

Conspectus

The functional group transformations carried out by the palladium-catalyzed Wacker and Heck reactions are radically different, but they are both alkenyl C-H bond functionalization reactions that have found extensive use in organic synthesis. The synthetic community depends heavily on these important reactions, but selectivity issues arising from control by the substrate, rather than control by the catalyst, have prevented the realization of their full potential. Because of important similarities in the respective selectivity-determining nucleopalladation and β-hydride elimination steps of these processes, we posit that the mechanistic insight garnered through the development of one of these catalytic reactions may be applied to the other. In this Account, we detail our efforts to develop catalyst-controlled variants of both the Wacker oxidation and the Heck reaction to address synthetic limitations and provide mechanistic insight into the underlying organometallic processes of these reactions.
In contrast to previous reports, we discovered that electrophilic palladium catalysts with non-coordinating counterions allowed for the use of a Lewis basic ligand to efficiently promote TBHP-mediated Wacker oxidation reactions of styrenes. This discovery led to the mechanistically guided development of a Wacker reaction catalyzed by a palladium complex with a bidentate ligand. This ligation may prohibit coordination of allylic heteroatoms, thereby allowing for the application of the Wacker oxidation to substrates that were poorly behaved under classical conditions.
Likewise, we unexpectedly discovered that electrophilic Pd-σ-alkyl intermediates are capable of distinguishing between electronically inequivalent C–H bonds during β-hydride elimination. As a result, we have developed E-styrenyl selective oxidative Heck reactions of previously unsuccessful electronically non-biased alkene substrates using arylboronic acid derivatives. The mechanistic insight gained from the development of this chemistry allowed for the rational design of a similarly E-styrenyl selective classical Heck reaction using aryldiazonium salts and a broad range of alkene substrates. The key mechanistic findings from the development of these reactions provide new insight into how to predictably impart catalyst control in organometallic processes that would otherwise afford complex product mixtures. Given our new understanding, we are optimistic that reactions that introduce increased complexity relative to simple classical processes may now be developed based on our ability to predict the selectivity-determining nucleopalladation and β-hydride elimination steps through catalyst design.
INTRODUCTION
In the late 1950’s, Smidt and co-workers,1 in a series of pioneering reports,2,3 described what is now recognized as the Wacker oxidation process, whereby ethylene is converted to acetaldehyde under aerobic oxidative Pd(II) catalysis and the co-catalysis of copper salts. The impact of this discovery rapidly captured the attention of the scientific community, ultimately providing the foundation for decades of research in organometallic catalysis applied to organic synthesis.4,5 Two formative contributions, both from 1964, have proven particularly influential: 1) Clement and co-workers found that using N,N-dimethylformamide (DMF) as a co-solvent with water allowed for extensive expansion of the substrate scope of the Wacker process,6 which provided the basis for Tsuji to develop his widely used modification of the Wacker oxidation,4,5,7,8 and 2) Henry’s investigation of the kinetics of the process, which resulted in the proposal that the Wacker oxidation proceeds by a syn-oxypalladation process (Figure 1).9 This latter report by Henry has proven to be a contentious contribution to the Wacker mechanism story.10,11 Regardless, it has had enduring impact on how one thinks about Pd(II) catalysis.
Figure 1.

Similarities of the Wacker oxidation and the oxidative Heck reaction.
In our opinion, a crucial impact of the Henry proposal is its relationship to the Heck reaction. The seminal report by Heck, who overlapped with Henry at the Research Center of Hercules in Wilmington, Delaware, describes the use of various organometallic reagents in combination with an alkene under Wacker-type conditions, leading to coupled products in what is now regarded as an oxidative Heck reaction.12 The mechanistic proposals are undeniably similar between the Wacker and Heck reactions, wherein Pd(II) promotes nucleopalladation of a coordinated olefin followed by β-hydride elimination although the nature of nucleopalladation for these processes (i.e., cis vs. trans addition, Markovnikov vs. anti-Markovnikov, and potentially reversible addition) is clearly dependent on the nucleophile being added and the reaction conditions.11 Both processes are early examples of metal-catalyzed C–H bond functionalization reactions, in which an alkenyl C–H bond is transformed into a C–X (X = N, O) or C–C bond (although, in the Wacker process, the number of C–H bonds in the product is conserved relative to the alkene starting material).
These classical processes share another common feature, namely that the reaction outcome is generally dictated by the substrate: the product distribution results from substrate-induced selectivity of either the initial nucleopalladation or the subsequent β-hydride elimination process (or both). In the Wacker oxidation, this is highlighted by substrate-dependent formation of mixtures of Markovnikov (methyl ketone) and anti-Markovnikov (aldehyde) products in unpredictable ratios as the result of indiscriminate oxypalladation (Figure 2).13 The Heck reaction also suffers from substrate control: the alkene substrate is commonly electronically biased (typically, α,β–unsaturated carbonyls and styrenes) so as to facilitate selective carbopalladation, leading to a scenario whereby β-hydride elimination forms a single product (Figure 2). Unfortunately, the use of electronically non-biased alkenes leads to complex mixtures arising from undiscerning carbopalladation followed by non-selective β-hydride elimination. These limitations have severely limited the generality of these widely-used synthetic methods. As described in this Account, a goal of our research program is to use a mechanistically-oriented approach to overcome the inherent substrate control of these fundamental processes to afford predictable and useful catalyst-controlled synthetic methods. Herein, we detail our efforts to develop catalyst-controlled variants of both the Wacker oxidation and the Heck reaction in order to address synthetic limitations, while providing mechanistic insight into the underlying organometallic processes.
Figure 2.

Substrate control in classical Pd-catalyzed C–H bond functionalization reactions.
Initial Goals and Approach
As mentioned above, a key goal of our program is to impart catalyst control upon classical Pd-catalyzed oxidation reactions. We planned to accomplish this by using a carefully designed ligand environment around Pd. However, when we initiated this program in 2002, most of the conditions reported for both the Wacker and oxidative Heck reactions required the use of a redox co-catalyst (generally salts of Cu) for effective turnover with molecular oxygen as the terminal oxidant. We initially considered Cu salts to be incompatible with our desired direct O2-coupled, ligand-controlled variant of these reactions due to the promiscuity of Pd(II) complexes to ligand substitution. Since many Cu salts are generally more Lewis acidic, this would lead to ligation of Cu instead of Pd for many ligands14 (although we have found the NHC-ligands are not readily prone to ligand substitution).15 Additionally, as discussed in more detail below, Cu is not necessarily merely involved in redox chemistry as is often assumed.16 Similarly, benzoquinone, another common oxidant, has been proposed to be directly involved in catalysis17 in addition to redox co-catalysis. Thus, we also wanted to avoid the use of this common additive. Direct O2-coupled, ligand-modulated variants of the Wacker oxidation had been reported before we initiated our efforts, namely by Sheldon using water-soluble bathophenanthroline ligands at high pressures,18 and later by Uemura who employed pyridine as the ligand and an alcohol to promote the formation of H2O2 in situ.19 While these are clearly exciting proof-of-concept studies, neither was aimed at the development of a highly useful synthetic method.
Based on our concurrent interest in Pd-catalyzed aerobic alcohol oxidations at the time,20 our initial plan was to use N-heterocyclic carbene (NHC) and bidentate amine ligand sets, which were found to be excellent templates for aerobic alcohol oxidations that also eliminated the requirement of redox co-catalysts. Initial evaluation of these ligands revealed the unanticipated finding that THF, in the absence of copper salts, was the only competent solvent for the NHC-Pd(II)-mediated Wacker oxidation of styrenes (Figure 3).21 In situ reaction monitoring provided evidence for the oxidation of THF to butyrolactone, presumably through the hydroperoxide. Gratifyingly, the procedurally simpler (and safer!) use of aqueous tert-butylhydroperoxide (TBHP)21 proved to be an exceptional oxidant for the Markovnikov-selective Wacker oxidation of diverse styrenes, which were previously reported to be poorly behaved substrates.7
Figure 3.

Initial discovery and proposed mechanism of a Pd-catalyzed TBHP-mediated Wacker oxidation of styrenes.
While the development of this system was serendipitous in many regards, the use of peroxides in Wacker oxidations had been reported in important contributions by Mimoun and coworkers.22,23 A surprising feature of our system is that the use of strongly Lewis basic ligands is allowed, which contrasts Mimoun’s results.22 We attribute this to the necessary use of relatively non-coordinating counterions, which has become an imperative aspect of our catalyst design efforts for various alkene functionalization reactions (vide infra). Additionally, preliminary mechanistic analysis through isotopic labeling studies confirms that the oxygen atom in the Wacker product arises from TBHP, and the hydrogen atom from the C–H bond that is cleaved is conserved in the product. Mimoun initially proposed the mechanism depicted in Figure 3 whereby O–O bond cleavage promotes a “hydride” migration, analogous to the Baeyer-Villiger oxidation mechanism, although initial kinetic studies did not reveal any pertinent details of this step.
Direct O2-Coupled Wacker Oxidation
Unfortunately, this initial system was limited to styrene substrates: the oxidation of simple terminal alkenes was fraught with isomerization and oxidation of the in situ-generated internal olefins. Several reports disclose that olefin isomerization is enhanced in alcoholic solvents24 and, considering the fact that methanol was our initial solvent of choice, solvents were reevaluated using aqueous TBHP as the oxidant with various Pd-complexes. Not surprisingly, in light of the historical significance of amide solvents in Wacker-type reactions, N,N-dimethylacetamide (DMA) was found to allow for the effective oxidation of decene to the methyl ketone product without observation of alkene isomerization or internal ketone products using Pd[(–)-sparteine]Cl2 as the catalyst.25 However, a completely unexpected aspect of this discovery was that the oxidant is O2, not TBHP, as revealed by a series of control experiments. This system is capable of oxidizing a wide range of simple alkene substrates at ambient pressures of O2. Of particular interest to target-oriented synthesis, homoallylic alcohols are excellent substrates for this oxidation and enantiomerically enriched substrates retain their enantiomeric excesses. It should be noted that Kaneda and coworkers have described closely related, highly useful direct-O2 coupled Wacker oxidations in DMA at modestly elevated pressures of O2.26,27
The Wacker oxidation of allylic alcohols commonly produce mixtures attributed to substrate control and to overcome this limitation alternative methods are required. As an example of this challenge, Crimmins and coworkers, in the synthesis of ophirin B, utilize a stoichiometric oxymercuration/Wacker oxidation to synthesize a key early stage synthetic intermediate.28 Indeed, effective scaling of this compound required a three step process in order to avoid using stoichiometric amounts of three toxic metals.29 Of note, our use of catalytic Pd[(–)-sparteine]Cl2 in the absence of additives in the Wacker oxidation of this substrate is effective albeit in modest yield and using modified conditions.25 This result inspired us to design alternative systems to effectively oxidize this challenging substrate class, as described in the succeeding section.
While the method has several synthetic limitations, the discovery of a direct O2-coupled Wacker oxidation provided us with a unique opportunity to investigate the mechanism of Wacker-type oxidations in the absence of copper. An underlying hypothesis in our group over the last decade is that Cu salts do not play an innocent role in Pd(II)-oxidase-type reactions. Considerable evidence exists, although mainly circumstantial,14,30 that this is indeed the case, and we could not pass up the opportunity to investigate the mechanism of this copper-free process. To do so, we equipped our laboratory with an instrument to measure gas uptake so as to streamline kinetic analysis.
Not surprisingly, the kinetics of this reaction are complex.31 The proposed mechanism based on these studies is depicted in Figure 5. In highly polar solvents, Pd[(–)-sparteine]Cl2 readily dissociates a chloride ion as determined electrochemically leading to B, the precatalyst in solution. Subsequent substitution of the remaining chloride to form C and reversible coordination of the alkene leads to the penultimate kinetically-relevant proposed intermediate D. In previous mechanistic studies of the Wacker oxidation, water was the solvent, but in our system the use of DMA, which allows us to probe the rate dependence on [H2O]. This experiment reveals an approximate third order dependence on [H2O], and a k(H2O)/k(D2O) of 2.6 ± 0.1 at high [decene] is also observed. Taken together, these experiments suggest a turnover-limiting step that involves the hydrogen bonding of three water molecules.
Figure 5.

Proposed mechanism of the direct O2-coupled Wacker oxidation using Pd[(–)-sparteine]Cl2
The computational community has undertaken substantial studies of the Wacker oxidation and several proposals involve the formation of a hydrogen bonding network of water in the oxypalladation step as depicted in intermediate E.32-34 (It should be noted that in all reports, the role of copper is ignored.) To further corroborate the reports from the computational community, we explored the feasibility of our proposed turnover limiting step through independent computations, which revealed an analogous transition state (see insert). Based on this model, our reaction presumably proceeds via an anti-oxypalladation process. Finally, we have briefly explored the role of copper in this particular variant of the Wacker oxidation and, not surprisingly, the outcome of the kinetic studies is complex, suggesting that copper is not innocent, which is consistent with our initial hypothesis.
TBHP-Mediated Wacker Oxidation: A General System
In the process of developing two distinct systems for the Wacker oxidation, we recognized the need for a general, catalyst-controlled method with broad applications in synthesis. Specifically, as cited above, substrates with allylic heteroatom substitution are notoriously truculent in that mixtures of the Markovnikov (methyl ketone) and anti-Markovnikov (aldehyde) products are formed under Tsuji-Wacker conditions.13,35 To overcome substrate control, we hypothesized that chelation of the allylic heteroatom must be avoided. To accomplish this, the use of TBHP as the oxidant was proposed to promote syn-oxypalladation, which should proceed at the Markovnikov position. Also, utilization of a bidentate ligand on Pd was thought to be necessary to leave one accessible coordination site for the alkene while preventing coordination of allylic heteroatoms.
This hypothesis was probed by evaluating a broad range of bidentate diamine ligands. Common ligands complexed with Pd(II) salts, such as bipyridine, were ineffective at the oxidation reaction of an allylic acetate.22 However, the parent quinoline oxazoline (quinox), which is now commercially-available and easily synthesized in a two-step, one-pot procedure,36 was found to be highly effective. It should be noted that an electrophilic catalyst is required, and is formed in situ via counterion metathesis with silver salts.37 Subsequently, we have found that this system is quite general, with various classes of allylic alcohols,37 simple alkenes,37 unprotected homoallylic alcohols,38 and protected allylic amines performing exceptionally well in this reaction.36 To highlight the catalyst-controlled nature of the method, the allylic and homoallylic amine derivatives depicted in Figure 7 were oxidized using both the quinox system and a standard Tsuji-Wacker method. In these cases, the quinox system provides high yields and excellent selectivity under more practical conditions (lower catalyst loadings and shorter reaction times) than those of the Tsuji-Wacker method, which affords a significant amount of aldehyde product. Additionally, applications of this method to target-oriented synthesis are forthcoming based on personal communications with several research groups.39
Figure 7.

Selected scope of the TBHP-mediated Wacker oxidation using the Pd-quinox catalyst.
A significant question that we have recently probed pertains to the unique ability of quinox to promote this reaction as compared to other simple bidentate diamine-type ligands.40 The primary hypothesis is that the ligand has a well-defined coordination sphere, leading to uniquely effective catalysis due to its electronic asymmetry as has also been implicated by Stahl and coworkers in enantioselective aza-Wacker reactions.41 Specifically, it is believed that the “electronically rich” oxazoline module promotes coordination of the electron rich TBHP in a trans fashion. As such, the relatively electron poor quinoline module supports trans coordination of the alkene. Kinetic experiments are consistent with reversible coordination of TBHP followed by coordination of the alkene and the turnover-limiting event. To explore the electronic asymmetry hypothesis, a series of systematically modified ligands at the quinoline module were prepared and the resultant Hammett analysis (ρ = 0.88) revealed that a more electron poor substituent on the quinoline leads to much faster rates. Conversely, using a series of systematically modified pyridyl ligands (in place of the oxazoline module) with an electron-poor quinoline revealed a Hammett correlation with ρ = −0.60, consistent with an electron-donating group promoting a faster reaction. In all, these data support the hypothesis and, more importantly, these studies reveal a new approach to catalyst design in Pd(II)-oxidation catalysis by using simple electronically asymmetric ligand classes to promote well-defined ligand environments about the metal center.
Heck Reactions: Initial Goals - Alkene Difunctionalization
Our interest in the development of catalyst-controlled variants of classical organometallic transformations has recently extended to the Heck reaction. The foundation of this project is serendipitous as outlined below.
Our group and others have recognized the potential of promoting multiple bond forming events by intercepting a Pd-σ-alkyl intermediate X fashioned via syn-carbopalladation or Heck insertion (Figure 9A). Specifically, controlled functionalization of this intermediate and diversion from the usual β-hydride elimination pathway leading to Heck products could afford alkene difunctionalization products. However, as expected, these intermediates normally exhibit short lifetimes and in order to allow for further functionalization, must be either stabilized or functionalized more rapidly than the rate of β-hydride elimination. A strategy for increasing the lifetime of Pd(II)-alkyl intermediates is to stabilize them via π-allyl or π-benzyl interactions.42 As an example of each of these strategies, Sanford and coworkers were able to generate arylhalogenation products (Figure 9B) by taking advantage of the stability of π-benzyl interactions arrived at via β-hydride elimination and Pd-hydride reinsertion affording 1,1-alkene difunctionalization products. Conversely, using an oxidant reactive enough to compete with β-hydride elimination yields the 1,2-alkene difunctionalization products.43,44
Figure 9.
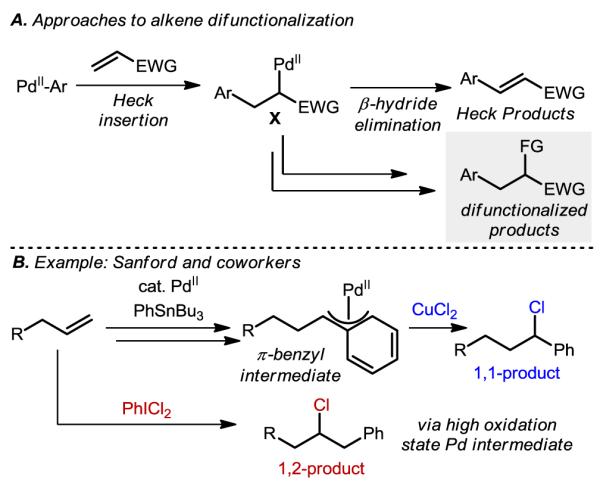
Approach and example of Heck insertion initiated alkene difunctionalization.
Concurrently, our group initiated a project to install two identical aryl groups in a 1,2-fashion upon styrenes using a similar strategy.45 In this case, the Heck insertion directly results in a benzylic Pd-alkyl, which is proposed to be stabilized by a π-benzyl intermediate (Figure 10). The success of this reaction was attributed to the cationic nature of the Pd(II) catalyst, which is believed to have a high affinity for this type of stabilizing interaction. Interestingly, a 1,1-diarylated minor byproduct was observed, and was proposed to arise from Pd-migration via β-hydride elimination and Pd-hydride reinsertion to the newly installed benzylic position similar to the results of Sanford and coworkers.43,44 It was noted that the amount of the 1,1-diarylation product relative to the 1,2-diarylation product increased with decreasing styrene electron density, which was attributed to mitigated π-benzyl stabilization. This result suggests that simple aliphatic olefins should deliver the 1,1-diarylated products through a process of Heck insertion followed by β-hydride elimination and Pd-hydride reinsertion at the newly installed benzylic position. This indeed proved to be the case (Figure 10), and the resulting methodology exhibited excellence functional group tolerance.46 It should be noted that the major byproduct of this reaction, especially when using electron-deficient organostannanes, was the oxidative Heck product, which is again suggestive of the importance of π-benzyl stabilization and served as a prologue of the following key discovery.
Figure 10.

Results leading to the discovery of an E-styrenyl selective oxidative Heck reaction of electronically non-biased alkenes.
At the outset, we realized that the utility of a Pd-catalyzed 1,1-alkene difunctionalization reaction is relatively limited, especially when using somewhat unappealing organostannanes and installing the same arene twice. Therefore, we explored the use of arylboronic acid derivatives in place of the organostannanes, but, to our surprise, no alkene diarylation was observed.47 Instead, the reaction resulted in the exclusive formation of the oxidative Heck product albeit in modest yield (Figure 10). At first glance, the discovery of an additional method to access well-known oxidative Heck products does not add considerably to the synthetic community. However, several observations compelled us to further explore this method. The first was the observation of a single E-isomeric-styrenyl product when using an allylic alcohol derivative although substrate chelation, as nicely demonstrated by the White group,48 may be responsible for the observed selectivity. Therefore, at a relatively early stage of development, we evaluated several unfunctionalized terminal alkenes and found nearly exclusive formation of the (E)-styrenyl products, suggesting that chelation was not responsible for the observed selectivity. As noted in the introduction, the vast majority of Heck reactions are performed on electronically-biased substrates. In our case, neither electronic bias nor chelation was found to be required, which provided a clear impetus for us to extensively explore this unanticipated result.
Heck reactions of electronically non-biased olefins
After modest optimization, scale-up provided oxidative Heck products in generally high yields and excellent selectivity for the desired E-styrenyl products using a variety of electronically non-biased olefins (Figure 11).47 Various functional groups proved compatible with the mild, base-free conditions, including free- or protected alcohols, esters, ketones, and weakly Lewis-basic nitrogen atoms. In many cases, substrates bearing heteroatoms distal to the olefin resulted in high selectivity, suggesting that the system is under catalyst– rather than substrate–control. Adding to the synthetic usefulness and providing evidence that the styrenyl product is not arrived at via equilibration, a protected enantiomerically-enriched allylic alcohol suffered no erosion in enantiomeric excess over the course of the reaction.
Figure 11.
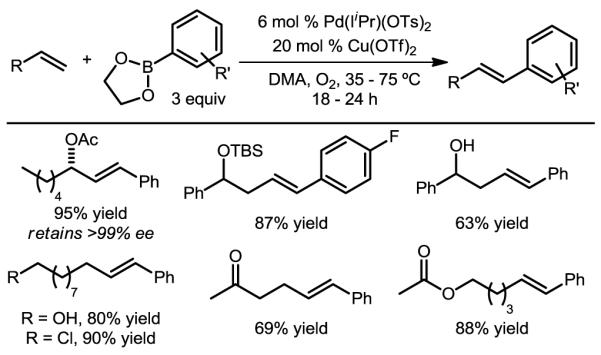
Selected scope of electronically non-biased alkenes as substrates in catalyst controlled oxidative Heck reactions.
To begin exploring the possible origin of selectivity for this reaction, several experiments were designed to probe the observed selectivity. To ascertain the relative importance of the catalyst components, a substrate susceptible to olefin isomerization was subjected to a variety of catalytic conditions (Figure 12).48 To this end, we established that catalyst-controlled β-hydride elimination is dependent on a highly electrophilic catalyst stabilized by a strongly coordinating NHC ligand. For example, the introduction of more coordinating counterions such as acetate (not pictured) or chloride, or the omission of the NHC ligand resulted in significantly reduced selectivity.
Figure 12.


Impact of counterion and ligand combination on selectivity in the oxidative Heck reaction.
Probing selectivity-determining β-hydride elimination through Hammett partitioning analysis.
Having determined what is required to allow for the observed high selectivity, we then wished to probe how the catalyst distinguishes between β-hydride elimination at either the allylic or styrenyl C–H bond. In order to accomplish this, a β,γ-unsaturated ester was subjected to the reaction conditions using a variety of electronically disparate arylboronic esters. The ratio of the allylic to styrenyl products was quantified and the log of this ratio for a given arene was then plotted vs Hammett α parameters revealing an unusual break in linearity (Figure 12). Typically, observations such as this are attributed to a change in mechanism over the range of electronic values evaluated. Applying this analysis would suggest that the catalyst selects for the more hydridic hydrogen atom when the arene substituent is electron-donating (stabilizing the developing positive charge) and leading to relatively more styrenyl product. When increasingly electron deficient arenes are used (which stabilize partial negative charge), the catalyst would select for the more protic hydrogen, also leading to greater amounts of the styrenyl product. This interpretation was ruled out because protons α to an ester are orders of magnitude more acidic than benzylic hydrogens, which would lead to overwhelming favoring of the allylic product if the deprotonation mechanism was operative. Instead, the observed break in linearity was attributed to the ability of the catalyst to distinguish between β-hydrogens on the basis of relative C–H bond strength, since both electron-donating and -withdrawing substituents weaken C–H bonds. These studies demonstrate that a highly electrophilic palladium catalyst is capable of distinguishing between β-hydrogens, unexpectedly based upon relative bond strength. In retrospect, it is interesting to consider that our 1,1-alkene diarylation method was likely also dependent on selective β-hydride elimination in order to arrive exclusively at the stabilized π-benzyl intermediates required to undergo the second functionalization event.45,46
Catalyst-Controlled β-Hydride Elimination using Pd(0) – Rational Design
The unforeseen discovery and development of a catalyst-controlled oxidative Heck reaction provided us with the knowledge that electrophilic Pd-σ-alkyl intermediates are capable of distinguishing between β-hydrogens based on their electronic nature. Armed with this knowledge, we anticipated that it may be possible to utilize this mechanistic insight in order to develop a complementary Pd(0)-catalyzed variant with similarly high levels of catalyst control. In order to do this, we envisioned that the oxidative addition event should proceed at low temperatures, and that the counterion on the resulting Pd(II)-aryl species should be non-coordinating. With these considerations in mind, aryldiazonium salts seemed uniquely well suited to meet these requirements.49-51 Indeed, after brief optimization using these salts, conditions for the desired highly selective classical Heck reaction were identified.52 All substrates tested in the oxidative Heck reaction performed well (representative examples are depicted in Figure 13), delivering high yields and selectivity rapidly at room temperature. In addition to this broad functional group tolerance, several substrates bearing functional groups such as carboxylic acids and nitriles, which are incompatible or untested under oxidative conditions, delivered excellent results. The high selectivity observed when submitting dodecene provides evidence that the β-hydride elimination event in this reaction is also under catalyst control.
Figure 13.
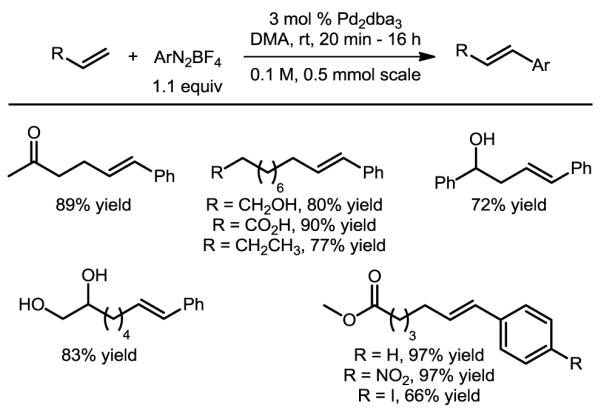
Selected scope of the Pd(0)-catalyzed Heck reaction of electronically non-biased olefins.
Having identified a highly selective, simple, and general classical Heck reaction counterpart to our oxidative Heck conditions, we wished to determine the origin of selectivity in the β-hydride elimination step of this variant. Initial studies focused on the same β,γ-unsaturated ester utilized previously, but product distribution was surprisingly unaffected by arene electronic effects giving good selectivity for the E-styrenyl product (Figure 12).47,52 This result strongly suggests that the mechanism by which the catalyst distinguishes between β-hydrogens is different from the previous conditions utilizing NHC-Pd(II) as the catalyst.
To further probe the origin of selectivity under these conditions, a more telling experiment was designed whereby allyl benzene was submitted to the reaction with several electronically disparate aryldiazonium salts.52 In this case, the catalyst must select between one of two types of benzylic β-hydrogens, which differ only by virtue of the electronic nature of the adjacent arenes. This experiment resulted in a more conventional Hammett plot (Figure 12), demonstrating that in this case, the relative hydridic nature of the β-hydrogens determines the selectivity under these conditions. The initial mechanistic studies performed on these two complementary systems demonstrate that not only is catalyst-controlled β-hydride elimination possible, but high selectivity may be achieved based upon different properties of the C–H bonds undergoing the selectivity-determining β-hydride elimination step.
Conclusions and Outlook
In summary, we have detailed our recent efforts to develop catalyst-controlled variants of classical Pd-catalyzed alkenyl C–H bond functionalization processes. To achieve catalyst control, ligand design efforts were essential in Wacker-type oxidations; the use an electronically asymmetric ligand (quinox) was found to be crucial for both reactivity and selectivity. Of note, this catalyst system has been found to promote the reaction of diverse substrates with excellent selectivity for the methyl ketone product under mild conditions. We hope that the mechanistic understanding garnered in the development of this reaction will provide us with insight to design catalysts that can achieve challenging aldehyde selective variants of the Wacker oxidation.53
A less obvious discovery during the development of the TBHP-mediated Wacker oxidation reaction was the need for relatively non-coordinating counterions for effective catalysis. This concept, while not new, has significantly influenced our approach to Pd-catalyzed alkene functionalization reaction development. Indeed, the discovery of conditions for alkene diarylation discussed above was highly dependent on the use of the correct counterion. This was highlighted in the development of both oxidative Heck and classical Heck reactions of electronically non-biased alkenes where counterions were also found to be critical. These reactions show considerable substrate scope under simple and mild conditions. Preliminary mechanistic studies suggest that the electrophilic nature of the catalyst allows for predictable determination of which C–H bond is cleaved. This revelation is clearly impacting our program, as we have recently reported a Pd-catalyzed 1,1-arylvinylation of terminal alkenes,54 which also relies on selective C–H bond cleavage to achieve the titled reaction by employing an electrophilic Pd-catalyst derived in situ from the oxidative addition of a vinyl triflate. Our current efforts are focused on using the concepts elucidated from these studies to develop new reactions where selective C–H bond cleavage of Pd-alkyl intermediates is involved.
Figure 4.
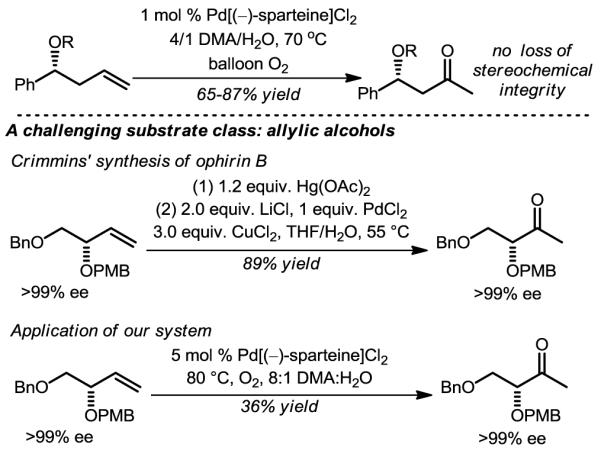
A direct O2-coupled Wacker oxidation at ambient pressures, and its application to the preparation of an intermediate in the synthesis of ophirin B.
Figure 6.
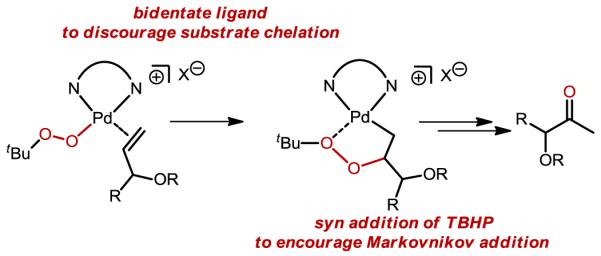
Catalyst design for the selective Wacker oxidation of allylic alcohols.
Figure 8.

Mechanistic proposal and studies of the TBHP-mediated Wacker oxidation using modified quinox-type ligands.
ACKNOWLEDGMENTS
I have been blessed with a highly talented and motivated group of coworkers to whom I am indebted for their contributions to this work. Special thanks goes to Candace Cornell, Brian Michel, Erik Werner, Brian Anderson, Laura Steffens, Kaveri Balan, and Jessica McCombs. I also would like to thank my collaborator Dr. John Keith for his contributions and insightful discussions. This project has received generous support from the National Institutes of Health. MSS thanks the American Chemical Society for support (Cope Scholar).
Biography
Sigman Biography: Matt Sigman was born in Los Angeles, California in 1970. He received a B.S. in chemistry from Sonoma State University in 1992 before obtaining his Ph.D. at Washington State University with Professor Bruce Eaton in 1996 in organometallic chemistry. He then moved to Harvard University to complete an NIH funded postdoctoral stint with Professor Eric Jacobsen. In 1999, he joined the faculty of the University of Utah where his research group has focused on the development of new synthetic methodology with an underlying interest in reaction mechanism. His research program explores the broad areas of oxidation catalysis, asymmetric catalysis, and the biological interactions of small molecules in breast cancer models.
Werner Biography: Erik W. Werner grew up in Olympia, Washington and received his B.Sc. in chemistry from Western Washington University in 2005. He stayed in Bellingham, WA until 2007 to complete his M.Sc. in chemistry working under Professor James Vyvyan. He is currently a graduate student studying catalyst-controlled β-hydride elimination within the mechanistic manifold of the Pd-catalyzed Heck reaction under the supervision of Professor Matthew Sigman at the University of Utah.
REFERENCES
- (1).Smidt J, Hafner W, Jira R, Sedlmeier J, Sieber R, Ruttinger R, Kojer H. Catalytic reactions of olefins on compounds of the platinum group. Angew. Chem. 1959;71:176–182. [Google Scholar]
- (2).Smidt J. Oxidation of Olefins with Palladium Chloride Catalysts. Chem. Ind. 1962:54–61. [Google Scholar]
- (3).Smidt J, Hafner W, Jira R, Sieber R, Sedlmeier J, Sabel A. Palladium Chloride-Catalyzed Oxidation of Olefins. Angew. Chem. 1962;74:93–102. [Google Scholar]
- (4).Cornell CN, Sigman MS. Recent Progress in Wacker Oxidations: Moving toward Molecular Oxygen as the Sole Oxidant. Inorg. Chem. 2007;46:1903–1909. doi: 10.1021/ic061858d. [DOI] [PubMed] [Google Scholar]
- (5).Takacs JM, Jiang X.-t. The Wacker Reaction and Related Alkene Oxidation Reactions. Curr. Org. Chem. 2003;7:369–396. [Google Scholar]
- (6).Clement WH, Selwitz CM. Improved Procedures for Converting Higher alpha-Olefins into Methyl Ketones with Palladium Chloride. J. Org. Chem. 1964;29:241–3. [Google Scholar]
- (7).Tsuji J. Synthetic Applications of the Palladium-Catalyzed Oxidation of Olefins to Ketones. Synthesis. 1984:369–84. [Google Scholar]
- (8).Tsuji J, Nagashima H, Nemoto H. A general synthetic method for the preparation of methyl ketones from terminal olefins: 2-decanone. Org. Synth. 1984;62:9–13. [Google Scholar]
- (9).Henry PM. Kinetics of the oxidation of ethylene by aqueous palladium(II) chloride. J. Am. Chem. Soc. 1964;86:3246–3250. [Google Scholar]
- (10).Keith JA, Henry PM. The Mechanism of the Wacker Reaction: A Tale of Two Hydroxypalladations. Angew. Chem. Int. Ed. 2009;48:9038–9049. doi: 10.1002/anie.200902194. [DOI] [PubMed] [Google Scholar]
- (11).McDonald RI, Liu G, Stahl SS. Palladium(II)-Catalyzed Alkene Functionalization via Nucleopalladation: Stereochemical Pathways and Enantioselective Catalytic Applications. Chem. Rev. 2011;111:2981–3019. doi: 10.1021/cr100371y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Heck RF. Acylation, methylation, and carboxyalkylation of olefins by Group VIII metal derivatives. J. Am. Chem. Soc. 1968;90:5518–5526. [Google Scholar]
- (13).Kang S-K, Jung K-Y, Chung J-U, Namkoong E-Y, Kim T-H. Complete Reverse Regioselection in Wacker Oxidation of Acetonides and Cyclic Carbonates of Allylic Diols. J. Org. Chem. 1995;60:4678–9. [Google Scholar]
- (14).Jensen KH, Webb JD, Sigman MS. Advancing the Mechanistic Understanding of an Enantioselective Palladium-Catalyzed Alkene Difunctionalization Reaction. J. Am. Chem. Soc. 2010;132:17471–17482. doi: 10.1021/ja108106h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Jensen DR, Sigman MS. Palladium Catalysts for Aerobic Oxidative Kinetic Resolution of Secondary Alcohols Based on Mechanistic Insight. Org. Lett. 2003;5:63–65. doi: 10.1021/ol027190y. [DOI] [PubMed] [Google Scholar]
- (16).Hosokawa T, Nomura T, Murahashi S-I. Palladium-Copper-DMF Complexes Involved in the Oxidation of Alkenes. J. Organomet. Chem. 1998;566:293. [Google Scholar]
- (17).Bäckvall JE, Bystroem SE, Nordberg RE. Stereo- and regioselective palladium-catalyzed 1,4-diacetoxylation of 1,3-dienes. J. Org. Chem. 1984;49:4619–4631. [Google Scholar]
- (18).ten Brink G-J, Arends IWCE, Papadogianakis G, Sheldon RA. Catalytic conversions in water. Part 10. Aerobic oxidation of terminal olefins to methyl ketones catalyzed by water soluble palladium complexes. Chem. Comm. 1998:2359–2360. [Google Scholar]
- (19).Nishimura T, Kakiuchi N, Onoue T, Ohe K, Uemura S. Palladium(II)-Catalyzed Oxidation of Terminal Alkenes to Methyl Ketones Using Molecular Oxygen. J. Chem. Soc., Perkin Trans. 1. 2000:1915–1918. [Google Scholar]
- (20).Sigman MS, Jensen DR. Ligand-Modulated Palladium-Catalyzed Aerobic Alcohol Oxidations. Acc. Chem. Res. 2006;39:221–229. doi: 10.1021/ar040243m. [DOI] [PubMed] [Google Scholar]
- (21).Cornell CN, Sigman MS. Discovery of and Mechanistic Insight into a Ligand-Modulated Palladium-Catalyzed Wacker Oxidation of Styrenes Using TBHP. J. Am. Chem. Soc. 2005;127:2796–2797. doi: 10.1021/ja043203m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Mimoun H, Charpentier R, Mitschler A, Fischer J, Weiss R. Palladium(II) tert-Butyl Peroxide Carboxylates. New Reagents for the Selective Oxidation of Terminal Olefins to Methyl Ketones. The Role of Peroxymetalation in Selective Oxidative Processes. J. Am. Chem. Soc. 1980;102:1047–54. [Google Scholar]
- (23).Roussel M, Mimoun H. Palladium-Catalyzed Oxidation of Terminal Olefins to Methyl Ketones by Hydrogen Peroxide. J. Org. Chem. 1980;45:5387–90. [Google Scholar]
- (24).Tang HG, Sherrington DC. Polymer-Supported Pd(II) Wacker-Type Catalysts. Part III. Isomerization of Alkene Double Bond. J. Mol. Catal. 1994;94:7–17. [Google Scholar]
- (25).Cornell CN, Sigman MS. Discovery of a Practical Direct O2−Coupled Wacker Oxidation with Pd[(−)-sparteine]Cl2. Org. Lett. 2006;8:4117–4120. doi: 10.1021/ol061662h. [DOI] [PubMed] [Google Scholar]
- (26).Mitsudome T, Umetani T, Nosaka N, Mori K, Mizugaki T, Ebitani K, Kaneda K. Convenient and Efficient Pd-Catalyzed Regioselective Oxyfunctionalization of Terminal Olefins by Using Molecular Oxygen as Sole Reoxidant. Angew. Chem. Int. Ed. 2006;45:481–485. doi: 10.1002/anie.200502886. [DOI] [PubMed] [Google Scholar]
- (27).Mitsudome T, Mizumoto K, Mizugaki T, Jitsukawa K, Kaneda K. Wacker-Type Oxidation of Internal Olefins Using a PdCl2/N,N-Dimethylacetamide Catalyst System under Copper-Free Reaction Conditions. Angewandte Chemie International Edition. 2010;49:1238–1240. doi: 10.1002/anie.200905184. [DOI] [PubMed] [Google Scholar]
- (28).Crimmins MT, Brown BH. An Intramolecular Diels-Alder Approach to the Eunicelins: Enantioselective Total Synthesis of Ophirin B. J. Am. Chem. Soc. 2004;126:10264–10266. doi: 10.1021/ja046574b. [DOI] [PubMed] [Google Scholar]
- (29).Crimmins MT, Brown BH, Plake HR. An Intramolecular Diels-Alder Approach to the Eunicellins: Enantioselective Total Syntheses of Ophirin B and Astrogorgin. J. Am. Chem. Soc. 2006;128:1371–1378. doi: 10.1021/ja056334b. [DOI] [PubMed] [Google Scholar]
- (30).Hosokawa T, Takano M, Murahashi S-I. The First Isolation and Characterization of a Palladium-Copper Heterometallic Complex Bearing μ4-Oxo Atom Derived from Molecular Oxygen. J. Am. Chem. Soc. 1996;118:3990–1. [Google Scholar]
- (31).Anderson BJ, Keith JA, Sigman MS. Experimental and Computational Study of a Direct O2-Coupled Wacker Oxidation: Water Dependence in the Absence of Cu Salts. J. Am. Chem. Soc. 2010;132:11872–11874. doi: 10.1021/ja1057218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Siegbahn PEM. Two, Three, and Four Water Chain Models for the Nucleophilic Addition Step in the Wacker Process. J. Phys. Chem. 1996;100:14672–14680. [Google Scholar]
- (33).Beyramabadi SA, Eshtiagh-Hosseini H, Housaindokht MR, Morsali A. Mechanism and Kinetics of the Wacker Process: A Quantum Mechanical Approach. Organometallics. 2008;27:72–79. [Google Scholar]
- (34).Comas-Vives A, Stirling A, Lledos A, Ujaque G. The Wacker Process: Inner- or Outer-Sphere Nucleophilic Addition? New Insights from Ab Initio Molecular Dynamics. Chemistry--A European Journal. 2010;16:8738–8747. S8738/1–S8738/12. doi: 10.1002/chem.200903522. [DOI] [PubMed] [Google Scholar]
- (35).Muzart J. Aldehydes from Pd-catalyzed oxidation of terminal olefins. Tetrahedron. 2007;63:7505–7521. [Google Scholar]
- (36).Michel BW, McCombs JR, Winkler A, Sigman MS. Catalyst-controlled Wacker-type oxidation of protected allylic amines. Angew. Chem. Int. Ed. 2010;49:7312–7315. doi: 10.1002/anie.201004156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Michel BW, Camelio AM, Cornell CN, Sigman MS. A General and Efficient Catalyst System for a Wacker-Type Oxidation Using TBHP as the Terminal Oxidant: Application to Classically Challenging Substrates. J. Am. Chem. Soc. 2009;131:6076–6077. doi: 10.1021/ja901212h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).McCombs JR, Michel BW, Sigman MS. Catalyst-controlled Wacker-type oxidation of homoallylic alcohols in the absence of protecting groups. J. Org. Chem. 2011;76:3609–3613. doi: 10.1021/jo200462a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Katcher MH, Sha A, Doyle AG. Palladium-Catalyzed Regio- and Enantioselective Fluorination of Acyclic Allylic Halides. J. Am. Chem. Soc. 2011;133:15902–15905. doi: 10.1021/ja206960k. [DOI] [PubMed] [Google Scholar]
- (40).Michel BW, Steffens LD, Sigman MS. On the Mechanism of the Palladium-Catalyzed tert-Butylhydroperoxide-Mediated Wacker-Type Oxidation of Alkenes Using Quinoline-2-Oxazoline Ligands. J. Am. Chem. Soc. 2011;133:8317–8325. doi: 10.1021/ja2017043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).McDonald RI, White PB, Weinstein AB, Tam CP, Stahl SS. Enantioselective Pd(II)-Catalyzed Aerobic Oxidative Amidation of Alkenes and Insights into the Role of Electronic Asymmetry in Pyridine-Oxazoline Ligands. Org. Lett. 2011;13:2830–2833. doi: 10.1021/ol200784y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Jensen KH, Sigman MS. Mechanistic approaches to palladium-catalyzed alkene difunctionalization reactions. Organic & Biomolecular Chemistry. 2008;6:4083–4088. doi: 10.1039/b813246a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Kalyani D, Sanford MS. Oxidatively Intercepting Heck Intermediates: Pd-Catalyzed 1,2- and 1,1-Arylhalogenation of Alkenes. J. Am. Chem. Soc. 2008;130:2150–2151. doi: 10.1021/ja0782798. [DOI] [PubMed] [Google Scholar]
- (44).Kalyani D, Satterfield AD, Sanford MS. Palladium-Catalyzed Oxidative Arylhalogenation of Alkenes: Synthetic Scope and Mechanistic Insights. J. Am. Chem. Soc. 2010;132:8419–8427. doi: 10.1021/ja101851v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Urkalan KB, Sigman MS. Palladium-Catalyzed Oxidative Intermolecular Difunctionalization of Terminal Alkenes with Organostannanes and Molecular Oxygen. Angew. Chem. Int. Ed. 2009;48:3146–3149. S3146/1–S3146/64. doi: 10.1002/anie.200900218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Werner EW, Urkalan KB, Sigman MS. PdII-Catalyzed Oxidative 1,1-Diarylation of Terminal Olefins. Org. Lett. 2010;12:2848–2851. doi: 10.1021/ol1009575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Werner EW, Sigman MS. A Highly Selective and General Palladium Catalyst for the Oxidative Heck Reaction of Electronically Nonbiased Olefins. J. Am. Chem. Soc. 2010;132:13981–13983. doi: 10.1021/ja1060998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Delcamp JH, Brucks AP, White MC. A General and Highly Selective Chelate-Controlled Intermolecular Oxidative Heck Reaction. J. Am. Chem. Soc. 2008;130:11270–11271. doi: 10.1021/ja804120r. [DOI] [PubMed] [Google Scholar]
- (49).Kikukawa K, Nagira K, Wada F, Matsuda T. Reaction of Diazonium Salt with Transition Metals--III: Palladium(0)-Catalyzed Arylation of Unsaturated Compounds with Arenediazoium Salts. Tetrahedron. 1981;37:31–36. [Google Scholar]
- (50).Sabino AA, Machado AHL, Correia CRD, Eberlin MN. Probing the Mechanism of the Heck Reaction with Arene Diazonium Salts by Electrospray Mass and Tandem Mass Spectrometry. Angew. Chem. Int. Ed. 2004;43:2514–2518. doi: 10.1002/anie.200353076. [DOI] [PubMed] [Google Scholar]
- (51).Taylor JG, Moro AV, Correia CRD. Evolution and Synthetic Applications of the Heck-Matsuda Reaction: The Return of Arenediazonium Salts to Prominence. Eur. J. Inorg. Chem. 2011:1403–1428. [Google Scholar]
- (52).Werner EW, Sigman MS. Operationally Simple and Highly (E)-Styrenyl-Selective Heck Reactions of Electronically Nonbiased Olefins. J. Am. Chem. Soc. 2011;133:9692–9695. doi: 10.1021/ja203164p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Dong G, Teo P, Wickens ZK, Grubbs RH. Primary Alcohols from Terminal Olefins: Formal Anti-Markovnikov Hydration via Triple Relay Catalysis. Science. 2011;333:1609–1612. doi: 10.1126/science.1208685. [DOI] [PubMed] [Google Scholar]
- (54).Liao L, Jana R, Urkalan KB, Sigman MS. A palladium-catalyzed three-component cross-coupling of conjugated dienes or terminal alkenes with vinyl triflates and boronic acids. J. Am. Chem. Soc. 2011;133:5784–5787. doi: 10.1021/ja201358b. [DOI] [PMC free article] [PubMed] [Google Scholar]