

Abstract
Introduction
The cannabinoid receptor type 2 (CB2) is an important target for development of drugs and imaging agents for diseases, such as neuroinflammation, neurodegeneration, and cancer. Recently we reported synthesis and results of in vitro receptor binding of a focused library of fluorinated 2-oxoquinoline derivatives as CB2 receptor ligands. Some of the compounds demonstrated as good CB2-specific ligands with Ki values in the nanomolar to sub-nanomolar concentrations; therefore, we pursued the development of their 18F-labeled analogues that should be useful for PET imaging of CB2 receptor expression. Here, we report the radiosynthesis of two 18F-labeled 2-oxoquinoline derivatives, and preliminary in vitro and ex-vivo evaluation of one compound as a CB2-specific radioligand.
Methods
4-[18F]Fluorobenzyl amine [18F]-3 was prepared by radiofluorination of 4-cyano-N,N,N-trimethylanilinium triflate salt followed by reduction with LiAlH4 and then coupled with acid chlorides 11 and 12 to afford [18F]-13 and [18F]-14. In vitro CB2 receptor binding assay was performed using U87 cells transduced with CB2- and CB1-receptor. Ex-vivo autoradiography was performed with [18F]-14 on spleen, CB2- and CB1-expressing and wild type U87 subcutaneous tumors grown in mice.
Results
The radiochemical yields of [18F]-13 and [18F]-14 were 10%-15.0% with an average of 12% (n=10); radiochemical purity was > 99% with specific activity 1200 mCi/μmole. The dissociation constant Kd for [18F]-14 was 3.4 nM. Ex-vivo autoradiography showed accumulation of [18F]-14 in the CB2-expressing tumor.
Conclusion
Two new [18F]-labeled CB2 ligands have been synthesized. Compound [18F]-14 appears to be a potential PET imaging agent for the assessment of CB2 receptor expression in vivo.
Keywords: Cannabinoid receptor CB2, [18F]-2-oxoqunoline derivatives, positron emission tomography
1. Introduction
The cannabinoid receptors are members of the superfamily G-protein coupled receptors. This family of receptors plays a key role in several biological processes involving the central nervous system, immune system, and metabolism [1, 2]. The main two distinct subtype receptors in mammalian tissue were identified as CB1 and CB2 [3, 4]. The CB1 receptor is located mainly in the central nervous system while the CB2 receptor is predominantly located in the peripheral nervous system and is associated with the immune response; however, it has been recently demonstrated that the CB2 receptor is expressed at low levels in the central nervous system [5, 6]. CB2 receptor overexpression is associated with various pathological conditions, such as neurodegenaration, multiple sclerosis, Alzheimer’s disease and human astrocytic tumors, in which the level of CB2 receptor up-regulation correlates with the level of tumor malignancy [7]. CB2 receptor up-regulation in the brain has been linked to neuroinflammation and neurodegeneration. The activation of CB2 receptor is involved in the mechanism that induces apoptotic cancer cell death, including several tumor cell lines such as human leukemia cell lines, prostate cancer PC3, and DLD-1 and HT29 colon cancer cells [8, 9]. Thus, there is a strong need of radioligands for in vivo PET imaging of the CB2 receptor expression.
Although, the pharmacologic and therapeutic potential of the selective CB2 receptor has been studied and reviewed extensively in the literature [10–13], very little progress has been made toward the synthesis of radioligands for in vivo PET imaging of this receptor. In the past decade, substantial progress has been made in the synthesis and evaluation of radiotracers for PET imaging of the CB1 receptor [14–16]; however, a very limited number of radioligands have been synthesized and tested for PET imaging of the CB2 receptor [17].
Recently, there has been a growing interest in developing radioligands for noninvasive PET imaging of the CB2 receptor in neurological diseases and cancer, and in monitoring therapeutic efficacy of anti-inflammatory drugs. These radioligands are derivatives of a novel class of 2-oxoquinoline derivatives that have shown high potencies at nanomolar concentrations and high selectivity for CB2 as inverse agonists [17–20]. Although the in vitro biological results of these radioligands were promising, the in vivo data were not quite satisfactory, because the compounds were metabolically unstable in vivo leaving only 13%–17% of the parent compound in circulation within 30 min [17]. These results suggest that there is a need for compounds with in vivo metabolic stability for PET imaging of CB2 receptor expression in neuroinflammation, multiple sclerosis and Alzheimer’s disease. The introduction of 18F into the phenyl ring could lead to a series of 2-oxo-1,2-dihydroquinoline-3-carboxamide derivatives that will significantly enhance the in vivo stability of the target molecule, because fluorobenzene is known to be metabolically more stable than the fluoroethyl ether group in the aliphatic side chain or in the aromatic ring [17, 21]. Most recently, two [11C]-labeled compounds, 2,2,3,3-tetramethylcyclopropanecarboxylic acid [3-(2-methoxyethyl)-4,5-dimethyl-3H-thiazol-(2Z)-ylidene]amide [22] and WG405833 [23] have been reported to be potential PET imaging agents for CB2 receptor, although these compound do not belong to the 2-oxoquinoline derivatives.
We have recently synthesized a focused library of 12 fluorinated 2-oxoquiniline derivatives as potential CB2-specific ligands and evaluated them in vitro by receptor binding assay on genetically engineered human U87-CB2 and U87-CB1 cell membrane using a known CB2 specific radioligand [3H]-CP55-940 [24]. Our in vitro receptor binding results demonstrated that several of these compounds are CB2-specific ligands with low nanomolar and subnanomolar binding concentrations. Consequently, we have developed a radiosynthesis method for two compounds, radiolabeled them with fluorine-18 and performed preliminary studies on in vitro cell binding assay and ex-vivo autoradiography using one compound, which had a low nM Ki value. Here, we report the 18F-labeling procedures of two compounds, 7-methoxy-8-alkoxy-2-oxo-1,2-dihydroquinoline-3-carboxylic-acid-(4-[18F]fluoro-benzyl)amide [18F]-13 and [18F]-14; and results from preliminary in vitro binding assay and ex-vivo quantitative autoradiographic studies of compound [18F]-14. Our preliminary results suggest that compound [18F]-14 may be a good candidate for in vivo PET imaging of the CB2 receptor expression.
2. Materials and methods
2.1. Chemistry
2.1.1 Reagents and instrumentation
All reagents and solvents were purchased from Aldrich Chemical Co. (Milwaukee, WI), and used without further purification. Solid-phase extraction (SPE) cartridges (silica gel, 900 mg) and reverse phase C18 cartridges were purchased from Alltech Associates (Deerfield, IL).
Thin layer chromatography (TLC) was performed on pre-coated Kieselgel 60 F254 (Merck, Darmstadt, Germany) glass plates. Proton, 13C, and 19F NMR spectra were recorded on either a Bruker 300 or 600 MHz spectrometer with tetramethylsilane used as an internal reference and hexafluorobenzene as an external reference at The University of Texas M. D. Anderson Cancer Center.
High-Performance Liquid Chromatography (HPLC) was performed with an 1100 series pump, (Agilent Technologies, Stuttgart, Germany), with a built-in UV detector operated at 254 nm, a radioactivity detector with a single-channel analyzer (Bioscan, Washington D C) using a semipreparative C18 reverse-phase column (10×250 mm, Econosil, Alltech) and an analytical C18 column (4.6×250 mm, Econosil, Alltech).
Synthesis of 7-methoxy-8-alkoxy-2-oxo-1,2-dihydroquinoline-3-carboxylic acid-(4-fluorobenzyl)amide (13 and 14)
Non-radioactive compounds 13 and 14 were synthesized by reaction of 4-fluorobenzyl amine 3 with the acid chlorides 11 and 12, and synthesis of 11 and 12 was performed following previously reported methods [17–19]. A detailed description for synthesis of 13 and 14 has been reported recently [24]. Briefly, to a solution of 3 (0.187 mmol) in dichloromethane (DCM, 3.0 mL) was added triethylamine (37 μL), and the mixture was cooled to 0°C. The acid chloride 11 or 12 (0.280 mmol) in DCM (0.5 mL) was added slowly and the mixture was stirred for 10 min, and then stirred for an additional 20 min at room temperature (RT). The solvent was removed under reduced pressure and the residue was purified by flash chromatography on a silica gel column using 30% hexane/ethyl acetate (EtOAc). The solvent was evaporated on a rotary evaporator and the product 13 was obtained as a white solid in 76% yield. The product 14 was obtained as a white solid in 50% yield.
2.2. Radiochemistry
2.2.1. Preparation of 4-[18F]-fluorophenyl cyanide ([18F]-2)
Both compounds [18F]-2 and [18F]-3 were prepared following literature methods [25, 26] with major modifications. The aqueous [18F]-fluoride was produced in the cyclotron by the reaction 18O(p, n)18F and trapped on an ion-exchange cartridge (Chromafix 30-PS-HCO3, ABX), then eluted with an aqueous solution of K2CO3 (0.4 mL, 2.75 mg/mL) into a V-vial containing kryptofix 2.2.2 solution (0.8 mL, 12.0 mg/mL) in acetonitrile (MeCN). Radiosyntheses were performed using 140–150 mCi of activity by manual procedure. Water was removed by an azeotropic evaporation with MeCN (1.0 mL) at 90°C under a stream of argon. A solution of 4-cyano-N,N,N-trimethylanilinium trifluoromethanesulfonate 1 (5.0–7.0 mg) in MeCN (0.5 mL) was added to the dried K18F/kryptofix in a sealed V-vial and the reaction mixture was heated at 110°C for 20 min. The mixture was cooled down in an ice bath for 1.0 min, passed through a SPE cartridge (silica) and eluted with either EtOAc (1 mL) or 7% methanol/dichloromethane (DCM, 1 mL) into a sealed V-vial. The solvent was removed under a slow stream of argon to obtain the compound [18F]-2.
2.2.2. Preparation of 4-[ 18F]-fluorobenzyl amine ([18F]-3)
A solution of LiAlH4 in THF (20.0 mg in 1.0 mL) was added to the reaction vial, which contains [18F]-2, and the reaction mixture was heated at 120°C for 10 min. An aliquot of the reaction mixture was removed, diluted with MeCN and injected into the analytical HPLC to confirm the completion of the reduction of the cyano group of [18F]-2 to the amine [18F]-3. The reaction mixture was quenched with water (0.3 mL) and the product [18F]-3 extracted with DCM (2×1.0 mL) into another V-vial for subsequent coupling with the acid chlorides 11 or 12.
2.2.3. Synthesis of 7-methoxy-8-alkoxy-2-oxo-1,2-dihydroquinoline-3-carboxylic acid-(4-[18F]-fluorobenzyl)amide ([18F]-13 and [18F]-14)
Triethylamine (0.2 mL) was added to the vial containing 4-[18F]-fluorobenzylamine ([18F]-3) then cooled to 0°C. A solution of the acid chloride 11 or 12 (5.0 mg) in DCM (0.3 mL) was added to the above solution of [18F]-3. The reaction mixture was shaken well and kept at RT for 10 min. An aliquot of the reaction mixture was withdrawn, diluted with MeCN and injected into the analytical HPLC to monitor the reaction, which showed that the reaction was complete. The solvent was evaporated and the residue was dissolved in MeCN (1 mL) and purified by HPLC on a semi-preparative column. Compound [18F]-13 was purified using 45% MeCN/water at a flow of 4 mL/min and compound [18F]-14 was purified using 50% MeCN/water at a flow of 4 mL/min. The radioactive material was collected within 16–19 min and 15–17.5 min for [18F]-13 and [18F]-14, respectively. The solvent was evaporated under reduced pressure and the product was diluted with 50% DMSO in saline. The final products [18F]-13 and [18F]-14 were further analyzed by HPLC for their purity and identity.
2.2.4. Partition coefficient
Partition co-efficient was determined following a previously reported method [17]. Briefly, twenty-five micro-liters of [18F]-14 collected from the HPLC, was added to a test tube containing 2 ml of 1-octanol and 2 ml of 0.025 M phosphate buffer pH 7.4. The test tube was vortexed at RT for 2 min and then centrifuged at 2700×g for 10 min. A 100-μl aliquot was taken from the 1-octanol phase and a 900-μl aliquot from the aqueous phase, avoiding cross contamination between the phases. The aliquots were transferred into tarred vials and the accurate volume added was calculated from the mass of the aliquots and the density (ρ) of the phase, assuming that ρ buffer=1.000 g/ml and ρ 1-octanol=0.827 g/ml. The radioactivity of the aliquots was counted on a γ-counter. The partition coefficient (P) was calculated as [radioactivity (cpm/mL) in 1-octanol]/[radioactivity (cpm/mL) in phosphate buffer].
3. Biology
3.1. Engineering of human CB2-expressing U87 glioblastoma cells
The detailed description of the cell transduction has been described in a previous paper [24]. Briefly, the human glioblastoma cell line U87 was transduced with lentivirus carrying human cannabinoid receptor 2 and green fluorescent protein. CB2 was subcloned into pDONR222 via BP reaction following amplification with attB1 and attB2 flanked primers, then recombined into a Gateway-adapted lentivirus encoding an internal mscv LTR and the green fluorescent protein eGFP downstream of an emcv IRES. Virus was packaged in 293-FT cells using pMD2.G and pCMV-deltaR8.91 and concentrated to 50-times. Concentrated viral supernatants were used to stably transduce the human glioblastoma cell line U87 (ATTC, Manassas, VA) via spinfection for 2 hr at 2200 RPM/30°C. CB1+/mKateS158A+ and CB2+/mKateS158A+ U87 cells were sorted on a FACSAria cell sorter (BD Biosciences, San Jose, CA) based on expression of the co-reporter mKateS158A. Cell surface expression of hCB1 and hCB2 was assessed via flow cytometry on a FACSCalibur (BD Biosciences, San Jose, CA). The resulting U87-CB2 cell line was >98% CB2+ and U87-CB1 cell line was >94% CB1+. These U87CB2- and U87-CB1 cells were used for receptor binding assay.
3.2. In vitro receptor binding assay
This study was performed using the compound [18F]-14 as a radiolabeled CB2 receptor ligand following a literature method [27]. Briefly, U87 cells transduced with CB2 receptor were cultured on Petri dishes until 80–90% of confluence was achieved. Then cells were washed with PBS, scraped and centrifuged at 1500 rpm for 5 min. Pellet was homogenized in binding buffer (50mM TRIS-HCl, 5mM MgCl2, 2.5mM EDTA, 0.5% BSA, pH=7.4) in Dounce homogenizer on ice. Aliquots (30–50 μg of protein) were incubated with [18F]-14 (0.025nM – 25nM) for 1 h at 4°C. Nonspecific binding was determined in the presence of 10μM of cold CP-55940. After incubation, aliquots were filtered through GF/C filters (presoaked in 0.1% polyethylenimine for 1 h) and washed 3×5 ml of cold 50mM TRIS-HCl. Radioactivity was counted in a gamma-counter (Cobra Quantum, PerkinElmer, MA, USA). Kd and Bmax were calculated by GraphPad Prism software using Scatchard plot analysis.
3.3. Ex-vivo quantitative autoradiography (QAR)
Animal study was performed under an approved animal protocol at the University of Texas M D Anderson Cancer Center. A group of nude mice (n=3) were injected with U87-wild type, U87CB2 and U87CB1 cells to grow tumors. When the tumor size was approximately 0.5 cm in diameter, the animals were sacrificed and the spleen and tumors were rapidly extracted, frozen, and embedded in a mounting medium M1 (Shandon-Lipshaw, Pittsburg, PA). Serial 20 mm thick coronal sections of frozen tumors and tissue were obtained at −13°C using a cryomicrotome (CM3050S, Leica, Germany). Tissue sections were thaw-mounted on poly-A lysine coated glass slides and heat-fixed for 5 min at 65°C on a slide warmer (Fischer Scientific, PA). For ex-vivo QAR, 150 ml of saline containing 50% DMSO and [18F]-14 (240 μCi/15 ml) was gently applied directly onto glass slides containing frozen tumor and spleen tissue sections. The tissue sections were placed in a humidified black box to reduce evaporation at room temperature for 60 min. The excess radioactivity was washed thrice by consecutive dipping in ice-cold PBS/Triton X-100 (0.01%) at pH 7.4. To assess the specificity of [18F]-14 binding, adjacent tissue sections were pre-blocked with cold compound 14 (1 mmol) for 30 min and then incubated with [18F]-14, as described above. Both sets of slides were dried and exposed to phosphor plate (FLA5100, Fuji Photo Film Co., Tokyo, Japan) for 6–8 h. The intensity of ex-vivo autoradiographic images was measured using the MCID software, version 7.0 (Interfocus Imaging Ltd., Cambridge, UK) and plotted as photostimulated luminescence (PSL).
4. Results and discussion
4.1. Chemistry and radiochemistry
The nonradioactive compounds 13 and 14 were synthesized following literature methods [17–20] (Synthetic Scheme 1) and detailed description on synthesis of these compounds has been reported recently [24].
Synthetic Scheme 1.

For synthesis of 13 and 14 the acid chlorides 11 and 12 were produced in situ according to the previously published methods [20, 24], and coupled with 4-fluorobenzylamine as described in Scheme 1. Compounds 13 and 14 were fully characterized by 1H and 19F NMR spectroscopy and the spectra were consistent with the literature results [24]. However, this method is not suitable for radiosynthesis to introduce [18F] in the aromatic ring. Therefore, we developed a new methodology and started with a precursor compound, 4-cyano-N,N,N-trimethylanilinium trifluoromethanesulfonate 1 for radiosynthesis of [18F]-3 and finally, [18F]-13 and [18F]-14 (Scheme 2).
Synthetic Scheme 2.

a) MeCN, K18F/kryptofix, 110°C, 20 min; b) THF, LiAlH4, 120°C, 10 min; c) CH2Cl2, Et3N, RT, 10 min.
A synthesis of nonradioactive compound 3 was performed first to adapt for radiosynthesis. Thus, fluorination of 1 was performed using Bu4NF in MeCN heated at 95°C for 20 min to produce 2. The chemical yield in this step was 50%. Compound 2 was reduced with LiAlH4 in THF by heating at 120°C for 30 min. After work up and purification, compound 3 was obtained in 90% yield. Compound 3 was characterized by 1H NMR spectroscopy and the spectrum was consistent with that of the commercial compound. Coupling of the acid chlorides 11 and 12 with synthetic 3 produced 13 and 14 with similar spectroscopic characterestics to those obtained by using commercial 4-fluorobenzylamine [24].
Radiofluorination of 1 was performed using K18F/kryptofix at 110°C for 20 min. The radiochemical yields in this step were 70%–75% (d. c), which was much higher than the non-radioactive fluorination. We optimized the radiofluorination conditions, 110°C for 20 min heating was observed as optimal for radiofluorination of 1. Under these reaction conditions we obtained 70%–75% yields consistently.
The work up procedure of [18F]-2 in our experiments was quite different than that reported earlier [25, 26]. Use of a C18 cartrdige for trapping the product [18F]-2 after removing the unreacted free 18F-fluoride was not efficient. Furthermore, reduction of [18F]-2 with LiAlH4 was not clean, several unidentified radioactive by-products were observed by HPLC. The clean product [18F]-2 was extremely important, because purity of [18F]-2 determines the yield of the coupling step with the acid chlorides to obtain the final desired products [18F]-13 and [18F]-14. Therefore, we changed the work up procedure for [18F]-2 to improve the quality and efficiency of reduction and the yield of the coupled target compounds. In our method, the reaction mixture (after radiofluorination) was passed through a silica gel SPE cartridge and the crude product eluted with either EtOAC (1 mL) or a mixture 7% MeOH in CH2Cl2 (1 mL). The solvent was evaporated by a slow stream of argon at RT. Approximately 45%–50% of the radioactivity was lost to get a solvent free product. With this loss of [18F]-2 we got consistently clean compound [18F]-3 as observed by HPLC. The presence of a small amount of any solvent, EtOAC or MeOH produced unidentified radioactive products in addition to [18F]-3 during reduction, and as a result, the overall yield of the coupled product went down.
Product [18F]-3 was extracted with CH2Cl2 (1.2 mL) into a clean V-vial for coupling with 11 or 12. In this step the efficiency of extraction was approximately 80%, about 20% activity retained in the LiAlH4 reaction vial. Coupling of [18F]-3 with 11 and 12 were complete at room temparature within 10 min, as determined by HPLC. After solvent evapoaration, the crude product was diluted with MeCN (1.2 mL) and purified by HPLC. It should be noted that in a clean reduction of [18F]2, approximately 60% of the radioactivity was recovered during HPLC purification of the coupled products. On the other hand, when the reduction was not clean, only 15%–20% of the radioactivity could be recovered from the HPLC column during purification.
We attempted to purify the radiolabeled compounds [18F]-13 and [18F]-14 using 65% EtOH in Na2PO4 buffer, but in this purification method a significant amount of non-radioactive impurities were eluted with the desired product. Furthermore, the EtOH/buffer solvent system produced a very high back-pressure on the HPLC column. Therefore, we used MeCN/water solvent system for routine purification of these radioactive compounds.
The final products [18F]-13 and [18F]-14 were analyzed by HPLC for their chemical and radiochemical purity and identity. Figure 1 represents a typical quality control analysis of [18F]-14, co-injected with standard 14. Although in vitro binding of the nonradioactive compound 13 demonstrated to be a poor ligand for CB2, we performed the radiosynthesis to validate the methodology.
Figure 1.

HPLC chromatogram of [18F]-14, co-injected with standard 14, Anal column, 55% MeCN, 1mL/min.
The decay corrected yields of compounds [18F]-13 and [18F]-14 were in the range of 10% to 15% from the end of bombardment (EOB). The radiochemical purity was greater than 99% and specific activity was estimated to be 1200 mCi/μmole. The synthesis time was 140–150 min from the EOB, including HPLC purification and solvent evaporation.
3.2. Partition coefficient
The lipophilicity of 7-methoxy-8-butoxy-2-oxo-1,2-dihydroquinoline-3-carboxylic acid-(4-fluorobenzyl)amide [18F]-14 was determined by partitioning between 1-octanol and 0.025 M phosphate buffer pH 7.4. The log-P of [18F]-14 was 4.4. One major problem with this compound ([18F]-14) was its solubility in aqueous system; the product was not quite soluble in buffer or water; therefore, we dissolved the product in a solution of 50% DMSO in saline for in vitro and ex-vivo studies.
4.2. In vitro receptor binding study
Figure 2 represents the scatchard plot of [18F]-14 for in vitro receptor binding assay. The dissociation constant Kd for [18F]-14 was 3.4 nM and the Bmax was 1690 fmol/mg of protein in the CB2 enriched U87 cells. This low nM concentration of Kd suggests that this compound may be an efficient CB2 binding ligand for PET imaging of CB2 receptor expression.
Figure 2.
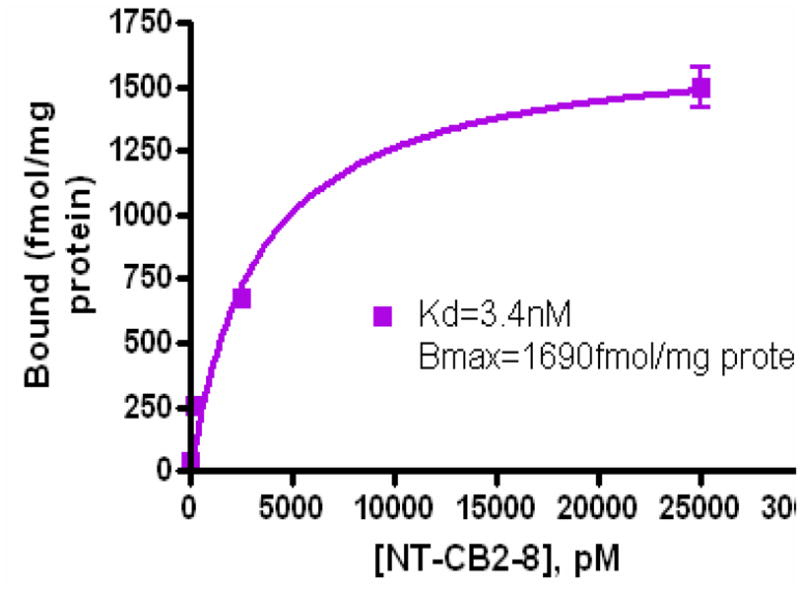
In vitro receptor binding assay of [18F]-14 in U87CB2-expressing cell lysate.
4.3. Ex-vivo quantitative autoradiography (QAR)
Figure 3 represents the results of ex-vitvo autoradiography of U87 wild type, U87CB2- and U87CB1-positive tumor xenografts and spleen. As Figure 3 shows, there was a high accumulation of radioactivity in the CB2-positive tumor compared with wild type- and CB1-positive tumors and spleen. The high accumulation of the compound [18F]-14 in the CB2-positive tumor (Fig. 3A) could be partially blocked with the non-radioactive compound 14 (Fig. 3B). The wild type U87 tumor, CB1-expressing tumor and spleen had a small and insignificant accumulation of the tracer. It was reported [17] that the CB2 receptor is over expressed in the spleen, however, in our study we did not observe any significant accumulation of the radiotracer in the spleen. Figure 3C represents a quantitative measurement of the binding and its inhibition in U87-wild type tumor, U87CB2- and U87CB1-expressing tumors and spleen. Approximatley 50% of binding in the CB2-positive tumor was inhibited by blocking the tumor tissue with the nonradioactive compound (Fig. 3C). No significant binding or inhibition of binding was observed in the wild type tumor, CB1-expressing tumor and spleen. Low inhibition of binding (50%) might be due to the insufficient amount of nonradioactive compound in the solution because of its poor solublity in the aqueous solvent synstem.
Figure 3.

Ex-vivo autoradiography of U87-wild type, U87CB2- and U87CB1-positive tumors and spleen; A: without blocking by cold compound; B: blocked with cold compound; C: quantification by photostimulated luminescence (PSL/mm3) in blocked and unblocked tissues.
The in vitro binding assay with the CB2 receptor expressing cell lysate and the preliminary ex-vivo autoradiography results demonstrate that compound [18F]-14 is a good CB2 specific lignad, which may be used for non-invasive assessment of CB2 receptor in tissues and organs with high expression of CB2. These results (in vitro and ex-vivo) with the radioactive compound [18F]-14 are consistent with our previously reported data of the nonradioactive compound 14, which exhibited the Ki value of 2.8 nM concentration [24]. These results also suggest that the nonradioactive compound may be a very good therapeutic agent for neuroinflammation and neurodegeneration and that the radioactive compound may be a very good PET imaging agent for assessment of the CB2 receptor expression. These results warrant further detailed investigations, including PET/CT imaging in normal mice, in the lipopolysaccharide (LPS)-induced mouse model of neuroinflammation and CB2 knockout mice. Further optimization of formulation of this compound is necessary to improve its solubility in aqueous solution for biological studies.
Conclusions
We have synthesized two [18F]-labeled radioligands in good yields, high purity and high specific activity for PET imaging of the CB2 receptor expression. The radiolabeling procedure can be extended for radiosyntheses of other 2-oxoquinoline derivatives. Preliminary in vitro receptor binding assay and ex-vivo autoradiography results suggest that the 7-methoxy-8-butoxy-2-oxo-1,2-dihydroquinoline-3-carboxylic acid-(4-fluorobenzyl)amide [18F]-14 may be a good agent for PET imaging CB2 receptors expression. These results warrant radiolabeling of the other potent 2-oxoquinoline analogues with subnanomolar binding efficiency to CB2 receptor and their investigation in vitro and in vivo, including PET/CT imaging in small animals.
Acknowledgments
This work was supported by the Developmental Projects Program of the Center for Advanced Biomedical Imaging Research (CABIR) at the MD Anderson Cancer Center, and grants: 1 U24 CA126577 01 (NIH) and CA 016672 (NIH).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Refererences
- 1.Frost JM, Dart MJ, Tietje KR, Garrison TR, Grayson GK, Daza AV, et al. Indol-3-ylcycloalkyl Ketones: Effects of N1 Substituted Indole Side Chain Variations on CB2 Cannabinoid Receptor Activity. J Med Chem. 2010;53:295–315. doi: 10.1021/jm901214q. [DOI] [PubMed] [Google Scholar]
- 2.Willis PG, Pavlova OA, Chefer SI, Vaupel DB, Mukhin AG, Horti AG. Synthesis and Structure—Activity Relationship of a Novel Series of Aminoalkylindoles with Potential for Imaging the Neuronal Cannabinoid Receptor by Positron Emission Tomography. J Med Chem. 2005;48:5813–5822. doi: 10.1021/jm0502743. [DOI] [PubMed] [Google Scholar]
- 3.Munro S, Thomas KL, Abushaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 4.Devane WA, Dysarz FA, Johnson MR, Melvin LS, Howlett AC. Determination and characterization of a cannabinoid receptor in rat brain. Mol Pharmcol. 1988;34:605–613. [PubMed] [Google Scholar]
- 5.Lozano-Ondoua AN, Wrigh C, Vardanyan A, King T, Largent-Milnes TM, Nelson M, Jimenez-Andrade JM, Mantyh PW, Vanderah TW, et al. A cannabinoid 2 receptor agonist attenuates bone cancer-induced pain and bone loss. Life Sci. 2010;86:646–653. doi: 10.1016/j.lfs.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Atwood BK, Mackie K. CB2: a cannabinoid receptor with an identity crisis. British J Pharmacol. 2010;160:467–479. doi: 10.1111/j.1476-5381.2010.00729.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reux B, Nevalainen T, Raitio KH, Koskinen AMP. Synthesis of quinolinyl and isoquinolinyl phenyl ketones as novel agonists for the cannabinoid. Bioorg Med Chem. 2009;17:4441–4447. doi: 10.1016/j.bmc.2009.05.013. [DOI] [PubMed] [Google Scholar]
- 8.Roche M, Finn DP. Receptors: Implications for Neuropsychiatric Disorders. Pharmaceuticals. 2010;3:2517–2553. doi: 10.3390/ph3082517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sarfaraz S, Afaq F, Adhami VM, Mukhtar H. Cannabinoid Receptor as a Novel Target for the Treatment of Prostate Cancer. Cancer Res. 2005;65:1635–1641. doi: 10.1158/0008-5472.CAN-04-3410. [DOI] [PubMed] [Google Scholar]
- 10.Raitio KH, Salo OMH, Nevalainen T, Poso A, Jaervinen T. Targeting the Cannabinoid CB2 Receptor: Mutations, Modeling and Development of CB2 Selective Ligands. Curr Med Chem. 2005;12:1217–1237. doi: 10.2174/0929867053764617. [DOI] [PubMed] [Google Scholar]
- 11.Muccioli GG, Lambert DM. Current Knowledge on the Antagonists and Inverse Agonists of Cannabinoid Receptors. Curr Med Chem. 2005;12:1361–1394. doi: 10.2174/0929867054020891. [DOI] [PubMed] [Google Scholar]
- 12.Goutopoulos A, Makriyannis A. From cannabis to cannabinergics: new therapeutic opportunities. Pharmacol Ther. 2002;95:103–117. doi: 10.1016/s0163-7258(02)00250-4. [DOI] [PubMed] [Google Scholar]
- 13.Huffman JW. The Search for Selective Ligands for the CB2 Receptor. Curr Pharm Des. 2000;6:1323–1337. doi: 10.2174/1381612003399347. [DOI] [PubMed] [Google Scholar]
- 14.Burns HD, Van Laere K, Sanabria-Bohórquez S, Hamill GT, Bormans G, Eng W-S, Hargreaves RJ, et al. [18F]MK-9470, a positron emission tomography (PET) tracer for in vivo human PET brain imaging of the cannabinoid-1 receptor. PNAS. 2007;104:9800–9805. doi: 10.1073/pnas.0703472104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Donohue SR, Halldin C, Schou M, Hong J, Phebus L, Chernet E, et al. Radiolabeling of a high potency cannabinoid subtype-1 receptor ligand, N-(4-fluoro-benzyl)-4-(3-(piperidin-1-yl)-indole-1-sulfonyl)benzamide (PipISB), with carbon-11 or fluorine-18. J Labelled Compd Radiopharm. 2008;51:146–152. [Google Scholar]
- 16.Liu P, Lin LS, Hamill TG, Jewell JP, Lanza TJ, Jr, Gibson RE, et al. Discovery of N-{(1S,2S)-2-(3-Cyanophenyl)-3-[4-(2-[18F]fluoroethoxy)phenyl]-1-methylpropyl}-2- methyl-2-[(5-methylpyridin-2-yl)oxy]propanamide, a Cannabinoid-1 Receptor Positron Emission Tomography Tracer Suitable for Clinical Use. J Med Chem. 2007;50:3427–3430. doi: 10.1021/jm070131b. [DOI] [PubMed] [Google Scholar]
- 17.Evens N, Muccioli GG, Houbrechts N, Lambert DM, Verbruggen AM, Van Laere K, et al. Synthesis and biological evaluation of carbon-11- and fluorine-18-labeled 2-oxoquinoline derivatives for type 2 cannabinoid receptor positron emission tomography imaging. Nucl Med Biol. 2009;36:455–465. doi: 10.1016/j.nucmedbio.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 18.Evens N, Bosier B, Lavey BJ, Kozlowski JA, Vermaelen P, Baudemprez L, et al. Labelling and biological evaluation of [11C]methoxy-Sch225336: a radioligand for the cannabinoid-type 2 receptor. Nucl Med Biol. 2008;35:793–800. doi: 10.1016/j.nucmedbio.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 19.Raitio KH, Savinainen JR, Vepsalainen J, Laitinen JT, Poso A, Jarvinen T, et al. Synthesis and SAR studies of 2-oxoquinoline derivatives as CB2 receptor inverse agonists. J Med Chem. 2006;49:2022–2027. doi: 10.1021/jm050879z. [DOI] [PubMed] [Google Scholar]
- 20.Gao M, Wang M, Miller KD, Hutchins GD, Zheng Q-H. Synthesis and in vitro biological evaluation of carbon-11-labeled quinoline derivatives as new candidate PET radioligands for cannabinoid CB2 receptor imaging. Bioorg Med Chem. 2010;18:2099–2106. doi: 10.1016/j.bmc.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 21.Wuest F. Aspects of positron emission tomography radiochemistry as relevant for food chemistry. Amino Acids. 2005;29:323–339. doi: 10.1007/s00726-005-0201-1. [DOI] [PubMed] [Google Scholar]
- 22.Horti AG, Gao Y, Ravert HT, Finley P, Valentine H, Wong DF, et al. Synthesis and biodistribution of [11C]A-836339, a new potential radioligand for PET imaging of cannabinoid type 2 receptors (CB2) Bioorg Med Chem. 2010;18:5202–5207. doi: 10.1016/j.bmc.2010.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vandeputte C, Evens N, Toelen J, Deroose CM, Bosier B, Ibrahimi A, et al. A PET Brain Reporter Gene System Based on Type 2 Cannabinoid Receptors. J Nucl Med. 2011;52:1102–1109. doi: 10.2967/jnumed.110.084426. [DOI] [PubMed] [Google Scholar]
- 24.Turkman N, Shavrin A, Ivanov RA, Rabinovich B, Volgin A, Gelovani JG, et al. Fluorinated cannabinoid CB2 receptor ligands: synthesis and in vitro binding characteristics of 2-oxo-quinoline derivatives. Bioorg Med Chem. 2011 doi: 10.1016/j.bmc.2011.07.062. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuhnast B, Dolle F, Terrazzino S, Rousseau B, Loc’h C, Vaufrey F, et al. General Method to Label Antisense Oligonucleotides with Radioactive Halogens for Pharmacological and Imaging Studies. Bioconj Chem. 2000;11:627–36. doi: 10.1021/bc990183i. [DOI] [PubMed] [Google Scholar]
- 26.Dolle F, Hinnen F, Vaufrey F, Tavitian B, Crouzel CA. A general method for labeling oligonucleotides with 18F for in vivo PET Imaging. J Labelled Comp Radiopharm. 1997;39:319–330. [Google Scholar]
- 27.Huffman JW, John Liddle J, Shu Yu S, Aung MM, Abood ME, Wiley JL, et al. 3-(10,10-Dimethylbutyl)-1-deoxy-Δ8-THC and Related Compounds: Synthesis of Selective Ligands for the CB2 Receptor. Bioor Med Chem. 1999;7:2905–2914. doi: 10.1016/s0968-0896(99)00219-9. [DOI] [PubMed] [Google Scholar]