duez C, Hoogsteden HC, Lambrecht BN. In vivo depletion of lung CD11c+ dendritic cells during allergen challenge abrogates the characteristic features of asthma. J Exp Med. 2005;201:981–991. doi: 10.1084/jem.20042311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murillo O, Dubrot J, Palazon A, Arina A, Azpilikueta A, Alfaro C, Solano S, Ochoa MC, Berasain C, Gabari I, Perez-Gracia JL, Berraondo P, Hervas-Stubbs S, Melero I. In vivo depletion of DC impairs the anti-tumor effect of agonistic anti-CD137 mAb. European Journal of Immunology. 2009;39:2424–2436. doi: 10.1002/eji.200838958. [DOI] [PubMed] [Google Scholar]
- 20.Birnberg T, Bar-On L, Sapoznikov A, Caton ML, Cervantes-Barragán L, Makia D, Krauthgamer R, Brenner O, Ludewig B, Brockschnieder D, Riethmacher D, Reizis B, Jung S. Lack of Conventional Dendritic Cells Is Compatible with Normal Development and T Cell Homeostasis, but Causes Myeloid Proliferative Syndrome. Immunity. 2008;29:986–997. doi: 10.1016/j.immuni.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 21.Teichmann LL, Ols ML, Kashgarian M, Reizis B, Kaplan DH, Shlomchik MJ. Dendritic cells in lupus are not required for activation of T and B cells but promote their expansion, resulting in tissue damage. Immunity. 2010;33:967–978. doi: 10.1016/j.immuni.2010.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H, Matte-Martone C, Tan HS, Venkatesan S, McNiff J, Demetris AJ, Jain D, Lakkis F, Rothstein D, Shlomchik WD. Graft-versus-host disease is independent of innate signaling pathways triggered by pathogens in host hematopoietic cells. Journal of Immunology. 2011;186:230–241. doi: 10.4049/jimmunol.1002965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, Wu S, Lang R, Iredale JP. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. Journal of Clinical Investigation. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Caton ML, Smith-Raska MR, Reizis B. Notch-RBP-J signaling controls the homeostasis of CD8- dendritic cells in the spleen. J Exp Med. 2007;204:1653–1664. doi: 10.1084/jem.20062648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ivanova A, Signore M, Caro N, Greene ND, Copp AJ, Martinez-Barbera JP. In vivo genetic ablation by Cre-mediated expression of diphtheria toxin fragment A. Genesis. 2005;43:129–135. doi: 10.1002/gene.20162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zheng H, Matte-Martone C, Li H, Anderson BE, Venketesan S, Sheng Tan H, Jain D, McNiff J, Shlomchik WD. Effector memory CD4+ T cells mediate graft-versus-leukemia without inducing graft-versus-host disease. Blood. 2008;111:2476–2484. doi: 10.1182/blood-2007-08-109678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blasius AL, Giurisato E, Cella M, Schreiber RD, Shaw AS, Colonna M. Bone marrow stromal cell antigen 2 is a specific marker of type I IFN-producing cells in the naive mouse, but a promiscuous cell surface antigen following IFN stimulation. Journal of immunology. 2006;177:3260–3265. doi: 10.4049/jimmunol.177.5.3260. [DOI] [PubMed] [Google Scholar]
- 28.Matte-Martone C, Wang X, Anderson B, Jain D, Demetris AJ, McNiff J, Shlomchik MJ, Shlomchik WD. Recipient B cells are not required for graft-versus-host disease induction. Biol Blood Marrow Transplant. 2010;16:1222–1230. doi: 10.1016/j.bbmt.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H, Matte-Martone C, Tan HS, Venkatesan S, McNiff J, Demetris AJ, Jain D, Lakkis F, Rothstein D, Shlomchik WD. Graft-versus-Host Disease Is Independent of Innate Signaling Pathways Triggered by Pathogens in Host Hematopoietic Cells. J Immunol. 2011;186:230–241. doi: 10.4049/jimmunol.1002965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sapoznikov A, Jung S. Probing in vivo dendritic cell functions by conditional cell ablation. Immunol Cell Biol. 2008;86:409–415. doi: 10.1038/icb.2008.23. [DOI] [PubMed] [Google Scholar]
- 31.Liu K, Victora GD, Schwickert TA, Guermonprez P, Meredith MM, Yao K, Chu FF, Randolph GJ, Rudensky AY, Nussenzweig M. In vivo analysis of dendritic cell development and homeostasis. Science. 2009;324:392–397. doi: 10.1126/science.1170540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reddy P, Maeda Y, Liu C, Krijanovski OI, Korngold R, Ferrara JL. A crucial role for antigen-presenting cells and alloantigen expression in graft-versus-leukemia responses. Nat Med. 2005;11:1244–1249. doi: 10.1038/nm1309. [DOI] [PubMed] [Google Scholar]
- 33.McIntyre KR, Seidman JG. Nucleotide sequence of mutant I-A beta bm12 gene is evidence for genetic exchange between mouse immune response genes. Nature. 1984;308:551–553. doi: 10.1038/308551a0. [DOI] [PubMed] [Google Scholar]
- 34.Li H, Kaplan DH, Matte-Martone C, Tan HS, Venkatesan S, Johnson K, Demetris AJ, McNiff J, Shlomchik MJ, Shlomchik WD. Langerhans cells are not required for graft-versus-host disease. Blood. 2011;117:697–707. doi: 10.1182/blood-2010-07-299073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chua ACL, Hodson LJ, Moldenhauer LM, Robertson SA, Ingman WV. Dual roles for macrophages in ovarian cycle-associated development and remodelling of the mammary gland epithelium. Development. 2010;137:4229–4238. doi: 10.1242/dev.059261. [DOI] [PubMed] [Google Scholar]
- 36.Garcia MR, Ledgerwood L, Yang Y, Xu J, Lal G, Burrell B, Ma G, Hashimoto D, Li Y, Boros P, Grisotto M, Van Rooijen N, Matesanz R, Tacke F, Ginhoux F, Ding Y, Chen SH, Randolph G, Merad M, Bromberg JS, Ochando JC. Monocytic suppressive cells mediate cardiovascular transplantation tolerance in mice. Journal of Clinical Investigation. 2010;120:2486–2496. doi: 10.1172/JCI41628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kono H, Karmarkar D, Iwakura Y, Rock KL. Identification of the cellular sensor that stimulates the inflammatory response to sterile cell death. Journal of Immunology. 2010;184:4470–4478. doi: 10.4049/jimmunol.0902485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lucas T, Waisman A, Ranjan R, Roes J, Krieg T, Müller W, Roers A, Eming SA. Differential roles of macrophages in diverse phases of skin repair. Journal of Immunology. 2010;184:3964–3977. doi: 10.4049/jimmunol.0903356. [DOI] [PubMed] [Google Scholar]
- 39.Lin SL, Li B, Rao S, Yeo EJ, Hudson TE, Nowlin BT, Pei H, Chen L, Zheng JJ, Carroll TJ, Pollard JW, McMahon AP, Lang RA, Duffield JS. Macrophage Wnt7b is critical for kidney repair and regeneration. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:4194–4199. doi: 10.1073/pnas.0912228107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Menke J, Iwata Y, Rabacal WA, Basu R, Yeung YG, Humphreys BD, Wada T, Schwarting A, Stanley ER, Kelley VR. CSF-1 signals directly to renal tubular epithelial cells to mediate repair in mice. Journal of Clinical Investigation. 2009;119:2330–2342. doi: 10.1172/JCI39087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Medoff BD, Seung E, Hong S, Thomas SY, Sandall BP, Duffield JS, Kuperman DA, Erle DJ, Luster AD. CD11b+ myeloid cells are the key mediators of Th2 cell homing into the airway in allergic inflammation. Journal of Immunology. 2009;182:623–635. doi: 10.4049/jimmunol.182.1.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bar-On L, Jung S. Defining in vivo dendritic cell functions using CD11c-DTR transgenic mice. Methods Mol Biol. 2010;595:429–442. doi: 10.1007/978-1-60761-421-0_28. [DOI] [PubMed] [Google Scholar]
- 43.Li H, Kaplan DH, Matte-Martone C, Tan HS, Venkatesan S, Johnson K, Demetris A, McNiff J, Shlomchik M, Shlomchik WD. Langerhans cells are not required for graft-versus-host disease. Blood. 2010 doi: 10.1182/blood-2010-07-299073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Segura E, Villadangos JA. Antigen presentation by dendritic cells in vivo. Curr Opin Immunol. 2009;21:105–110. doi: 10.1016/j.coi.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 45.Randolph GJ, Jakubzick C, Qu C. Antigen presentation by monocytes and monocyte-derived cells. Curr Opin Immunol. 2008;20:52–60. doi: 10.1016/j.coi.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Robinson MJ, Sancho D, Slack EC, LeibundGut-Landmann S, Reis e Sousa C. Myeloid C-type lectins in innate immunity. Nat Immunol. 2006;7:1258–1265. doi: 10.1038/ni1417. [DOI] [PubMed] [Google Scholar]
- 47.Joffre O, Nolte MA, Sporri R, Reis e Sousa C. Inflammatory signals in dendritic cell activation and the induction of adaptive immunity. Immunol Rev. 2009;227:234–247. doi: 10.1111/j.1600-065X.2008.00718.x. [DOI] [PubMed] [Google Scholar]
- 48.Bajenoff M, Egen JG, Qi H, Huang AY, Castellino F, Germain RN. Highways, byways and breadcrumbs: directing lymphocyte traffic in the lymph node. Trends Immunol. 2007;28:346–352. doi: 10.1016/j.it.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 49.Palm NW, Medzhitov R. Pattern recognition receptors and control of adaptive immunity. Immunol Rev. 2009;227:221–233. doi: 10.1111/j.1600-065X.2008.00731.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.