

A large number of existing and candidate pharmaceuticals contain perfluoroalkyl groups because these moieties can favorably affect the physical and biological properties of a compound.[1] Accordingly, the development of methods to introduce perfluoroalkyl groups into aromatic compounds has become increasingly important. To introduce the CF3 moiety, the simplest perfluoroalkyl group, most current industrial methods rely on the Swarts reaction,[2] wherein benzotrichlorides are treated with HF or SbF5 under forcing conditions. While effective for the bulk synthesis of simple commodity chemicals, the harshness of this reaction significantly limits its utility in complex-molecule synthesis, particularly for late-stage introduction of a trifluoromethyl group for studies on structure–activity relationships (SAR). Furthermore, this classical route to trifluoromethylarenes does not provide access to higher-order perfluoroalkylarenes.
In recent years, synthetic methods have been developed for the preparation of perfluoroalkyl arenes from aryl iodides[3] and aryl chlorides.[4] However, analogous methods to prepare these compounds from aryl bromides, which are particularly desirable starting materials, due to their ease of synthesis and wide commercial availability, are notably lacking. Similarly, methods to prepare perfluoroalkyl arenes directly from arenes would be valuable because the halogenation step is avoided altogether. Existing catalytic perfluoroalkylations of arenes require a directing group or occur with low selectivity,[5] and no methods currently exist for the perfluoroalkylation of bromoarenes with a broad substrate scope.[6, 7] Here, we report a general strategy for accessing perfluoroalkyl arenes from arenes and aryl bromides without directing groups, high temperatures, or acidic conditions.
Our strategy for the synthesis of perfluoroalkyl arenes from arenes and aryl bromides, shown in Scheme 1, starts from the formation of an arylboronate ester in situ, either by iridium-catalyzed borylation of arenes or palladium-catalyzed borylation of aryl bromides. The arylboronate ester is then converted to the perfluoroalkylarene by reaction with [(phen)CuRF] (1) in air.[8] These two sequences are complementary because bromination of arenes is typically controlled by the electronic properties of the arene and iridium-catalyzed borylation is controlled by the steric properties of the arene.[9]
Scheme 1.

Copper-mediated trifluoromethylation of arenes and aryl bromides.
The coupling of arylboronic acids with electrophilic sources of CF3 to give benzotrifluorides have been described recently, but arylboronate esters tend to be much less reactive than boronic acids.[10] We hypothesized that [(phen)CuCF3] (1a), a reagent we recently reported,[7] and its higher perfluoroalkyl congeners (1b, 1c) could convert arylboronate esters to the corresponding perfluoroalkylarenes under oxidative Chan–Lam-type conditions. Reactions of this reagent would circumvent the need for excess quantities (2–5 equiv) of TMSCF3 (Ruppert’s reagent) typically used to compensate for the decompostion of CF3−. Compound 1 is simple to use because it is a solid that is commercially available, stable indefinitely under nitrogen, and sufficiently stable to oxygen and moisture that it can be weighed in air.
We initiated our studies by examining conditions for the conversion of 4-fluorophenylboronates to the corresponding benzotrifluoride (Table 1). After surveying a range of bases, solvents and oxidants, we found that reactions conducted in DMF with air as the oxidant in conjunction with one equivalent of KF to activate the boronate ester occurred in higher yields than those conducted with other oxidants we tested. With air and added KF, 77% yield of the desired benzotrifluoride was formed from the pinacolatoboronate ester, as determined by 19F NMR spectroscopy (Table 1, entry 2). Reactions with pre-formed [(phen)CuCF3] occurred in higher yields than those conducted with the reagent generated in situ (entries 1 and 2).
Table 1.
Trifluoromethylation of arylboronate esters.
 | |||
|---|---|---|---|
| Entry | X[a] | Conditions | Yield [%][b] |
| 1 | Bpin | CuI, phen, KOtBu, CF3TMS, KF, air | 49 |
| 2 | Bpin | [(phen)CuCF3], KF, air | 77 |
| 3 | B(OH)2 | [(phen)CuCF3], KF, air | 36 |
| 4 | Bnpg | [(phen)CuCF3], KF, air | 67 |
| 5 | Bcat | [(phen)CuCF3], KF, air | 16 |
| 6 | BMIDA | [(phen)CuCF3], KF, air | 10 |
| 7 | BF3K | [(phen)CuCF3], KF, air | np |
| 8 | Bpin | 20 mol% [(phen)CuCF3] 1.2 equiv CF3TMS, KF, air |
42 |
pin=pinacolato, npg=neopentylglycolate, cat=catecholato, MIDA=N-methylimino diacetate, TMS=trimethylsilyl.
Reactions run on a 0.1 mmol scale; yields determined by 19F NMR spectroscopy with 4-trifluoromethoxyanisole as an internal standard.
In addition to studying the trifluoromethylation of pinacolatoboronate esters, we studied the trifluoromethylation of a variety of less hindered boronate esters and boronic acids. Reactions of boronic acids under the standard conditions gave the corresponding trifluoromethylated product, but the yields were lower than those of the reactions of pinacolatoboronate esters (Table 1, entry 3). Arylboronic acids containing electron-withdrawing substituents on the aryl group did, however, react in higher yields than those with a relatively electron-neutral fluorine substituent (see Supporting Information for additional substrates). Although reactions conducted with catecholboronate esters and trifluoroborate salts did not give benzotrifluoride products in high yield, reactions of the corresponding neopentylglycolboronate ester proceeded in good yield (Table 1, entry 4). Finally, we briefly examined the reaction of the pinacolatoboronate ester with Ruppert’s reagent in the presence of [(phen)CuCF3] as a catalyst (entry 8). Some turnover was observed, but higher yields were observed with [(phen)-CuCF3] as reagent, and the latter conditions were explored further.
Based on these conditions that we developed for the trifluoromethylation of aryl boronate esters, a one-pot sequence for the generation of benzotrifluorides from arenes was developed. This sequence consists of Ir-catalyzed borylation of the arene,[9] followed by removal of the volatile components and subsequent copper-mediated trifluoromethylation with [(phen)CuCF3]. Because the regioselectivity of the iridium-catalyzed borylation of arenes is controlled by steric effects, 1,3-disubstituted arenes generate 1,3,5-trisubstituted arylboronate esters and symmetric 1,2-disubstituted arenes give 1,2,4-trisubstiuted arylboronate esters.[9] Despite the presence of Ir and other by-products from the borylation process, the trifluoromethylation of the boronate ester occurred in the same reaction vessel, and good yields of benzotrifluorides were obtained over the two-step process.
The scope of the trifluoromethylation of arenes is shown in Table 2. Both yields determined by 19F NMR spectroscopy and yields of isolated product are reported; due to the volatility of the perfluoroalkylarene products and their similar low polarity to some of the side products, isolated yields tended to be much lower than the chemical yield determined by 19F NMR spectroscopy. Amines, pyridines, and esters were tolerated by the reaction conditions. Although silyl-protected alcohols were tolerated by the conditions of the Ir-catalyzed borylation, the silicon–oxygen bond was cleaved during the trifluoromethylation step. Aryl halides reacted to form benzotrifluorides by cleavage of a C–H bond, not by cleavage of the carbon–halogen bond. Based on the selectivity for Ir-catalyzed C–H borylation, symmetric 1,2-substitued arenes formed products containing a trifluoromethyl group at the 4-position. The aldehyde function was not tolerated by the conditions of the borylation step, but was tolerated by the conditions of the trifluoromethylation step (see below).
Table 2.
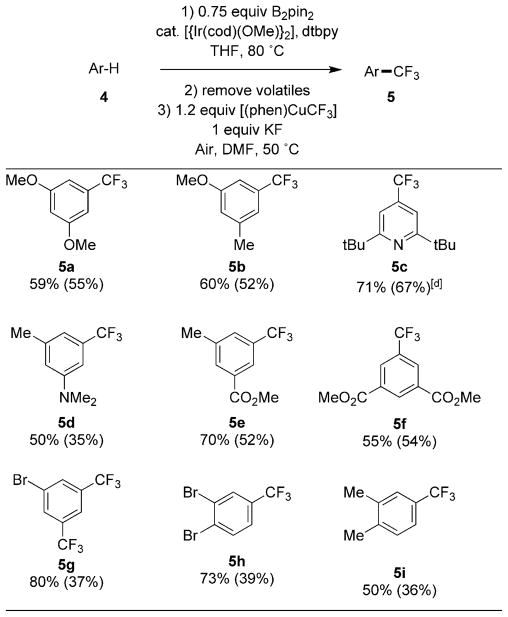
|
Reactions run on a 0.1 mmol scale to determine 19F NMR yields and run on a 0.5 mmol scale to obtain yields of isolated products. 19F NMR yields are listed first followed by yields of isolated products in parenthesis.
Reaction conditions: 0.75 equiv B2pin2, 0.1 mol% [{Ir-(cod)OMe}2] (cod=cyclooctadiene), 0.2 mol% dtbpy (4,4′-di-tert-butyl-2,2′-bipyridyl), 0.5M THF, 80°C.
Yields determined by 19F NMR spectroscopy with 4-trifluoromethoxyanisole as an internal standard. Compounds were isolated by chromatography on silica gel, see Supporting Information for details.
3 mol%[{Ir(cod)OMe}2], 6 mol% dtbpy.
Like the trifluoromethyl moiety, pentafluoroethyl and heptafluoropropyl moieties are valuable for modulating the properties of molecules of interest to the pharmaceutical and agrochemical industries.[1b] However, methods to prepare perfluoroalkyl arenes are limited because they cannot be prepared by halogenation of the corresponding alkylarene.[ 3c,11] Fortunately, perfluoroalkyl analogs of [(phen)-CuCF3], such as [(phen)CuCF2CF3] and [(phen)-CuCF2CF2CF3], are easily prepared.[7] These reagents react with arylboronate esters in a fashion similar to that of [(phen)CuCF3], giving perfluoroalkyl arenes in good yields. Table 3 shows the scope of the one-pot sequence of borylation and perfluoroalkylation. The reaction yields were generally unaffected by the nature of the [(phen)CuRF] reagent; sequences that lead to products of trifluoromethylation, perfluoroethylation and perfluoropropylation occurred in similar yields with the same arene.
Table 3.
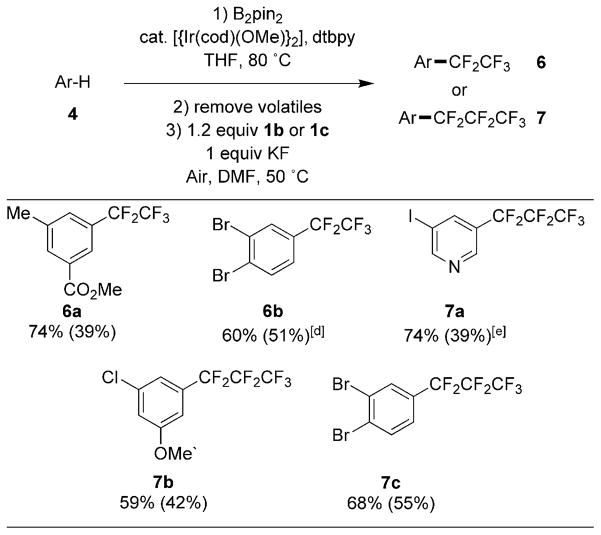
|
Reactions run on a 0.1 mmol scale to determine 19F NMR yields and run on a 0.5 mmol scale to obtain yields of isolated products.
Reaction conditions: 0.75 equiv B2pin2, 0.1 mol% [{Ir(cod)OMe}2], 0.2 mol% dtbpy, 0.5M THF, 80°C.
Yields determined by 19F NMR spectroscopy with 4-trifluoromethoxyanisole as an internal standard. Compounds were isolated by chromatography on silica gel, see the Supporting Information for details.
Approximately 5% 1,2-dibromobenzene from protodeborylation could not be separated from the desired product.
3 mol% [{Ir(cod)OMe}2], 6 mol% dtbpy.
The success of our one-pot sequence consisting of C–H borylation and trifluoromethylation prompted us to investigate the combination of the borylation and trifluoromethylation of aryl bromides. Despite the widespread utility of aryl bromides as building blocks for the synthesis of complex arenes, the development of trifluoromethylations of aryl bromides has been challenging. We anticipated that perfluoroalkyl arenes could be accessed from aryl bromides by first forming the arylboronate ester in situ. We sought to identify conditions for the borylation of aryl bromides that would be compatible with the perfluoroalkylation of arylboronate esters.
After examining several sets of conditions, we found that those of the palladium-catalyzed borylation of aryl bromides reported by Ishiyama, Murata, and Miyaura were most compatible with the subsequent trifluoromethylation.[8d] Three equivalents of KOAc were required for the borylation of the aryl bromide to proceed in high conversion. Unfortunately, the excess KOAc caused the trifluoromethylation step to occur in low yield. However, filtration of the crude reaction mixture through a small plug of Celite before the trifluoromethylation step allowed the formation of the trifluoromethylarene to occur in higher yields.
Several examples of the perfluoroalkylation of aryl bromides are shown in Table 4. Trifluoromethylarenes, pentafluoroethylarenes, and heptafluoropropylarenes were formed in good yield from the arylbromide precursor. Notably, electron-rich arylbromides, which are typically unreactive in other copper-mediated trifluoromethylation methods, gave good yields of the corresponding perfluroalkylarenes. Electrophilic functionality such as nitriles, esters, and aldehydes were tolerated. This method allows access to 1,2-, 1,3-, and 1,4-disubstituted arenes, complementing the selectivity observed for the Ir-catalyzed borylation of arenes (see above).
Table 4.
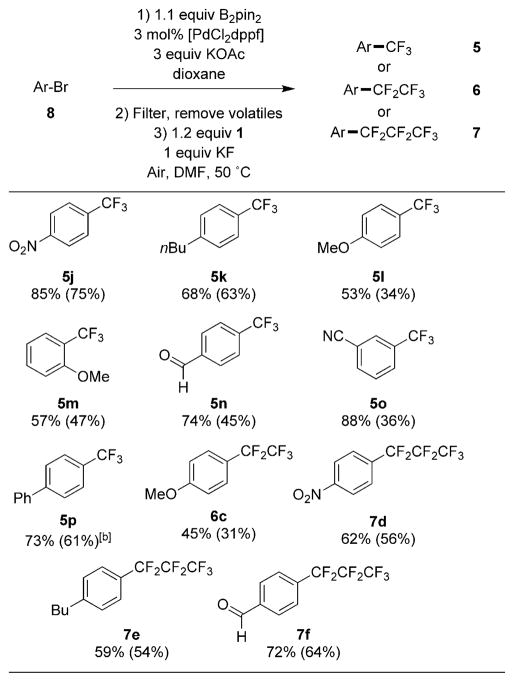
|
Reactions run on a 0.1 mmol scale to determine 19F NMR yields and run on a 0.5 mmol scale to obtain yields of isolated products.
A small amount of homodimer was inseparable from the desired product.
Yields determined by 19F NMR spectroscopy with 4-trifluoromethoxyanisole as an internal standard. Compounds were isolated by chromatography on silica gel, see the Supporting Information for details.
In summary, we have developed a versatile method for the synthesis of perfluoroalkyl arenes from two common classes of aromatic reactants. Disubstituted or trisubstituted arenes and a range of aryl bromides are converted regioselectively to an arylboronate ester in situ, and this ester readily undergoes perfluoroalkylation under mild conditions with a stable, preformed copper reagent. By this sequence, arenes are selectively functionalized with regioselectivity that contrasts that of directed arene perfluoroalkylations. In addition, a variety of aryl bromides are converted to benzotrifluorides for the first time under mild conditions. The two processes we report are complementary because the borylation of arenes is controlled by steric effects and the bromination that forms the aryl bromide reagent is controlled by electronic effects. Both methods are based on recently developed copper reagents that are thermally stable, easily handled solids. Studies to explore methods based on these reagents are ongoing.
Experimental Section
General procedure for one-pot generation of perfluoroalkyl arenes via Ir-catalyzed C–H borylation: In a nitrogen-filled glove box, the arene (0.500 mmol, 1 equiv) and a stock solution of B2pin2, [{Ir-(cod)OMe}2], and dtbpy were combined in a 20 mL vial. The stock solution contained B2pin2 (95.3 mg, 0.375 mmol, 0.75 equiv), [{Ir-(cod)OMe}2] (0.1–3.0 mol%) and dtbpy (0.2–6.0 mol%) per 1 mL of THF (0.5M). The reaction mixture was heated in a sealed vessel at 80°C for 18 h. The dark red solution was then cooled to room temperature, and the volatile materials were evaporated under reduced pressure for 2–4 h. The reaction vessel was returned to the glove box where [(phen)CuCF3] (188 mg, 0.600 mmol, 1.2 equiv) or [(phen)CuCF2CF3] (218 mg, 0.600 mmol, 1.2 equiv) or [(phen)-CuCF2CF2CF3] (248 mg, 0.600 mmol, 1.2 equiv), KF (29.1 mg, 0.500 mmol, 1.0 equiv) and DMF (5 mL, 0.1M) were added, and the vial was sealed with a screw cap fitted with a septum. Outside of the glove box, air from a balloon was bubbled into the DMF solution for 5–10 min. The balloon was removed, and the vial was placed in a 50 °C heating bath. After 18 h, the reaction mixture was cooled to room temperature and diluted with 5 mL of Et2O. The mixture was filtered over Celite, washed with an additional 20 mL of Et2O, and transferred to a separatory funnel. The mixture was washed with 4 × 40 mL of H2O and 1 × 25 mL brine, dried with MgSO4, filtered, and concentrated under vacuum. The crude product was purified by column chromatography on silica gel with pentane or pentane/Et2O mixtures as the eluent.
General procedure for one-pot generation of perfluoroalkyl arenes via palladium-catalyzed C–Br borylation: In a nitrogen-filled glove box, the aryl bromide (0.500 mmol, 1 equiv), B2pin2 (139 mg, 0.550 mmol, 1.1 equiv), KOAc (147 mg, 1.50 mmol, 3 equiv), [PdCl2dppf] (12.2 mg, 0.015 mmol, 3 mol%), and 2.5 mL dioxane (0.2M) were combined in a 20 mL vial. The reaction vessel was sealed with a Teflon-lined cap and heated at 80°C for 18 h, or until the starting material was consumed, as determined by GC-MS. The solution was then allowed to cool to room temperature and filtered through a plug of Celite. The Celite plug was rinsed with a minimal volume of EtOAc (ca. 2 mL), and the resulting solution was concentrated under vacuum for 2–4 h. The reaction vessel was returned to the glove box where [(phen)CuCF3] (188 mg, 0.600 mmol, 1.2 equiv) or [(phen)CuCF2CF3] (218 mg, 0.600 mmol, 1.2 equiv) or [(phen)CuCF2CF2CF3] (248 mg, 0.600 mmol, 1.2 equiv), KF (29 mg, 0.500 mmol, 1.0 equiv) and DMF (5 mL, 0.1M) were added, and the vial was sealed with a screw cap fitted with a septum. Outside of the glove box, air from a balloon was bubbled into the DMF solution for 5–10 min. The balloon was removed, and the vial was placed in a 50 °C heating bath. After 18 h, the reaction mixture was cooled to room temperature and diluted with 5 mL of Et2O. The mixture was filtered over Celite, washed with an additional 20 mL of Et2O, and transferred to a separatory funnel. The mixture was washed with 4 × 40 mL of H2O and 1 × 25 mL brine, dried with MgSO4, filtered, and concentrated under vacuum. The crude product was purified by column chromatography on silica gel with pentane or pentane/Et2O mixtures as the eluent.
Supplementary Material
Footnotes
We thank the NIH for funding (GM-58108 to J.F.H. and 1F32M093540-01 to N.D.L.). Carl Liskey is acknowledged for helpful discussions. phen=phenanthroline; RF=perfluoroalkylated residue
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201106668.
References
- 1.a) Mueller K, Faeh C, Diederich F. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]; b) Purser S, Moore PR, Swallow S, Gouverneur V. Chem Soc Rev. 2008;37:320–330. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]; c) Furuya T, Kamlet AS, Ritter T. Nature. 2011;473:470–477. doi: 10.1038/nature10108. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Schlosser M. Angew Chem. 2006;118:5558–5572. [Google Scholar]; Angew. Chem. Int. Ed. 2006, 45, 5432–5446; e) Tomashenko OA, Grushin VV. Chem Rev. 2011;111:4475–4521. doi: 10.1021/cr1004293. [DOI] [PubMed] [Google Scholar]; f) Roy S, Gregg BT, Gribble GW, Le V-D, Roy S. Tetrahedron. 2011;67:2161–2195. [Google Scholar]
- 2.Swarts F. Bull Acad R Belg. 1892;24:309. [Google Scholar]
- 3.Dubinina GG, Furutachi H, Vicic DA. J Am Chem Soc. 2008;130:8600–8601. doi: 10.1021/ja802946s.Oishi M, Kondo H, Amii H. Chem Commun. 2009:1909–1911. doi: 10.1039/b823249k.Popov I, Lindeman S, Daugulis O. J Am Chem Soc. 2011;133:9286–9289. doi: 10.1021/ja2041942.Knauber T, Arikan F, Röschenthaler GV, Gooßen LJ. Chem Eur J. 2011;17:2689–2697. doi: 10.1002/chem.201002749.Weng Z, Lee R, Jia W, Yuan Y, Wang W, Feng X, Huang KW. Organometallics. 2011;30:3229–3232.Kondo H, Oishi M, Fujikawa K, Amii H. Adv Synth Catal. 2011;353:1247–1252.Tomashenko OA, Escudero-Adán EC, Belmonte MM, Grushin VV. Angew Chem. 2011;123:7797–7801. doi: 10.1002/anie.201101577.Angew Chem Int Ed. 2011;50:7655–7659. doi: 10.1002/anie.201101577.Li Y, Chen T, Wang H, Zhang R, Jin K, Wang X, Duan C. Synlett. 2011:1713–1716.See also ref. [1e].
- 4.Cho EJ, Senecal TD, Kinzel T, Zhang Y, Watson DA, Buchwald SL. Science. 2010;328:1679–1681. doi: 10.1126/science.1190524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Wang X, Truesdale L, Yu JQ. J Am Chem Soc. 2010;132:3648–3649. doi: 10.1021/ja909522s. [DOI] [PubMed] [Google Scholar]; b) Langlois B, Laurent E, Roidot N. Tetrahedron Lett. 1991;32:7525–7528. [Google Scholar]; c) Ji Y, Brueckl T, Baxter RD, Fujiwara Y, Seiple IB, Su S, Blackmond DG, Baran PS. Proc Natl Acad Sci USA. 2011;108:14411–14415. doi: 10.1073/pnas.1109059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For examples of methods which address the trifluoromethylation of bromoarenes, see the following and reference [7]: Chen QY, Wu SW. J Chem Soc Chem Commun. 1989:705–706.Dubinina GG, Brennessel WW, Miller JL, Vicic DA. Organometallics. 2008;27:3933–3938.Samant BS, Kabalka GW. Chem Commun. 2011;47:7236–7238. doi: 10.1039/c1cc12098k.
- 7.Morimoto H, Tsubogo T, Litvinas ND, Hartwig JF. Angew Chem. 2011;123:3877–3882. doi: 10.1002/anie.201100633. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2011;50:3793–3798. doi: 10.1002/anie.201100633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Chen H, Schlecht S, Semple T, Hartwig J. Science. 2000;287:1995–1997. doi: 10.1126/science.287.5460.1995. [DOI] [PubMed] [Google Scholar]; b) Boller T, Murphy J, Hapke M, Ishiyama T, Miyaura N, Hartwig J. J Am Chem Soc. 2005;127:14263–14278. doi: 10.1021/ja053433g. [DOI] [PubMed] [Google Scholar]; c) Ishiyama T, Takagi J, Ishida K, Miyaura N, Anastasi N, Hartwig J. J Am Chem Soc. 2002;124:390–391. doi: 10.1021/ja0173019. [DOI] [PubMed] [Google Scholar]; d) Ishiyama T, Murata M, Miyaura N. J Org Chem. 1995;60:7508–7510. [Google Scholar]
- 9.Marder TB, Mkhalid IAI, Barnard JH, Murphy JM, Hartwig JF. Chem Rev. 2010;110:890–931. doi: 10.1021/cr900206p. [DOI] [PubMed] [Google Scholar]
- 10.a) Chu L, Qing FL. Org Lett. 2010;12:5060–5063. doi: 10.1021/ol1023135. [DOI] [PubMed] [Google Scholar]; b) Senecal TD, Parsons AT, Buchwald SL. J Org Chem. 2011;76:1174–1176. doi: 10.1021/jo1023377. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Liu T, Shen Q. Org Lett. 2011;13:2342–2345. doi: 10.1021/ol2005903. [DOI] [PubMed] [Google Scholar]; d) Xu J, Luo DF, Xiao B, Liu ZJ, Gong TJ, Fu Y, Liu L. Chem Commun. 2011;47:4300–4302. doi: 10.1039/c1cc10359h. [DOI] [PubMed] [Google Scholar]
- 11.Loy RN, Sanford MS. Org Lett. 2011;13:2548–2551. doi: 10.1021/ol200628n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.