

Abstract
Adenosine monophosphate-activated protein kinase (AMPK) regulates lipid, cholesterol and glucose metabolism in specialized metabolic tissues, such as muscle, liver and adipose tissue. Agents that activate AMPK, such as metformin and AICAR, have beneficial effects on liver glucose and lipid metabolism. Additionally, AMPK activation in proliferating hepatic stellate cells (HSCs) induces growth arrest and inhibits hepatic fibrosis. As metformin and AICAR act in different ways to achieve their effects, our aim was to examine the effects of AMPK activation in quiescent HSCs with these two agents on HSC function. We found that phospho-AMPK levels were markedly up-regulated by both AICAR and metformin in quiescent HSCs. However, while AICAR treatment induced cell death, cells treated with metformin did not differ from untreated controls. AICAR-mediated HSC cell death was paralleled by loss of expression of the TGF-β decoy receptor Bambi, while metformin increased Bambi expression. Transfection of siRNA-Bambi into HSCs also induced cell death, mimicking the effects of AICAR, while over expression of Bambi partially rescued AICAR-treated cells. As Bambi has previously been shown to promote cell survival through Wnt/β-catenin signalling, a reporter incorporating binding sites for a downstream target of this pathway was transfected into HSCs and was induced. We conclude that while AICAR and metformin both activate AMPK in quiescent HSCs, AICAR rapidly induced cell death while metformin-treated cells remained viable. The finding that metformin increases Bambi expression and activates Wnt/β-catenin signaling provides a possible mechanistic explanation for this observation. These results suggest that AICAR and metformin may confer disease specific therapeutic benefits.
Keywords: Hepatic stellate cell, liver fibrosis, AMPK, Bambi, Wnt/β-catenin signalling
Introduction
Hepatic fibrosis underlies most forms of chronic liver disease (1, 2), the 12th most common cause of death in the United States (3). The main causes of hepatic fibrosis in industrialized countries include chronic hepatitis C virus (HCV) infection, alcohol abuse, and nonalcoholic fatty liver disease (NAFLD) (4). NAFLD has emerged as a considerable public health concern, as its major risk factors, obesity and insulin resistance, have reached epidemic proportions worldwide. Furthermore, conditions that induce hepatic fibrosis, such as HCV and hepatitis B infection, alcoholism, and NAFLD, are the most common risk factors for the development of hepatocellular carcinoma (HCC) (5), suggesting that therapeutic intervention for liver fibrosis may hold great value in the prevention of HCC.
Independent of etiology, chronic liver disease follows a common progression from inflammation to fibrosis, and finally to cirrhosis. Hepatocyte damage often serves as a triggering event in the pathogenesis of liver fibrosis, which initiates a complex crosstalk between hepatocytes and non-parenchymal liver cells. Injured or apoptotic hepatocytes can directly activate HSCs (reviewed in (6)) and also release reactive oxygen species (ROS) and cytokines, leading to recruitment of inflammatory immune cells and to activation of resident macrophages (Kupffer cells) (7). Activated Kupffer cells and injured hepatocytes also secrete profibrogenic cytokines, including tumor necrosis factor-α (TNF-α), transforming growth factor-β1 (TGFβ1), and fibroblast growth factor (FGF), which stimulate the activation of hepatic stellate cells (HSC) (8). HSC reside in the space of Disse; in normal liver HSC are quiescent and are characterized in part by the presence of cytoplasmic lipid droplets, which serve as the major site of retinol storage. In response to hepatic injury, HSC undergo rapid activation, and transdifferentiate into myofibroblast-like cells with a multitude of acquired morphological and functional properties. Activated HSC lose lipid droplets, obtain proliferative and contractile capability, express myogenic markers such as α-smooth muscle actin and myocyte enhancer factor-2, and secrete pathological extracellular matrix (ECM) proteins such as collagen type I (4). In addition to matrix protein production, HSC contribute to tissue repair in the injured liver by secreting pro-inflammatory cytokines (such as TGF-β, RANTES, and MCP-1) and adhesion molecules (including integrins, VCAM-1, and ICAM-1) that promote immune cell recruitment and activation (9). While transient HSC activation promotes wound healing and tissue repair, chronic HSC activation results in excessive ECM accumulation, destruction of liver cytoarchitecture, and eventually to cirrhosis and liver failure (4). The extent and duration of HSC activation are naturally limited by senescence; in mice lacking key senescence mediators, HSCs undergo aberrant proliferation in response to hepatic damage, leading to excessive liver fibrosis (10). Pharmacologic means by which to limit the activation state of HSC, by inhibiting proliferation or promoting senescence or apoptosis, therefore represents an appealing therapeutic avenue in chronic liver disease. Further, given the established connection between HSC activation and hepatic fibrosis (6), and the implications of hepatic fibrosis in the development of HCC as described above, strategies to limit HSC activation may also be of value for cancer prevention.
While insulin resistance has long been implicated in the progression of hepatic fibrosis, the molecular mechanisms underlying this relationship remain unclear. Adiponectin, a hormone secreted by adipose tissue, is markedly decreased in plasma under conditions of obesity and type 2 diabetes and adiponectin levels are directly correlated with insulin sensitivity, and inversely with the presence of metabolic syndrome (11, 12). Administration of adiponectin ameliorates liver damage during experimental steatohepatitis and reduces the development of fibrosis (13, 14). In agreement with these findings, adiponectin has been shown to decrease proliferation, migration, and fibrogenic gene expression and to induce apoptosis in activated HSC (14, 15). Subsequent studies determined that adiponectin principally inhibits profibrogenic actions of HSC via adenosine monophosphate-activated protein kinase (AMPK) (16, 17), a known downstream effector through which adiponectin induces glucose utilization and fatty acid oxidation (18). AMPK is an energy sensor that maintains cellular energy homeostasis in response to various stimuli including stress conditions, nutrient deprivation, exercise, and adipocyte-derived hormones (19, 20). AMPK can also be activated pharmacologically by 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside (AICAR) and by metformin, a widely used anti-diabetic drug (21, 22). Drugs that activate AMPK have well-established beneficial effects on liver glucose and lipid metabolism, which may augment their antifibrogenic properties. In HSCs, AMPK is thought to oppose activation by inhibiting the mTOR pathway, a known AMPK target (23), and perhaps by inhibiting NF-κB (16). AICAR and metformin also inhibit platelet-derived growth factor (PDGF)-induced proliferation and migration in activated HSC (17). While activation of AMPK was shown to negatively regulate the activated HSC phenotype, the effect of AMPK activating compounds on quiescent HSC remains unknown. While quiescent HSC serve a physiological function in the liver, and excess apoptosis and cytokine release can exacerbate hepatic fibrosis, maintenance of quiescent HSC viability is a desirable outcome of therapeutic strategies for chronic liver disease.
The current study aimed to evaluate the role of AMPK in quiescent primary rat HSCs in vitro. While AICAR and metformin both activated AMPK in quiescent HSC to the same extent, these agents elicited distinct effects on HSC function. AICAR rapidly induced cell death, while HSCs treated with metformin remained viable. We found that activation of BMP and activin membrane-bound inhibitor (Bambi), a TGF-β pseudoreceptor, by metformin promotes the survival of quiescent HSC. Use of small interfering RNA against Bambi in quiescent primary rat HSCs resulted in a dramatic decrease in viable cells, and we further demonstrated that metformin-induced Bambi expression activates a pro-survival Wnt/β-catenin signaling pathway unique to quiescent HSC (24).
Materials and Methods
HSC isolation, cell culture and transfection
HSCs were isolated from male Sprague-Dawley rats by in situ pronase, collagenase perfusion and single-step Histodenz gradient as previously reported (25). Isolated HSCs were cultured in DMEM (GIBCO) containing 20% FCS (JRH) on plastic for 40 hours. This medium was removed and fresh medium without FCS but in the presence or absence of aminoimidazole carboxamide ribonucleotide (AICAR) or Metformin at concentrations as indicated in the figure legend, 1α,25(OH)2 vitamin D3 (calcitriol) (10 nM), LPS (15ng/ml), cycloheximide (1mg/ml) and TGF-β1 (2ng/ml) were added to the cells for various time points. For transfection, HSCs were grown to 50% confluence in 6-well plates and a human Bambi expression vector transfected using FuGENE-HD (Roche), as recommended by the manufacturer. For siRNA transfection (siRNA-Bambi or siRNA-AMPK), Lipofectamine RNAiMAX (Invitrogen) was used as recommended by the manufacturer. Six hours later, different treatments were given to cells as indicated in figure legends. Each experimental condition was measured in triplicate and the values given represent the mean±standard deviation from two to three experiments. Student's unpaired t test was performed to assess the significance of treatments versus controls. Statistically significant differences (p <0.05) are indicated by *.
RNA isolation and Quantitative real-time PCR (qPCR)
Total RNA from cultured HSCs was isolated using RNeasy (Qiagen, Valencia, CA) according to the manufacture's instructions. A260/280 ratios were determined by a NanoDrop ND-1000 spectrophotometer and the integrity of the total RNA was verified using a Bioanalyzer (Agilent Technologies). 500 ng of deoxyribonuclease-treated total RNA was reverse transcribed into cDNA using SuperScript III reverse transcriptase (Invitrogen). Quantification of mRNA expression was performed by quantitative real-time PCR (qPCR) using a StepOnePlus qPCR platform (Applied Biosystems). The following primer pairs were used for the amplification of genes of interest:
rat Bambi forward 5'-tcatctggctgcagttgg-3'
rat Bambi reverse 5'-catcacagtagcatctgatctcg-3'
ratIL-1β forward 5'-tacctatgtcttgcccgtggag- 3'
ratIL-1β reverse 5'-atcatcccacgagtcacagagg- 3'
ratTGFβ1 forward 5'-cctggaaagggctcaacac-3'
ratTGFβ1 reverse 5'-cagttcttctctgtggagctga-3'
rat TRAIL forward 5'-ccaaaatcggactagcttgc-3'
rat TRAIL reverse 5'-tcaaaggttctcaaagtcacctc-3'
human BAMBI forward 5'-tgcacgatgttctctctcct-3'
human BAMBI reverse 5'-gaagtcagctcctgcacctt-3'
rat MCP-1 forward 5'-agcatccacgtgctgtctc-3'
rat MCP-1 reverse 5'-gatcatcttgccagtgaatgag-3'
rat β-catenin forward 5'-cgaggactcaataccattcc-3′
rat β-catenin reverse 5'-agccgtttcttgtagtcctg-3′
rat Bcl-2 forward 5'-gtacctgaaccggcatctg-3'
rat Bcl-2 reverse 5'-ggggccatatagttccacaa-3'
Luciferase forward 5′-gcctgaagtctctgattaagt-3′
Luciferase reverse 5′-acacctgcgtcgaagt-3′
For standardization, rat Sp1 was amplified using the following primers:
rat Sp1 forward 5'-gctatagcaaacaccccaggt-3'
rat Sp1 reverse 5'-gatcagggctgttctctcctt-3'
Western Immunoblot Analysis
HSCs treated with or without AICAR (500μM), metformin (1mg/ml), calcitriol (10 nM), LPS (15ng/ml) or TGF-β1 (2ng/ml) for 2 h were dissolved in whole cell extraction buffer [25 mM Tris-Cl, pH 8.0, 10% (w/v) glycerol, 2 mM EDTA, 0.2 mM dithiothreitol (DTT), 1% Triton X-100, 1.5 mM MgCl2 and 200 mM NaCl)] and lysed on ice for 1 h, then centrifuged at 14,000 rpm for 15 min, 4°C. 20 μg of solubilized HSC extracts were analyzed on 10% SDS-PAGE gels. Gels were electroblotted onto Hybond-P Extra nitrocellulose membrane (Amersham Biosciences) and blocked for 4 h, 22°C with PBS containing 5% skim milk powder. To check for equal protein loading/transfer, the membrane was stained with Ponceau S solution (Sigma). After removing the stain by washing in water, the membrane was probed with monoclonal antibodies (phopho-AMPK or phosphor-S6RP at 1/250; Chemicon) in PBS overnight at 4°C, followed by anti-rabbit peroxidase-conjugate (1/10,000; Sigma) for 1 h at 22°C. Immunoreactive bands were detected by chemiluminesence (Lumi-LightPLUS; Roche Diagnostics). In addition to Ponceau S staining, the membranes were also probed with β-actin or CTGF antibodies for loading control.
Apoptosis assay
To measure apoptosis, Caspase-3/7 Assay Kit from AnaSpec (Campus Dr, Fremont, California) was used according to the manufacturer's instructions. Briefly, rat HSCs were treated with AICAR (500μM) or metformin (1mg/ml) for the time points indicated in the figure and lysed in the buffer provided. HSCs transfected with siRNA-Bambi were lysed 18h after transfection. Caspase activity was measured after the incubation of 125 μl of cell extract with 70 μl of caspase-3/7 substrate (60 min at room temperature) by fluorescence spectroscopy at Ex/Em=490nm/520nm.
Results
AICAR and metformin have distinct effects on quiescent hepatic stellate cell survival but identical impact on AMPK activation and inhibition of mTOR pathway
Previously published studies have focused on the effect of AMPK activation in activated HSC. The results from these studies were promising in that AMPK activating agents AICAR and metformin inhibited proliferation and profibrogenic activity while promoting apoptosis of activated HSC (16, 17). The effect of AMPK activation in quiescent HSC was the focus of this study. To determine the effect of AMPK activation on quiescent HSC survival, primary rat HSC maintained in culture for 48h were treated with AICAR or metformin, and cell viability was assessed. HSCs treated with AICAR for 24h showed dramatic induction of cell death even at the lowest concentration used (100μM), while metformin-treated HSCs remained viable with a normal appearance, even at the highest concentration employed (1000μg/ml) (Fig. 1A, Supplementary Fig. 1). We next examined the kinetics of AICAR-induced HSC cell death. For this purpose, HSC viability was assessed over a 24h time course following application of 500 μM AICAR. Cell density began to decrease as early as 3h after AICAR administration, with progressive loss of viable cells until near complete cell death occurred by 24h (Fig. 1B). This loss of cell viability occurred coincident with an increase in caspase 3/7 activity, consistent with an induction of cell apoptosis (Fig. 1C). These results suggest that AICAR and metformin have distinct impacts on quiescent HSC survival.
Figure 1.
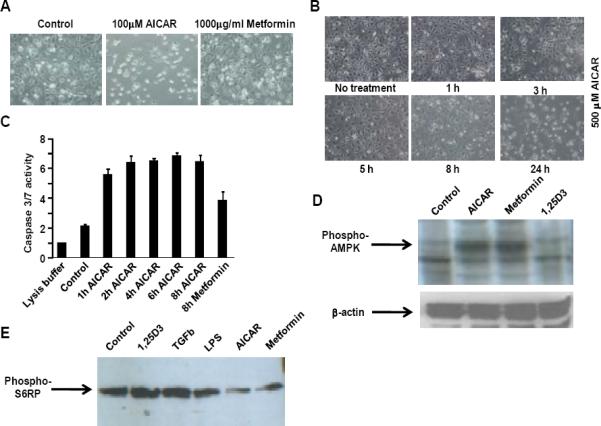
Effect of AICAR and metformin on quiescent HSCs. A, Light microscopy images of quiescent HSCs treated with 100μM AICAR or 1mg/ml metformin for 24 hrs. B, Light microscopy images of HSCs treated with 500μM AICAR for indicated times. Representative images are shown (n=3). C, Caspase activity in HSCs treated with AICAR (500 μM) or metformin (1 mg/ml). D, Western immunoblot analysis of phosphorylated AMPK with β-actin as a loading control and E, phosphorylated S6 ribosomal protein (S6RP) in HSCs after 2 hr treatment with indicated agents.
Previous studies have reported that treatment with AMPK activators AICAR and metformin results in a dose-dependent increase in Thr-172 phosphorylation of α-AMPK in activated HSC (17). A difference in AMPK phosphorylation could potentially account for the distinct responses to AICAR and metformin in quiescent HSC. We therefore compared the timecourse of α-AMPK Thr-172 phosphorylation in quiescent HSC treated with AICAR or metformin. Western immunoblot analysis demonstrates maximal induction of Thr-172 phosphorylation of α-AMPK in quiescent HSCs treated with either of these drugs after 2 hours (Fig. 1D). This result implies that the extent of AMPK activation does not explain the different outcomes of these treatments. Therefore, we reasoned that metformin may induce a downstream or AMPK-independent response that protects quiescent HSC from cell death or alternatively, AICAR may activate mechanisms that impede HSC survival.
AMPK regulates cellular metabolism in part by suppressing the mammalian target of rapamycin complex 1 (mTORC1) via direct phosphorylation of TSC2 (23) and mTORC1 subunit Raptor (26), consequently decreasing mTORC1-directed cell growth. AMPK activation results in rapid dephosphorylation of mTORC1 substrates 4EBP1 and S6K (23), leading to decreased phosphorylation of S6K substrate S6 ribosomal protein (S6RP). We postulated that differential mTORC1 inhibition by AICAR and metformin may underlie the varying responses to these drugs in quiescent HSC. To assess the degree of mTORC1 inhibition by AMPK activating agents in quiescent HSC, phosphorylated S6RP was measured after 2h treatment with these compounds (Fig. 1E). While HSC activation through AMPK-independent pathways (vitamin D signaling (27), TGF-β1 signaling (28) and TLR4/LPS signaling (29)) did not significantly affect S6RP phosphorylation, AICAR and metformin treatment induced similar reductions in S6RP phosphorylation. Given this similarity in AICAR- and metformin-induced inhibition of mTOR signaling, it is unlikely that differential mTORC1 inhibition was responsible for the observed AICAR-induced cell death.
AICAR suppresses inflammatory gene expression in primary hepatic stellate cells
During liver injury various inflammatory mediators induce cell damage and apoptosis (29, 30). We next explored the possibility that AICAR-induced cell death in HSCs was due to induction of proinflammatory chemokines/cytokines such as MCP-1 and IL-1β. Contrary to our expectation, MCP-1 and IL-1β gene expression progressively decreased in HSCs upon AICAR treatment (Fig. 2A). Expression levels of the metabolic genes PEPCK and LPL were found to increase with AICAR treatment, consistent with the know actions of AMPK activators (Supplementary Figure 2). In addition, AICAR treatment completely abrogated lipopolysaccharide (LPS)-induced expression of MCP-1 and IL-1β, suggesting that proinflammatory factors do not play a role in this event (Fig. 2B).
Figure 2.

Inflammatory gene modulation by AMPK-activation in HSCs. A, Relative IL1β and MCP1 mRNA expression in quiescent HSCs treated with 500 μM AICAR. B, Relative IL1β and MCP1 expression in HSCs treated with AICAR (500 μM) and/or LPS for 10 hrs.
AICAR-induced HSC cell death parallels loss of Bambi and silencing of AMPK restores Bambi expression and partially rescues HSCs from death
The TGF-β pseudoreceptor Bambi functions as a negative regulator of TGF-β signaling, a potent cytokine that is a prime mediator of hepatic fibrogenesis (31). Downregulation of Bambi and consequent sensitization to TGF-β signaling is an integral component of HSC activation (29). As Bambi may function not only to promote the quiescent HSC phenotype, but also to promote quiescent HSC survival we wished to determine whether AICAR and metformin could elicit differential effects on Bambi expression, thereby altering HSC survival. Interestingly, expression levels of Bambi progressively declined over time in AICAR treated cells (Fig. 3A). In contrast, metformin treatment induced an increase in Bambi expression for up to 10 h, followed by a return to baseline after 24–32 hrs (Fig. 3B). These results suggested that differential expression of Bambi may account for the differences in HSC survival. To further examine the role of AMPK in Bambi expression and HSC survival, the effects of AICAR and metformin treatment were examined after prior siRNA knockdown of AMPK expression. AICAR treated control cells (500 μm AICAR for 15h after transfected with a scambled siRNA) appeared poised to undergo apoptosis/necrosis and expressed markedly reduced levels of Bambi, consistent with our earlier findings (Fig. 3C & D). However, while HSCs lacking AMPK resembled control cells, these cells were partially rescued from AICAR-induced cell death and maintained normal levels of Bambi expression (Fig. 3C & 3D). In contrast, siRNA knockdown of AMPK had little effect on the response of HSCs to metformin. A similar induction of Bambi was seen in control (transfected with scrambled siRNA) and AMPK knockdown cells, and no effect on cell viability was observed (Fig. 3E and data not shown). Together these results suggest that AICAR-mediated HSC cell death is due in part to AMPK-dependent inhibition of Bambi expression, and that metformin-mediated HSC cell survival is attributable to an AMPK-independent increase in Bambi.
Figure 3.

AICAR and metformin conversely modulate Bambi. A, Induction of Bambi mRNA in HSCs treated with 500 μM AICAR and B, 1 mg/ml metformin. C, Bambi mRNA expression in HSCs after siRNA knockdown of AMPK in the presence or absence of AICAR and D, light microscopy images of HSCs after siRNA knockdown of AMPK and treated with AICAR. E, Bambi mRNA expression after siRNA knockdown of AMPK in the presence or absence of metformin.
Ectopic expression of Bambi improves HSCs survival upon treatment with AICAR
During the resolution phase of hepatic fibrosis, a crucial mechanism that operates to reduce injury is an increase in the apoptosis of activated HSC (32). Given our finding that AICAR induces cell death/apoptosis in quiescent HSCs, we next asked whether this effect was mediated by changes in Bambi expression. HSCs transfected with siRNA targeting Bambi showed reduced viability, similar to that seem with AICAR treatment (Fig. 4A). Furthermore, HSCs transfected with human BAMBI displayed increased resistance to AICAR-induced cell death (Fig. 4A). As activated HSCs have been shown to be sensitive to apoptosis induction by tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and subsequent activation of a protease cascade (32), we investigated whether a similar signaling pathway occurs in quiescent cells. The apoptotic response in quiescent HSCs after AMPK activation, with or without manipulation of Bambi expression, was measured by the expression of the pro-apoptotic gene TRAIL and anti-apoptotic gene Bcl-2. Results show that TRAIL levels were increased by 3 to 4-fold upon AICAR treatment while anti-apoptotic Bcl-2 levels fell 3-fold, consistent with induction of apoptosis (Fig. 4B). Ectopic overexpression of Bambi increased Bcl-2 expressio n 2-fold without significantly altering TRAIL expression. After AICAR treatment, ectopic Bambi expression partially increased HSC survival, and TRAIL expression was reduced by 50% compared to control (Fig. 4A & 4B), though Bcl-2 expression was diminished by AICAR independent of ectopic Bambi expression. In addition, siRNA-mediated reduction of Bambi had minor effects on TRAIL while significantly reducing the expression of anti-apoptotic Bcl-2 gene (Fig. 4B). In HSCs treated with metformin TRAIL levels gradually decreased with time (Fig 4C), suggesting that metformin functions differently to AICAR and is protective against TRAIL-dependent cell death. Furthermore, consistent with a pro-survival role for Bambi, siRNA knockdown of Bambi in HSCs resulted in a marked increase in caspase activity (Fig. 4D).
Figure 4.
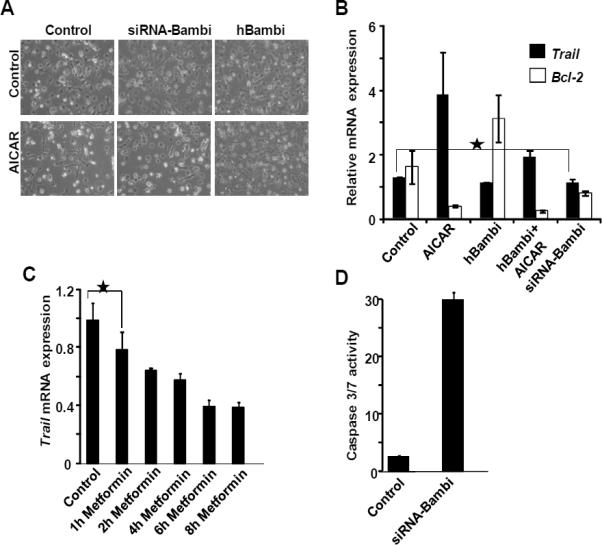
Ectopic expression of Bambi partially rescues AICAR-induced cell death. A, Light microscopy images of HSCs transfected with human Bambi or siRNA targeting Bambi treated with or without AICAR. Representative images from n=3. B, Trail and Bcl-2 expression in cells from (A). C, Trail expression in HSCs treated with metformin (1mg/ml). D, Caspase activity in HSCs after Bambi knockdown by siRNA.
Ectopic expression of Bambi induces the Wnt/β-catenin pathway in HSCs
Bambi has been shown to induce Wnt/β-catenin signaling in other cellular systems leading to cell proliferation and survival (33). This prompted us to investigate whether Bambi-dependent partial rescue of AICAR-induced cell death was in part due to induction of the Wnt/β-catenin signaling pathway. Supporting this hypothesis, transfection of HSCs with increasing amounts of Bambi significantly increased β-catenin mRNA and protein (Fig. 5A & 5B). To confirm that Bambi augments the functional Wnt/β-catenin signaling pathway, quiescent HSC were transfected with a β-catenin-dependent TCF-4/ELF-binding luciferase reporter from the fibronectin promoter/enhancer. This reporter was induced several-fold by Bambi, demonstrating that a true activation of Wnt/β-catenin signaling operates in quiescent HSCs in response to heightened Bambi expression (Fig. 5A). In addition, a reduced β-catenin expression was observed on Bambi knockdown (Fig. 5B).
Figure 5.

Bambi induce Wnt/β-catenin signaling in HSC. HSCs were transfected either with human Bambi expression plasmid or with siRNA against Bambi and after 24h RNA and protein extracted from the cells. A, Quantification of β-catenin and fire-fly luciferase mRNA was done by RT-PCR. B, Protein extracted from the cells were analysed by western immunoblot analysis for β-catenin expression. CTGF (connective tissue growth factor) was used as a loading control.
Bambi mRNA expression is stabilized by addition of cycloheximide in quiescent HSCs
In order to determine whether metformin acts directly to up-regulate Bambi, quiescent HSC were treated with metformin with and without cycloheximide, a de novo protein synthesis inhibitor. Inhibition of protein synthesis markedly enhanced the induction of Bambi by metformin (Fig. 6A), consistent with reported results (40). Interestingly, though the extent of stabilization of Bambi mRNA by cycloheximide in AICAR-treated cells was also significant, it was less robust, suggesting that different protein synthesis-independent regulatory mechanisms were operating. In addition, metformin-mediated Bambi induction was not found in activated HSCs (Fig. 6B), where metformin treatment reduced Bambi expression. Taken together these data imply that in quiescent HSC, metformin partially stabilizes Bambi mRNA, protecting cells from death, while AICAR fails to elicit Bambi expression and promotes apoptosis.
Figure 6.
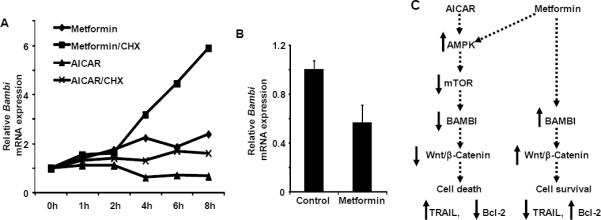
Post-transcriptional regulation of Bambi. A, Effects of cycloheximide on Bambi mRNA levels in quiescent HSCs. Cells were treated with metformin (1mg/ml) or AICAR (200 μM) in the presence or absence of cycloheximide (CHX, 1mg/ml). B, HSCs were activated in culture (7 days growth) prior to overnight treatment with metformin (1mg/ml). Bambi mRNA was quantified by RT-PCR. C, Proposed signaling events in quiescent HSCs upon AICAR and metformin treatment.
Discussion
Sustained AMPK activation by AICAR, metformin and adiponectin in activated HSCs has been linked to apoptosis (16, 34). Similarly, AMPK-mediated cell death has been reported in other cell types treated with these AMPK-activators (35). However, AICAR can induce apoptosis independent of AMPK through activation of the mitochondrial apoptotic pathway (36). The present study revealed contrasting effects of the AMPK activators AICAR and metformin in quiescent HSCs. While both drugs activated AMPK, AICAR selectively induced cell death through an AMPK dependent mechanism and reduced Bambi expression. AMPK knockdown partially rescued AICAR-induced HSC death and restored Bambi expression, consistent with an AMPK-dependent Bambi modulation. In contrast, metformin treatment induced Bambi expression independent of AMPK. These studies revealed a correlation between Bambi expression levels and quiescent HSC survival that was substantiated by studies in HSCs with targeted abrogation or ectopic expression of Bambi. Mechanistically, increased Bambi expression was linked to increased expression of anti-apoptotic factor Bcl-2, while AICAR induced expression of the pro-apoptotic factor TRAIL as well as enhanced caspase activity. Furthermore, we report that Bambi induces the Wnt/β-catenin signal transduction pathway, implicating this pathway in metformin-induced cell survival. Thus, the apoptotic response of quiescent HSCs depends on the balance of pro- and anti-apoptotic signals (32).
This study revealed that the AMPK activators AICAR and metformin operate differently in quiescent HSCs such that AICAR induces cell death in an AMPK-dependent manner while metformin, though inducing pro-survival factors, did not significantly effect overall cell survival. These findings may have implications on the therapeutic use of these AMPK activators as treatments for chronic liver disease and as preventative therapies for HCC.
Supplementary Material
Acknowledgments
The authors thank E. Ong and S. Ganley for administrative assistance, and Ruth Yu for useful discussion.
Grant Support This work was supported, in part, by grants from Ipsen/Biomeasure, The Helmsley Charitable Trust, Howard Hughes Medical Institute, NIH (DK062434, DK090962) and National Health and Medical Research Council of Australia Project grant 512354 and 632886. R. Evans is supported in part by a Stand Up to Cancer Dream Team Translational Cancer Research Grant, a Program of the Entertainment Industry Foundation (SU2C-AACR-DT0509). M.H. Sherman is supported by a NIH T32 training grant CA009370.
Footnotes
Disclosure of Potential Conflicts of Interest R.M. Evans is an investigator of the Howard Hughes Medical Institute and March of Dimes Chair in Molecular and Developmental Biology at the Salk Institute.
No potential conflicts of interest were disclosed.
References
- 1.Brenner DA. Molecular pathogenesis of liver fibrosis. Transactions of the American Clinical and Climatological Association. 2009;120:361–8. [PMC free article] [PubMed] [Google Scholar]
- 2.Friedman SL. Liver fibrosis -- from bench to bedside. Journal of hepatology. 2003;38(Suppl 1):S38–53. doi: 10.1016/s0168-8278(02)00429-4. [DOI] [PubMed] [Google Scholar]
- 3.Heron M, Hoyert DL, Murphy SL, Xu J, Kochanek KD, Tejada-Vera B. Deaths: final data for 2006. National vital statistics reports : from the Centers for Disease Control and Prevention, National Center for Health Statistics, National Vital Statistics System. 2009;57:1–134. [PubMed] [Google Scholar]
- 4.Bataller R, Brenner DA. Liver fibrosis. The Journal of clinical investigation. 2005;115:209–18. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang JD, Harmsen WS, Slettedahl SW, Chaiteerakij R, Enders FT, Therneau TM, et al. Factors that affect risk for hepatocellular carcinoma and effects of surveillance. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2011;9:617–23. e1. doi: 10.1016/j.cgh.2011.03.027. [DOI] [PubMed] [Google Scholar]
- 6.Rombouts K, Marra F. Molecular mechanisms of hepatic fibrosis in non-alcoholic steatohepatitis. Dig Dis. 2010;28:229–35. doi: 10.1159/000282094. [DOI] [PubMed] [Google Scholar]
- 7.Kisseleva T, Brenner DA. Role of hepatic stellate cells in fibrogenesis and the reversal of fibrosis. Journal of gastroenterology and hepatology. 2007;22(Suppl 1):S73–8. doi: 10.1111/j.1440-1746.2006.04658.x. [DOI] [PubMed] [Google Scholar]
- 8.Bataller R, Brenner DA. Hepatic stellate cells as a target for the treatment of liver fibrosis. Seminars in liver disease. 2001;21:437–51. doi: 10.1055/s-2001-17558. [DOI] [PubMed] [Google Scholar]
- 9.Knittel T, Dinter C, Kobold D, Neubauer K, Mehde M, Eichhorst S, et al. Expression and regulation of cell adhesion molecules by hepatic stellate cells (HSC) of rat liver: involvement of HSC in recruitment of inflammatory cells during hepatic tissue repair. The American journal of pathology. 1999;154:153–67. doi: 10.1016/s0002-9440(10)65262-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134:657–67. doi: 10.1016/j.cell.2008.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tilg H, Hotamisligil GS. Nonalcoholic fatty liver disease: Cytokine-adipokine interplay and regulation of insulin resistance. Gastroenterology. 2006;131:934–45. doi: 10.1053/j.gastro.2006.05.054. [DOI] [PubMed] [Google Scholar]
- 12.Matsuzawa Y. Therapy Insight: adipocytokines in metabolic syndrome and related cardiovascular disease. Nature clinical practice Cardiovascular medicine. 2006;3:35–42. doi: 10.1038/ncpcardio0380. [DOI] [PubMed] [Google Scholar]
- 13.Xu A, Wang Y, Keshaw H, Xu LY, Lam KS, Cooper GJ. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. The Journal of clinical investigation. 2003;112:91–100. doi: 10.1172/JCI17797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kamada Y, Tamura S, Kiso S, Matsumoto H, Saji Y, Yoshida Y, et al. Enhanced carbon tetrachloride-induced liver fibrosis in mice lacking adiponectin. Gastroenterology. 2003;125:1796–807. doi: 10.1053/j.gastro.2003.08.029. [DOI] [PubMed] [Google Scholar]
- 15.Ding X, Saxena NK, Lin S, Xu A, Srinivasan S, Anania FA. The roles of leptin and adiponectin: a novel paradigm in adipocytokine regulation of liver fibrosis and stellate cell biology. The American journal of pathology. 2005;166:1655–69. doi: 10.1016/S0002-9440(10)62476-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caligiuri A, Bertolani C, Guerra CT, Aleffi S, Galastri S, Trappoliere M, et al. Adenosine monophosphate-activated protein kinase modulates the activated phenotype of hepatic stellate cells. Hepatology. 2008;47:668–76. doi: 10.1002/hep.21995. [DOI] [PubMed] [Google Scholar]
- 17.Adachi M, Brenner DA. High molecular weight adiponectin inhibits proliferation of hepatic stellate cells via activation of adenosine monophosphate-activated protein kinase. Hepatology. 2008;47:677–85. doi: 10.1002/hep.21991. [DOI] [PubMed] [Google Scholar]
- 18.Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nature medicine. 2002;8:1288–95. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- 19.Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes & development. 2011;25:1895–908. doi: 10.1101/gad.17420111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nature cell biology. 2011;13:1016–23. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. The Journal of clinical investigation. 2001;108:1167–74. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hardie DG. AMP-activated protein kinase as a drug target. Annual review of pharmacology and toxicology. 2007;47:185–210. doi: 10.1146/annurev.pharmtox.47.120505.105304. [DOI] [PubMed] [Google Scholar]
- 23.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–90. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 24.Zhu NL, Wang J, Tsukamoto H. The Necdin-Wnt pathway causes epigenetic peroxisome proliferator-activated receptor gamma repression in hepatic stellate cells. J Biol Chem. 2010;285:30463–71. doi: 10.1074/jbc.M110.156703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weiskirchen R, Gressner AM. Isolation and culture of hepatic stellate cells. Methods Mol Med. 2005;117:99–113. doi: 10.1385/1-59259-940-0:099. [DOI] [PubMed] [Google Scholar]
- 26.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Molecular cell. 2008;30:214–26. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abramovitch S, Dahan-Bachar L, Sharvit E, Weisman Y, Ben Tov A, Brazowski E, et al. Vitamin D inhibits proliferation and profibrotic marker expression in hepatic stellate cells and decreases thioacetamide-induced liver fibrosis in rats. Gut. 2011;60:1728–37. doi: 10.1136/gut.2010.234666. [DOI] [PubMed] [Google Scholar]
- 28.Hellerbrand C, Stefanovic B, Giordano F, Burchardt ER, Brenner DA. The role of TGFbeta1 in initiating hepatic stellate cell activation in vivo. Journal of hepatology. 1999;30:77–87. doi: 10.1016/s0168-8278(99)80010-5. [DOI] [PubMed] [Google Scholar]
- 29.Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nature medicine. 2007;13:1324–32. doi: 10.1038/nm1663. [DOI] [PubMed] [Google Scholar]
- 30.Pradere JP, Troeger JS, Dapito DH, Mencin AA, Schwabe RF. Toll-like receptor 4 and hepatic fibrogenesis. Semin Liver Dis. 2010;30:232–44. doi: 10.1055/s-0030-1255353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wanninger J, Neumeier M, Bauer S, Weiss TS, Eisinger K, Walter R, et al. Adiponectin induces the transforming growth factor decoy receptor BAMBI in human hepatocytes. FEBS Lett. 2011;585:1338–44. doi: 10.1016/j.febslet.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 32.Shen H, Fan J, Minuk G, Gong Y. Apoptotic and survival signals in hepatic stellate cells. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2007;32:726–34. [PubMed] [Google Scholar]
- 33.Shin HW, Park SY, Lee KB, Jang JJ. Down-regulation of Wnt signaling during apoptosis of human hepatic stellate cells. Hepatogastroenterology. 2009;56:208–12. [PubMed] [Google Scholar]
- 34.Canto C, Auwerx J. AMP-activated protein kinase and its downstream transcriptional pathways. Cell Mol Life Sci. 2010;67:3407–23. doi: 10.1007/s00018-010-0454-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mihaylova MM, Shaw RJ, Carling D, Mayer FV, Sanders MJ, Gamblin SJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism AMP-activated protein kinase: nature's energy sensor. Nat Cell Biol. 2011;13:1016–23. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garcia-Garcia C, Fumarola C, Navaratnam N, Carling D, Lopez-Rivas A. AMPK-independent down-regulation of cFLIP and sensitization to TRAIL-induced apoptosis by AMPK activators. Biochem Pharmacol. 2010;79:853–63. doi: 10.1016/j.bcp.2009.10.022. [DOI] [PubMed] [Google Scholar]
- 37.Friedman SL. A deer in the headlights: BAMBI meets liver fibrosis. Nat Med. 2007;13:1281–2. doi: 10.1038/nm1107-1281. [DOI] [PubMed] [Google Scholar]
- 38.Behari J. The Wnt/beta-catenin signaling pathway in liver biology and disease. Expert Rev Gastroenterol Hepatol. 2010;4:745–56. doi: 10.1586/egh.10.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sekiya T, Adachi S, Kohu K, Yamada T, Higuchi O, Furukawa Y, et al. Identification of BMP and activin membrane-bound inhibitor (BAMBI), an inhibitor of transforming growth factor-beta signaling, as a target of the beta-catenin pathway in colorectal tumor cells. J Biol Chem. 2004;279:6840–6. doi: 10.1074/jbc.M310876200. [DOI] [PubMed] [Google Scholar]
- 40.Xavier S, Gilbert V, Rastaldi MP, Krick S, Kollins D, Reddy A, et al. BAMBI is expressed in endothelial cells and is regulated by lysosomal/autolysosomal degradation. PLoS One. 2010;5:e12995. doi: 10.1371/journal.pone.0012995. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.