

Abstract
The gamma-(γ)-herpesviruses are characterized by their ability to establish life-long latency. Subsequent immune suppression leads to viral reactivation from latency and the onset of a variety of pathologies, including lymphoproliferative disease and cancers. CD8 T cells play a key role in preventing reactivation of latent virus. Therefore, to develop effective therapeutic immune strategies, it is essential to understand the maintenance of CD8 T cell responses during latency. Because the γ-herpesviruses are highly species-specific and mice cannot be infected with the human pathogens, Epstein-Barr virus or Kaposi’s sarcoma-associated herpesvirus, we have utilized a natural rodent γ-herpesvirus experimental infection model, γHV68. In this report, we show that during long-term latent infection, naïve CD8 T cells are recruited into the ongoing immune response in an epitope-specific manner. When virus reactivation is induced in vivo, the recruitment of CD8 T cells for some, but not all, epitopes is enhanced. The variation in recruitment is not due to differences in epitope presentation. We also show that CD8 T cells that are newly stimulated during reactivation are functionally impaired compared with acutely-stimulated cells in terms of cytokine production. Thus, our results demonstrate unexpected complexity in the response of CD8 T cells specific for different viral epitopes that were stimulated during acute infection, quiescent latency, and reactivation.
Introduction
After a brief acute infection, the human gamma-(γ)-herpesviruses Epstein-Barr virus (EBV) and Kaposi’s sarcoma-associated herpesvirus (KSHV) are maintained in a latent state for the life of the host. EBV and KSHV are strongly species-specific and do not readily infect mice. The rodent pathogen murine γ-herpesvirus-68 (γHV68) is closely related to EBV and KSHV and infection of mice with γHV68 provides an experimental animal model to study γ-herpesvirus pathogenesis and immunity. As antiviral T cell activity is critical for the control of latent herpesvirus infections and the loss of T cell memory numbers or function can lead to viral reactivation and recurrent disease (1-3), it is of utmost importance to understand how antiviral CD8 T cell responses are maintained during γHV68 infection.
Memory T cells in persistent infections can be maintained by at least two mechanisms. First, a subset of memory T cells that retains the capacity for self-renewal can replenish the memory pool as non-replicating memory T cells are eliminated. In this model, all of the memory T cells required for controlling the latent pathogen are derived from cells stimulated during the initial acute infection (4, 5). Alternatively, new naïve cells with specificity for viral epitopes can be recruited into the ongoing T cell response, supplying a population of cells to replace pre-existing memory cells. In this model, over time the memory T cell population accumulates cells that were not stimulated during the acute infection, but rather were recruited into the response by presentation of antigens during viral latency or persistence (4, 6, 7). These two models are not mutually exclusive, and antiviral T cell populations may be maintained by both the replication of a subset of memory T cells and the recruitment of naïve T cells (4, 6, 8). Which of these two mechanisms is used, and/or to what extent, might depend on the specific antigen involved, as viral antigens can be differentially expressed at various stages of viral persistence.
We and others have recently identified a panel of γHV68-specific CD8 epitopes that detect CD8 T cell responses with differential kinetics throughout the infection (9, 10). Here we have used tetramers to track the response of T cells specific for individual epitopes and have used adoptive transfers and generation of partial hematopoietic chimeras in infected mice (4, 6, 7) in order to track the response of naïve and memory CD8 T cells during quiescent latency and during viral reactivation. We found that only naïve CD8 T cells specific for ORF61524Kb were recruited into the ongoing response during quiescent latency. However, following induction of virus reactivation from latency by depleting the pre-existing memory pool, naïve donor CD8 T cells for additional, but not all, epitopes, entered the response. Importantly, the ability of adoptively-transferred memory cells to respond to all epitopes following viral reactivation showed that the failure of naïve CD8 T cells specific for some epitopes to respond was not a consequence of poor antigen expression during reactivation. There was also a functional difference of antigen stimulation following primary infection or viral reactivation in that the ability to generate dual IFNγ/TNFα cytokines was impaired following stimulation of naïve CD8 T cells by reactivating virus. Taken together, our data show that γHV68-specific CD8 T cell memory is differentially maintained during latent infection in an epitope-specific manner, there is little stimulation of new T cells during quiescent latency and that viral reactivation from latency stimulates naïve CD8 T cells specific for some, but not all, epitopes. Furthermore, this epitope specificity could not be explained by differences in antigen presentation, as all epitopes are expressed during reactivation. Finally, polyfunctional cytokine secretion analysis shows that CD8 T cells stimulated during viral reactivation are functionally impaired compared to CD8 T cells stimulated during acute infection.
Materials and Methods
Mice and viruses
Male or female 8- to 12-week-old C57BL/6 (B6; CD45.2+Thy1.2+), B6.SJL-PtprcaPepcb/Boy (CD45.1+Thy1.2+), and B6.PL-Thy1a/Cy (CD45.2+Thy1.1+) were obtained from the Trudeau Institute animal facility and maintained under specific-pathogen free conditions. Mice were anesthetized with 2,2,2,-tribromoethanol and infected intranasally (i.n.) with 400 PFU of wild-type (WT) γHV68 (strain WUMS) or latency-deficient AC-RTA (11). All experiments were approved by the Trudeau Institute Institutional Animal Care and Use Committee.
Tetramers and flow cytometry
Allophycocyanin-conjugated MHC class I-restricted tetramers specific for γHV68 epitopes ORF6487-495Db (AGPHNDMEI), ORF8604-612Kb (KNYIFEEKL), ORF39167-174Kb (LVLFYRPI), ORF48148-155Kb (TNYKFSLV), ORF54253-260Kb (AVVQFIRV), ORF61524-531Kb (TSINFVKI), ORF75c176-184Db (SAIENYETF), and ORF75c940-947Kb (KSLTYYKL) were obtained from the Trudeau Institute Molecular Biology Core Facility. Cells were treated with Fc block (BD Biosciences) and then stained with tetramers for 1 hour at room temperature. For intracellular cytokine staining, cells were incubated with congenic splenocytes, 10 μg/ml of the relevant peptide, and brefeldin-A (Epicentre Biotechnologies) for 5 hours at 37°C, then washed, labeled, and permeabilized using the BD Cytofix/Cytoperm kit according to the manufacturer’s instructions. Fluorochrome-conjugated antibodies against CD8, CD19, CD44, CD45.1, CD45.2, CD62L, IFNγ, Thy1.1, Thy1.2, and TNFα were purchased from BioLegend, BD Biosciences, or eBiosciences as needed. Samples were collected on a BD FACSCanto II cytometer and analyzed by FlowJo software (TreeStar).
Generation of partial hematopoietic chimeras
WT γHV68- or AC-RTA-infected CD45.1+Thy1.2+ mice 2 months p.i. received 0.6 mg busulfan i.p. (Busulfex; Otsuka America Pharmaceuticals). One day later, 2 × 107 cells isolated from the bone marrow of naïve CD45.2+Thy1.1+ mice were injected intraveneously (i.v.). Reconstitution in B cells was previously shown to be 63.32 ± 6.585, in CD4 T cells, 43.38 ± 5.658, and in CD8 T cells 33.06 ± 5.331 (3). Engraftment was in this range for the current studies.
Anti-Thy1.2 mAb treatment
Mice were treated with 0.25 mg monoclonal antibody (mAb) to Thy1.2 (clone 30H12; BioXcell), administered intraperitoneally (i.p.) every 2-3 days for 12 days. T cell depletion efficiency was assessed by flow cytometry.
Quantitative real-time PCR
Host (CD45.1+) and donor (CD45.2+) CD19+ B cells were sorted via flow cytometry from spleens of WT γHV68-infected partial mixed bone marrow chimeras by CD45 marker expression. DNA was isolated from the purified B cell populations and quantitative real-time PCR for γHV68 ORF50 gene copy number was performed essentially as described previously (12).
Adoptive transfers
To measure memory recall responses to reactivation, CD44hi CD8 T cells were positively sorted by flow cytometry from spleens of WT γHV68-infected CD45.2+Thy1.1+ mice 2 months p.i. and 1 × 106 cells were transferred i.v. into WT γHV68- or AC-RTA-infected B6 mice. Recipient mice were then injected i.p. with PBS or anti-Thy1.2 mAb. Spleens were harvested 12 days after transfer and analyzed for donor tetramer-positive cells by flow cytometry. To measure naïve T cell responses to reactivation, CD44lo CD8 T cells were sorted by flow cytometry from spleens of naïve CD45.2+Thy1.1+ mice 2 months p.i. and 1.8 × 106 cells were transferred i.v. into WT γHV68- or AC-RTA-infected B6 mice. Recipient mice were then injected i.p. with anti-Thy1.2 mAb. Spleens were harvested 12 days after transfer and analyzed for donor tetramer-positive cells by flow cytometry.
Statistical Analysis
Data were analyzed for normality using the D’Agostino and Pearson omnibus normality test and compared using Student’s t test or one-way ANOVA with Bonferroni’s post test where appropriate. All analyses were performed using Prism 5 software (GraphPad). P-values ≤0.05 were considered statistically significant.
Results
Epitope-specific stimulation of naive T cells during γHV68 latency
To determine if naïve CD8 T cells could be recruited into an ongoing antiviral immune response during latent γHV68 infection, we treated mice with busulfan and then transferred congenic bone marrow to establish partial hematopoietic chimeras in latently-infected mice (Figure 1A). This technique has been used to demonstrate that new naïve T cells can respond to persistent viral infections (4, 7). We have recently shown that busulfan treatment of γHV68-infected mice does not affect the latent viral load or impair the pre-existing humoral and cellular immunity (3). We allowed latently-infected mice to reconstitute for up to 28 weeks then measured the phenotype of the host and donor CD8 T cells (Figure 1B). The donor CD8 T cell population was less activated than the host T cells, consistent with the donor cells being mostly naïve, despite existing in the presence of a latent viral infection. Using MHC class I-restricted tetramers for two well-characterized epitopes ORF6487Db and ORF61524Kb, we demonstrated that donor-derived CD8 T cells specific for ORF61524Kb, but not ORF6487Db, could be detected in the chimeric mice 28 weeks after reconstitution (Figure 1C). These data demonstrate that new naïve T cells can contribute to an ongoing immune response during γHV68 latency, and, interestingly, the level of contribution varies depending on the epitope. Using the detection of tetramer-positive cells shown in Figure 1D, we measured the specific response of host (Figure 1E, left) and donor (Figure 1E, right) CD8 T cell populations specific for 8 epitopes. Only the immunodominant ORF61524Kb-specific naïve T cells expanded in the latently-infected mice.
Figure 1.
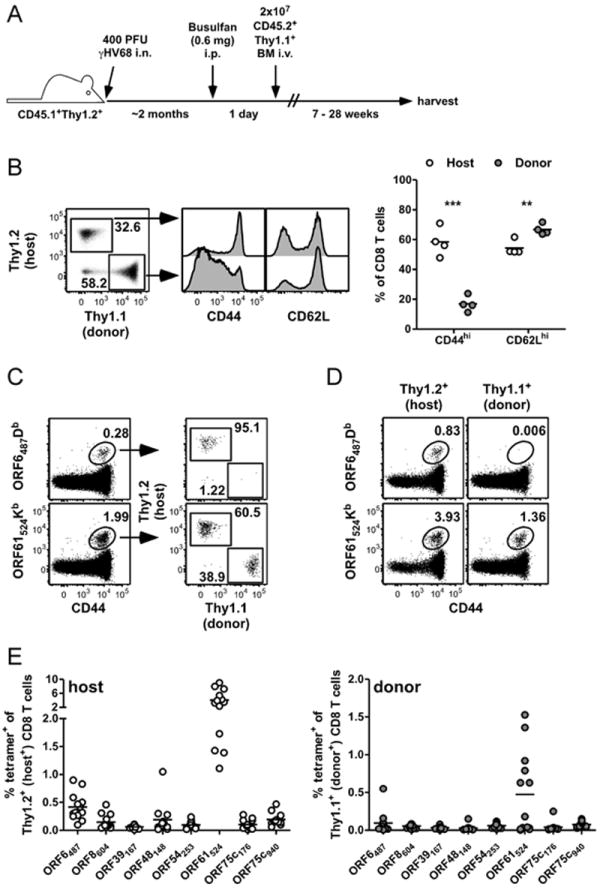
Restricted priming of naïve antiviral CD8 T cells during γHV68 latency. A, Experimental design to establish partial hematopoietic chimerism. BM, bone marrow; i.n., intranasal; i.p., intraperitoneal; i.v., intraveneous. B, 28 weeks after busulfan treatment and reconstitution, the expression of CD44 and CD62L was determined on Thy1.1+ (donor) and Thy1.2+ (host) CD8 T cells in the spleen by flow cytometry. Dot plot and histograms are from a representative mouse from the experiment depicted in the graph. Numbers in the dot plot indicate the percent of CD8 T cells in each gate (n=4, representative of 3 experiments) **P≤0.01; ***P≤0.001; Student’s t test). C, 28 weeks after busulfan treatment and reconstitution, MHC class I-restricted tetramer binding of CD8 T cells specific for ORF6487Db or ORF61524Kb from spleens and the percent of each tetramer population that is host- or donor-derived was determined for a representative mouse. D, Numbers in plots indicate percent of host or donor CD8 T cells that are tetramer-positive, from the same representative mouse as in panel C (representative of 3 experiments). E, Combined data from 3 experiments showing the percent of Thy1.2+ host (left graph) or Thy1.1+ donor (right graph) CD8 T cells that bind indicated γHV68-specific tetramers 20-28 weeks after busulfan treatment and reconstitution (n=13).
Epitope-specific stimulation of naïve T cells during viral reactivation
In our recent report, we demonstrated that depletion of the host T cell response by anti-Thy1.2 mAb injections led to substantial reactivation of latent virus, leading to enhanced infection of the donor B cells (3). It is unknown, however, whether viral reactivation also stimulates the recruitment of naïve T cells into the ongoing response. To test this, latently-infected mice that had been treated with busulfan and reconstituted about 6 weeks previously were treated with anti-Thy1.2 mAb injections over a period of 12 days (Figure 2A). We chose to treat mice at 6 weeks after reconstitution as there is not yet recruitment of naïve donor-derived ORF61524Kb-specific CD8 T cells at that time. Anti-Thy1.2 mAb treatment profoundly depleted the host T cells but not the Thy1.1+ donor T cells (Figure 2B). In addition, it led to reactivation, evidenced by the acquisition of viral genomes in the donor B cells (Figure 2C). Anti-Thy1.2 mAb treatment led to the stimulation and expansion of several epitope-specific donor CD8 T cell populations, although some specificities were not expanded significantly (Figure 2D and E).
Figure 2.
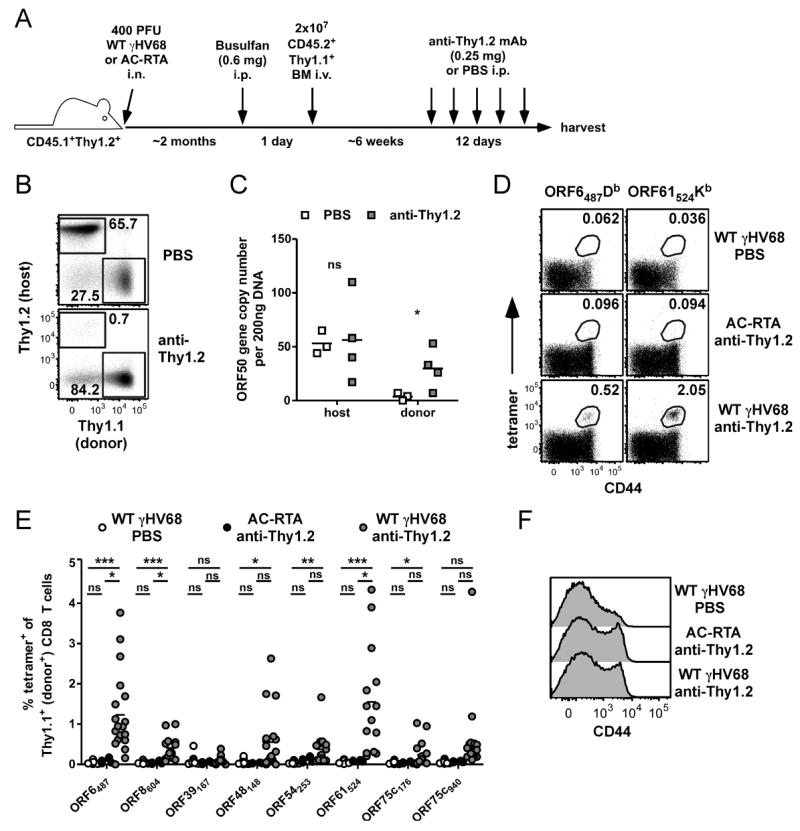
Enhanced recruitment of donor CD8 T cells into the antiviral response following host T cell depletion to induce viral reactivation. A, Experimental design for host T cell depletion in partial hematopoietic chimeras. B, Representative plots showing the efficiency of Thy1.2+ CD8 T cell depletion following anti-Thy1.2 mAb (30H12) treatment. Spleens were harvested and labeled with antibodies to CD8, Thy1.1, and Thy1.2 (clone 53.2.1). Numbers in plots indicate the percent of CD8 T cells in each gate (n=17 to 19/group; representative of 6 experiments) C, 12 days after the start of anti-Thy1.2 mAb (or PBS control) treatment, 200 ng DNA was isolated from sorted host and donor B cell populations from the spleen, and analyzed by quantitative real-time PCR for the γHV68 ORF50 gene (n=3-4/group, representative of 3 experiments; ns, not significant; *P≤0.05, Student’s t test). D-F, Spleens were harvested 12 days after the start of anti-Thy1.2 mAb (or PBS control) treatment in WT γHV68- or AC-RTA-infected chimeras and analyzed by flow cytometry. D, Representative dot plots show tetramer staining. E, Combined data from 2 to 6 experiments showing the percent of Thy1.1+ donor CD8 T cells that bind indicated tetramers under given infection and treatment conditions. F, Representative histograms show expression of CD44 on donor CD8 T cells. (n=4 to 19/group; ns, not significant; *P≤0.05; **P≤0.01; ***P≤0.001; one-way ANOVA with Bonferroni’s post test).
Depleting the majority of host T cells from the chimeric animals can lead to lymphopenia and drive antigen-independent proliferation of the donor T cells (13, 14). To assess whether this occurred, we compared CD44 expression on donor CD8 T cells after anti-Thy1.2 mAb treatment in chimeric mice that were latently-infected with WT γHV68 or had been infected with AC-RTA, a recombinant γHV68 that causes acute infection but is unable to establish latency (10, 11). A similar proportion of donor CD8 T cells increased expression of CD44 regardless of the presence of viral reactivation (Figure 2F), consistent with conversion of cells to a transient memory-like phenotype during lymphopenia (13-17). Notably however, lymphopenia alone did not elicit the expansion of tetramer-positive donor T cells, as assessed following depletion of host CD8 T cells in mice infected with AC-RTA virus compared with wild-type virus (Figure 2D and E). Thus, viral reactivation from latency differentially stimulates donor CD8 T cell responses in an antigen-driven, epitope-specific manner, which can be specifically detected above a background of lymphopenia-induced stimulation.
Expression of CD8 T cell epitopes during viral reactivation
Interestingly, ORF39167Kb-specific T cells did not seem to respond at all to anti-Thy1.2 treatment. This was surprising, as the ORF39167 epitope is one of the strongest stimulators of IFNγ production, as measured by ELISpot assay 12 days after γHV68 infection (10). The ORF39 gene encodes the viral glycoprotein M (gM), which forms a complex with viral gN protein and is essential for viral lytic replication (18, 19). Whether gM is required for virus reactivation from latency is unknown, and as such, gM might not be expressed (or expressed at a very low level) during reactivation. Since memory CD8 T cells are more sensitive to cognate antigen-expression than naïve cells due to their reduced requirement for costimulation (20-22), we used memory T cells to determine if the ORF39167Kb epitope was being expressed during reactivation by transferring congenic (Thy1.1+) memory T cells into latently-infected mice coupled with depletion of the host T cells by anti-Thy1.2 mAb treatment (Figure 3A). Prior to transfer, ORF39167Kb-specific cells represented a small but detectable proportion of the donor T cell pool (Figure 3B). After transfer, ORF39167Kb-specific cells expanded in WT γHV68-infected anti-Thy1.2 mAb-treated mice 19.3 ± 6.36-fold over latency-deficient AC-RTA-infected anti-Thy1.2 mAb-treated controls, indicating the ORF39167Kb antigen is expressed during virus reactivation. Indeed, all 8 tetramer-specific populations we measured significantly expanded after transfer of memory CD8 T cells and anti-Thy1.2 mAb-induced viral reactivation (Figure 3C), demonstrating that all the epitopes were expressed during reactivation.
Figure 3.
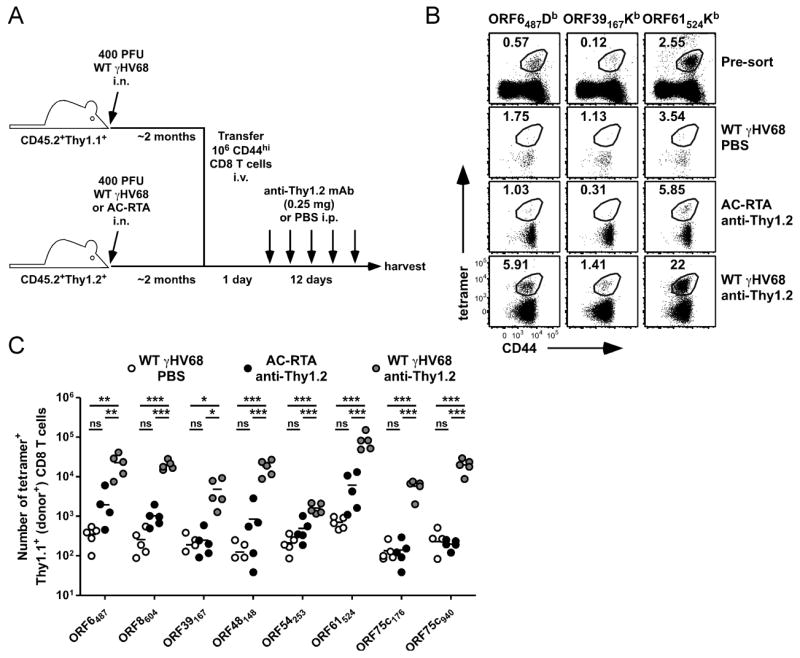
Virus reactivation stimulates memory CD8 T cell responses. A, Experimental design for adoptive transfer of memory CD8 T cells. B, Representative dot plots of CD8 T cells from spleens of WT γHV68-infected CD45.2+Thy1.1+ mice 2 months p.i. prior to sorting (Pre-sort), or of donor CD8 T cells from spleens of WT γHV68- or AC-RTA-infected B6 recipient mice 13 days after transfer of donor CD44hi CD8 T cells (12 days after PBS control or anti-Thy1.2 mAb treatment). Numbers indicate the percent of the donor CD8 T cell population that was tetramer-positive. C, The number of tetramer-positive Thy1.1+ donor CD8 T cells recovered from spleens of recipient mice (n=4 to 5/group; **P≤0.01; ***P≤0.001; one-way ANOVA with Bonferroni’s post test; representative of 3 experiments).
Viral reactivation can stimulate naïve epitope-specific CD8 T cell responses
Given that all the epitopes, including ORF39167Kb were expressed during reactivation, it is very interesting that reactivation did not elicit an ORF39167Kb-specific response in the anti-Thy1.2 mAb-treated partial bone marrow chimeras. This result could be due to several reasons, including a fundamental difference in the ability of naïve cells that mature during latency to mount an antiviral T cell response or antigen presentation during reactivation being insufficient to stimulate naïve CD8 T cells. As mentioned above, memory CD8 T cells have a lower threshold for stimulation by cognate antigen-expression than naïve cells (20-22). Therefore, even though virus reactivation induces expression of each epitope enough to stimulate memory T cells after transfer (Figure 3C), naïve cells might not expand in response to reactivation. To determine if viral reactivation could elicit the priming and expansion of naïve CD8 T cells specific for ORF39167Kb, we transferred 2 × 106 Thy1.1+ naïve CD8 T cells into Thy1.2+ mice that were infected with either WT γHV68 or AC-RTA 2 months previously, then depleted the recipient mice of T cells by anti-Thy1.2 mAb treatment (Figure 4A). We chose to transfer this number of cells because it resulted in robust epitope-specific T cell responses in an in vivo limiting dilution analysis assay, and it is roughly the number of donor-derived CD8 T cells we recover from the spleens of partial mixed bone marrow chimeras (data not shown). Notably, even at this number of transferred cells, it appears some epitope-specific responses were not stimulated by viral reactivation, which may reflect precursor frequencies below 0.5 × 10-6 or a lack of antigen presentation; however, naïve cells specific for ORF39167Kb were significantly stimulated by anti-Thy1.2 mAb treatment in WT γHV68 infected mice (2.2 ± 0.31-fold expansion over AC-RTA controls; P=0.0087, Student’s t test)(Figure 4B). So, although this assay does not conclusively demonstrate that all epitopes are expressed during reactivation at a level that stimulates naïve T cell responses, it does confirm that viral gM protein is expressed, processed, and presented at a level sufficient to stimulate naïve ORF39167Kb-specific cells. This is in particular contrast to the donor-derived ORF39167Kb-specific response after anti-Thy1.2 mAb treatment of partial bone marrow chimeras, suggesting that there may be a qualitative difference in naïve cells which matured in an uninfected animal compared to naïve cells which matured in a latently-infected animal.
Figure 4.
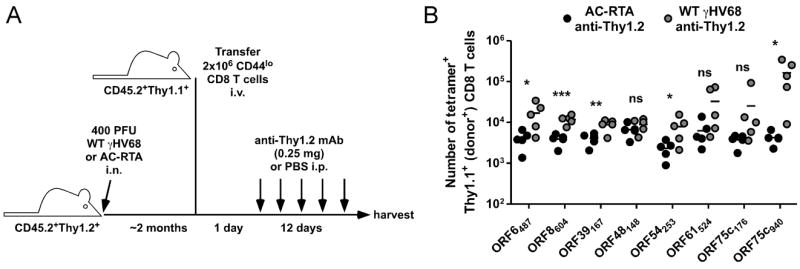
Stimulation of naïve antiviral CD8 T cells by virus reactivation. A, Experimental design for adoptive transfer of naïve CD8 T cells. B, The number of tetramer-positive Thy1.1+ donor CD8 T cells recovered from spleens of WT γHV68- or AC-RTA-infected B6 recipient mice treated with anti-Thy1.2 mAb for 12 days beginning 1 day after transfer of donor CD44lo CD8 T cells (n=4 to 5/group; representative of 3 experiments; ns, not significant; *P≤0.05; **P≤0.01; ***P≤0.001; Student’s t test).
Functional differences in T cells primed during acute infection and reactivation
We next sought to determine if CD8 T cell responses induced by viral reactivation were functionally similar to responses primed during acute lytic infection. Considering there is no ORF39167Kb-specific response to measure after anti-Thy1.2 mAb-induced reactivation, we focused our efforts on the two responses that are co-dominant early in infection, ORF6487Db and ORF61524Kb (23). The quality of CD8 T cell priming is highly dependent on the amount of antigen, the duration of antigen presentation, the expression of costimulatory molecules on the presenting APC, and the inflammatory milieu (24). Thus, it is likely that priming of new naïve T cells during γHV68 reactivation would differ substantially from priming during acute infection, with considerably less costimulation and inflammation during reactivation. Such differences could result in functional differences of the reactivation-primed cells compared to their lytic infection-primed counterparts (4, 6, 7, 25-28). To address this, we assayed the abilities of lytic infection-primed T cells and donor-derived T cells primed during anti-Thy1.2 mAb-induced reactivation (Figure 5A) to produce the antiviral cytokines IFNγ and TNFα in response to cognate antigen presentation. Although the different priming conditions resulted in similar frequencies of ORF6487Db- and ORF61524Kb-specific cells (Figure 5B), the frequency of cells that co-expressed IFNγ and TNFα was significantly reduced for the reactivation-primed responses compared to responses 12 d after primary infection (Figure 5B and 5C). These data suggest that the CD8 T cell response generated by reactivating virus is functionally impaired compared with the response generated during acute infection.
Figure 5.
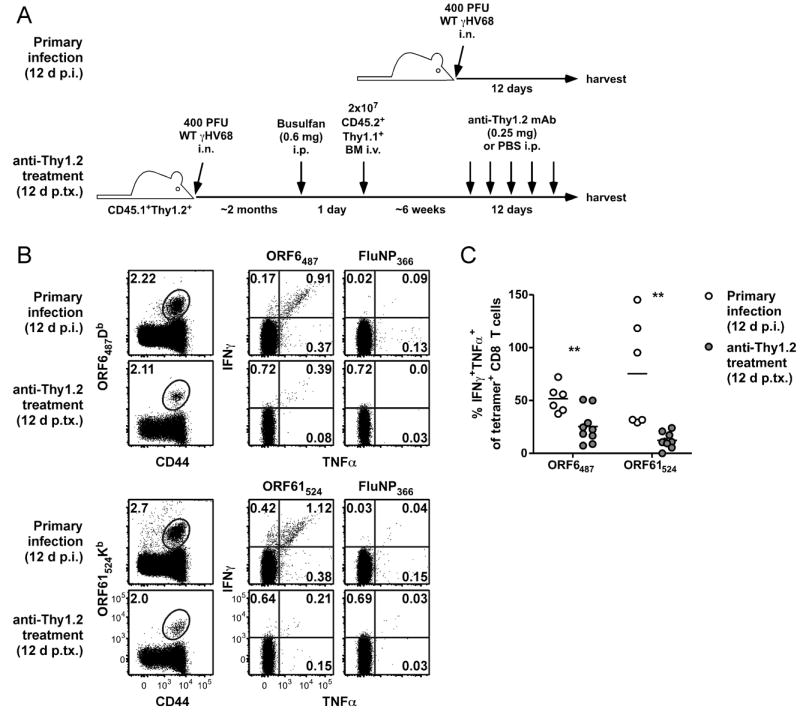
Impaired antigen-driven cytokine production by reactivation-induced CD8 T cells. A, Experimental design. Spleens were harvested either from B6 mice 12 days after primary infection or from partial hematopoietic chimeras 12 days after the start of anti-Thy1.2 mAb treatment and analyzed by flow cytometry for tetramer-binding or intracellular cytokine production. B, Representative dot plots showing CD8 T cells from B6 mice or Thy1.1+ donor CD8 T cells from partial hematopoietic chimeras (left column) or cytokine production by CD8 T cell populations after 5 h stimulation with indicated peptides (middle and right columns). Numbers in plots indicate the percent of CD8 T cells (or donor CD8 T cells, where appropriate) that are in the given gate or quadrant. C, Percent of CD8 T cells (white circles) or Thy1.1+ donor CD8 T cells (grey circles) that are IFNγ+TNFα+ (double-positive) after stimulation with indicated peptides expressed as a percent of the CD8 T cells that were tetramer-positive for each condition, as determined in a separate stain of the sample (n=6-9/group, representative of at least 2 experiments; p.tx, post treatment; **P≤0.01; Student’s t test)
Discussion
The regulation of T cell responses during persistent infections may vary depending on several factors, including the quality of T cell priming, the anatomical sites of viral persistence, the inflammatory microenvironment, and the timing and duration of cognate antigen presentation (29-34). It is unclear how the antiviral T cell response is maintained long-term during persistent γ-herpesvirus infections. Using the mouse γHV68 infection model, it has been shown that virus-specific CD8 T cells proliferate rapidly during latency, yet the majority of antiviral CD8 T cells express markers of terminal differentiation and replicative senescence (5, 10, 35-37). In order to design rational therapeutic vaccine strategies that target epitope-specific CD8 T cell responses, it is important to determine how antiviral memory T cell is maintained long-term. The goals of these studies were to identify whether naïve virus-specific CD8 T cells could contribute to an ongoing immune response in latently-infected animals and whether reactivation of virus from latency could stimulate naïve CD8 T cells to enter the response.
In mice that are persistently-infected with polyoma virus or lymphochoriomeningitis virus, naïve T cells were shown to be recruited into the ongoing antiviral immune response (6, 7). However, the extent of recruitment of naïve T cells to the maintenance of these T cell responses is unclear (7, 8, 38). During latent murine cytomegalovirus infection, optimal maintenance of memory T cell responses was shown to require both the recruitment of naïve T cells and the proliferation and differentiation of a population of cells primed early in infection (4). We have recently demonstrated that virus-specific naïve CD4 T cells can enter the immune response during γHV68 latency (39). However, another recent report has suggested that the CD8 T cell response to γHV68 is maintained mainly by the continuous turnover of activated T cells that were primed during acute infection (5). Importantly, that report did not rule out the possibility that naïve T cells could contribute to the ongoing antiviral T cell response, and did not examine epitope-specific T cell responses. In addition, it has been shown that naïve CD8 T cells can respond to DCs from latently-infected mice in vitro (40), suggesting that they might contribute to the ongoing immune response. In the studies presented here, we assessed whether naïve CD8 T cells could enter the immune response during latent γHV68 infection in vivo. We observed a profound epitope-specific limitation in their recruitment – only cells specific for the immunodominant epitope ORF61524Kb exhibited any appreciable recruitment into the antiviral T cell response during quiescent latency. This result supports the contention that antigen presentation drives recruitment of naive T cells, as it is likely that the ORF61524Kb epitope is expressed substantially during latent infection given its immunodominance in the presence of latency and its loss of dominance in the absence of latency (10, 36).
Anti-γHV68 immunity is long-lasting and highly functional, yet latent infection is maintained for life. This is due in part to the ability of the virus to continually spread to naïve B cells throughout infection, even in the presence of immunity (3). Should the immune system become compromised, however, γHV68 can reactivate from latency leading to recrudescent disease, morbidity, and even mortality (1, 3, 41). Whether the immune system can alleviate the detrimental effects of viral reactivation by generating a new T cell response to the reactivating virus is unknown. Here, we induced reactivation by depleting host T cells in latently-infected partial hematopoietc chimeric mice (3) and assessed whether reactivating virus could stimulate donor-derived naïve T cells to expand. We observed significant expansion of several epitope-specific responses after induction of viral reactivation following anti-Thy1.2 mAb treatment. The hierarchy of expanded responses 12 d after T cell depletion did not directly compare with the response hierarchy 12 d after primary infection (9, 10), and anti-Thy1.2 mAb treatment did not seem to induce a response to ORF39167Kb. Several factors that could influence the generation of T cell responses might be different between primary infection and anti-Thy1.2 mAb-induced reactivation. These include differences in APC type, co-stimulatory molecule expression, inflammatory milieu, and the timing of antigen expression. We sought to address the question of antigen presentation by transferring memory and naïve Thy1.1+ CD8 T cells into latently-infected Thy1.2+ animals and inducing reactivation by anti-Thy1.2 mAb treatment. Intriguingly, ORF39167Kb-specific memory CD8 T cells responded vigorously, consistent with expression of that epitope during reactivation. Somewhat surprisingly, naïve ORF39167Kb-specific cells also responded to reactivation. Our data therefore suggest that ORF39167Kb antigen expression during virus reactivation is sufficient to stimulate both memory and naïve CD8 T cell responses, yet is insufficient to stimulate responses when the cells have matured in a latently-infected mouse. Why this might be the case is of considerable interest as it may inform future therapeutic vaccine strategies designed to elicit or improve CD8 T cell responses in latently-infected animals. One possibility that is difficult to directly address experimentally is epitope-specific thymic tolerance, as the γ-herpesviruses are thought to infect the thymus. The finding, if true, that some epitopes but not others drive thymic tolerance could provide key insight into the pattern of epitope expression during latency.
Another important implication of our findings is that reactivation-induced T cell responses appear to be functionally impaired in their ability to produce cytokines. Specifically, there was a deficiency in the generation of cells secreting dual cytokines, IFNγ and TNFα. These data raise the intriguing possibility, currently under investigation, that successive reactivation may drive the generation of dysfunctional T cells, leading to progressive dampening of the overall antiviral immune response.
In conclusion, our findings identify a complex system in which virus-specific naïve T cells are highly restricted in their ability to enter the response during latency. Under conditions of immunosuppression, viral reactivation drives the expansion of dysfunctional T cells. This expansion is limited to only CD8 T cells of certain epitope specificities, even though the other epitopes that don’t drive the expansion of naïve T cells are capable of being expressed and presented to both memory and naïve cells during reactivation. Thus, CD8 T cells that are generated and mature during latent viral infection are deficient in their ability to recognize cognate antigen and develop into fully functional effector T cells.
Acknowledgments
We thank Dr. Jacob E. Kohlmeier for critically reading the manuscript and Otsuka America Pharmaceuticals for the generous gift of Busulfex.
Footnotes
This work was supported by NIH grants AI042927, AI082919, and CA148250 (to MAB), T32 AI049823 (to DLW), F32 AI084327 (to MLF), and funds from the Trudeau Institute.
References
- 1.Kim IJ, Flano E, Woodland DL, Blackman MA. Antibody-mediated control of persistent gamma-herpesvirus infection. J Immunol. 2002;168:3958–3964. doi: 10.4049/jimmunol.168.8.3958. [DOI] [PubMed] [Google Scholar]
- 2.Freeman ML, Sheridan BS, Bonneau RH, Hendricks RL. Psychological stress compromises CD8+ T cell control of latent herpes simplex virus type 1 infections. J Immunol. 2007;179:322–328. doi: 10.4049/jimmunol.179.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Freeman ML, Burkum CE, Yager EJ, Woodland DL, Blackman MA. De novo infection of B cells during murine gammaherpesvirus 68 latency. J Virol. 2011;85:10920–10925. doi: 10.1128/JVI.05027-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Snyder CM, Cho KS, Bonnett EL, van Dommelen S, Shellam GR, Hill AB. Memory inflation during chronic viral infection is maintained by continuous production of short-lived, functional T cells. Immunity. 2008;29:650–659. doi: 10.1016/j.immuni.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bannard O, Kraman M, Fearon DT. Cutting edge: Virus-specific CD8+ T cell clones and the maintenance of replicative function during a persistent viral infection. J Immunol. 2010;185:7141–7145. doi: 10.4049/jimmunol.1002537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kemball CC, Lee ED, Vezys V, Pearson TC, Larsen CP, Lukacher AE. Late priming and variability of epitope-specific CD8+ T cell responses during a persistent virus infection. J Immunol. 2005;174:7950–7960. doi: 10.4049/jimmunol.174.12.7950. [DOI] [PubMed] [Google Scholar]
- 7.Vezys V, Masopust D, Kemball CC, Barber DL, O’Mara LA, Larsen CP, Pearson TC, Ahmed R, Lukacher AE. Continuous recruitment of naive T cells contributes to heterogeneity of antiviral CD8 T cells during persistent infection. J Exp Med. 2006;203:2263–2269. doi: 10.1084/jem.20060995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin E, Kemball CC, Hadley A, Wilson JJ, Hofstetter AR, Pack CD, Lukacher AE. Heterogeneity among viral antigen-specific CD4+ T cells and their de novo recruitment during persistent polyomavirus infection. J Immunol. 2010;185:1692–1700. doi: 10.4049/jimmunol.0904210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gredmark-Russ S, Cheung EJ, Isaacson MK, Ploegh HL, Grotenbreg GM. The CD8 T-cell response against murine gammaherpesvirus 68 is directed toward a broad repertoire of epitopes from both early and late antigens. J Virol. 2008;82:12205–12212. doi: 10.1128/JVI.01463-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Freeman ML, Lanzer KG, Cookenham T, Peters B, Sidney J, Wu TT, Sun R, Woodland DL, Sette A, Blackman MA. Two kinetic patterns of epitope-specific CD8 T-cell responses following murine gammaherpesvirus 68 infection. J Virol. 2010;84:2881–2892. doi: 10.1128/JVI.02229-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jia Q, Freeman ML, Yager EJ, McHardy I, Tong L, Martinez-Guzman D, Rickabaugh T, Hwang S, Blackman MA, Sun R, Wu TT. Induction of protective immunity against murine gammaherpesvirus 68 infection in the absence of viral latency. J Virol. 2010;84:2453–2465. doi: 10.1128/JVI.01543-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Usherwood EJ, Ward KA, Blackman MA, Stewart JP, Woodland DL. Latent antigen vaccination in a model gammaherpesvirus infection. J Virol. 2001;75:8283–8288. doi: 10.1128/JVI.75.17.8283-8288.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldrath AW, Bevan MJ. Low-affinity ligands for the TCR drive proliferation of mature CD8+ T cells in lymphopenic hosts. Immunity. 1999;11:183–190. doi: 10.1016/s1074-7613(00)80093-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ernst B, Lee DS, Chang JM, Sprent J, Surh CD. The peptide ligands mediating positive selection in the thymus control T cell survival and homeostatic proliferation in the periphery. Immunity. 1999;11:173–181. doi: 10.1016/s1074-7613(00)80092-8. [DOI] [PubMed] [Google Scholar]
- 15.Goldrath AW, Bogatzki LY, Bevan MJ. Naive T cells transiently acquire a memory-like phenotype during homeostasis-driven proliferation. J Exp Med. 2000;192:557–564. doi: 10.1084/jem.192.4.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cho BK, Rao VP, Ge Q, Eisen HN, Chen J. Homeostasis-stimulated proliferation drives naive T cells to differentiate directly into memory T cells. J Exp Med. 2000;192:549–556. doi: 10.1084/jem.192.4.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamilton SE, Wolkers MC, Schoenberger SP, Jameson SC. The generation of protective memory-like CD8+ T cells during homeostatic proliferation requires CD4+ T cells. Nat Immunol. 2006;7:475–481. doi: 10.1038/ni1326. [DOI] [PubMed] [Google Scholar]
- 18.Virgin HW, Latreille P, Wamsley P, Hallsworth K, Weck KE, Dal Canto AJ, Speck SH. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J Virol. 1997;71:5894–5904. doi: 10.1128/jvi.71.8.5894-5904.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.May JS, Colaco S, Stevenson PG. Glycoprotein M is an essential lytic replication protein of the murine gammaherpesvirus 68. J Virol. 2005;79:3459–3467. doi: 10.1128/JVI.79.6.3459-3467.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ahmed R, Gray D. Immunological memory and protective immunity: understanding their relation. Science. 1996;272:54–60. doi: 10.1126/science.272.5258.54. [DOI] [PubMed] [Google Scholar]
- 21.Seder RA, Ahmed R. Similarities and differences in CD4+ and CD8+ effector and memory T cell generation. Nat Immunol. 2003;4:835–842. doi: 10.1038/ni969. [DOI] [PubMed] [Google Scholar]
- 22.Dutton RW, Swain SL, Bradley LM. The generation and maintenance of memory T and B cells. Immunol Today. 1999;20:291–293. doi: 10.1016/s0167-5699(98)01415-7. [DOI] [PubMed] [Google Scholar]
- 23.Stevenson PG, Belz GT, Altman JD, Doherty PC. Changing patterns of dominance in the CD8+ T cell response during acute and persistent murine gamma-herpesvirus infection. Eur J Immunol. 1999;29:1059–1067. doi: 10.1002/(SICI)1521-4141(199904)29:04<1059::AID-IMMU1059>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 24.Williams MA, Bevan MJ. Effector and memory CTL differentiation. Annu Rev Immunol. 2007;25:171–192. doi: 10.1146/annurev.immunol.25.022106.141548. [DOI] [PubMed] [Google Scholar]
- 25.Sarawar SR, Lee BJ, Reiter SK, Schoenberger SP. Stimulation via CD40 can substitute for CD4 T cell function in preventing reactivation of a latent herpesvirus. Proc Natl Acad Sci U S A. 2001;98:6325–6329. doi: 10.1073/pnas.101136898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.D’Souza WN, Hedrick SM. Cutting edge: latecomer CD8 T cells are imprinted with a unique differentiation program. J Immunol. 2006;177:777–781. doi: 10.4049/jimmunol.177.2.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lyon AB, Sarawar SR. Differential requirement for CD28 and CD80/86 pathways of costimulation in the long-term control of murine gammaherpesvirus-68. Virology. 2006;356:50–56. doi: 10.1016/j.virol.2006.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sarkar S, Teichgraber V, Kalia V, Polley A, Masopust D, Harrington LE, Ahmed R, Wherry EJ. Strength of stimulus and clonal competition impact the rate of memory CD8 T cell differentiation. J Immunol. 2007;179:6704–6714. doi: 10.4049/jimmunol.179.10.6704. [DOI] [PubMed] [Google Scholar]
- 29.van der Most RG, Murali-Krishna K, Lanier JG, Wherry EJ, Puglielli MT, Blattman JN, Sette A, Ahmed R. Changing immunodominance patterns in antiviral CD8 T-cell responses after loss of epitope presentation or chronic antigenic stimulation. Virology. 2003;315:93–102. doi: 10.1016/j.virol.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 30.Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol. 2003;77:4911–4927. doi: 10.1128/JVI.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wherry EJ, Barber DL, Kaech SM, Blattman JN, Ahmed R. Antigen-independent memory CD8 T cells do not develop during chronic viral infection. Proc Natl Acad Sci U S A. 2004;101:16004–16009. doi: 10.1073/pnas.0407192101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sheridan BS, Khanna KM, Frank GM, Hendricks RL. Latent virus influences the generation and maintenance of CD8+ T cell memory. J Immunol. 2006;177:8356–8364. doi: 10.4049/jimmunol.177.12.8356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shin H, Blackburn SD, Blattman JN, Wherry EJ. Viral antigen and extensive division maintain virus-specific CD8 T cells during chronic infection. J Exp Med. 2007;204:941–949. doi: 10.1084/jem.20061937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramachandran S, Davoli KA, Yee MB, Hendricks RL, Kinchington PR. Delaying the expression of herpes simplex virus type 1 glycoprotein B (gB) to a true late gene alters neurovirulence and inhibits the gB-CD8+ T-cell response in the trigeminal ganglion. J Virol. 2010;84:8811–8820. doi: 10.1128/JVI.00496-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Obar JJ, Crist SG, Leung EK, Usherwood EJ. IL-15-independent proliferative renewal of memory CD8+ T cells in latent gammaherpesvirus infection. J Immunol. 2004;173:2705–2714. doi: 10.4049/jimmunol.173.4.2705. [DOI] [PubMed] [Google Scholar]
- 36.Obar JJ, Fuse S, Leung EK, Bellfy SC, Usherwood EJ. Gammaherpesvirus persistence alters key CD8 T-cell memory characteristics and enhances antiviral protection. J Virol. 2006;80:8303–8315. doi: 10.1128/JVI.00237-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cush SS, Flano E. KLRG1+NKG2A+ CD8 T cells mediate protection and participate in memory responses during gamma-herpesvirus infection. J Immunol. 2011;186:4051–4058. doi: 10.4049/jimmunol.1003122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miller NE, Bonczyk JR, Nakayama Y, Suresh M. Role of thymic output in regulating CD8 T-cell homeostasis during acute and chronic viral infection. J Virol. 2005;79:9419–9429. doi: 10.1128/JVI.79.15.9419-9429.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Freeman ML, Burkum CE, Lanzer KG, Jensen MK, Ahmed M, Yager EJ, Flano E, Winslow GM, Woodland DL, Blackman MA. Cutting Edge: Activation of Virus-Specific CD4 T Cells throughout gamma-Herpesvirus Latency. J Immunol. 2011;187:6180–6184. doi: 10.4049/jimmunol.1102745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kupresanin F, Chow J, Mount A, Smith CM, Stevenson PG, Belz GT. Dendritic cells present lytic antigens and maintain function throughout persistent gamma-herpesvirus infection. J Immunol. 2007;179:7506–7513. doi: 10.4049/jimmunol.179.11.7506. [DOI] [PubMed] [Google Scholar]
- 41.Cardin RD, Brooks JW, Sarawar SR, Doherty PC. Progressive loss of CD8+ T cell-mediated control of a gamma-herpesvirus in the absence of CD4+ T cells. J Exp Med. 1996;184:863–871. doi: 10.1084/jem.184.3.863. [DOI] [PMC free article] [PubMed] [Google Scholar]