

Abstract
Sm proteins form the core of small nuclear ribonucleoprotein particles (snRNPs), making them key components of several mRNA-processing assemblies, including the spliceosome. We report the 1.75-Å crystal structure of SmAP, an Sm-like archaeal protein that forms a heptameric ring perforated by a cationic pore. In addition to providing direct evidence for such an assembly in eukaryotic snRNPs, this structure (i) shows that SmAP homodimers are structurally similar to human Sm heterodimers, (ii) supports a gene duplication model of Sm protein evolution, and (iii) offers a model of SmAP bound to single-stranded RNA (ssRNA) that explains Sm binding-site specificity. The pronounced electrostatic asymmetry of the SmAP surface imparts directionality to putative SmAP–RNA interactions.
Eukaryotic pre-mRNA processing is an intricate cellular task whose many steps include intron excision. This final maturation step occurs in the spliceosome, a large (≈60 S), transiently stable ribonucleoprotein particle that ligates two exons and releases an intron lariat. The major spliceosome contains several small nuclear ribonucleoprotein particles (snRNPs; e.g., U1, U2, and U4/U6⋅U5), and each U snRNP consists of a respective small nuclear RNA (snRNA; e.g., U1, U2) and many proteins (reviewed in refs. 1 and 2). The subset of proteins common to all spliceosomal U snRNPs is the Sm proteins. Discovered as a group of eight small antigens involved in autoimmune diseases such as systemic lupus erythematosus (3), these core snRNP Sm proteins have been found in most eukaryotes (4) and, recently, in a few archaeal species (ref. 5 and this report). Also, several sets of Sm-like (Lsm) proteins have been discovered in organisms with Sm proteins (4, 6). Biochemical characterization of Lsm proteins has verified their similarity to canonical Sm proteins (reviewed in ref. 7), and this report treats them as equivalent.
Together with U snRNAs, canonical Sm proteins form snRNP core complexes. The molecular structure and function(s) of Sm assemblies and Sm–RNA interactions within these core complexes are unknown, but are presumably more generic than those of snRNP-specific proteins. Cytoplasmic Sm proteins associate with exported snRNAs at short single-stranded regions that are usually flanked by stem-loop structures (8, 9). The consensus sequence for the Sm binding site is RAU4–6GR (R = purine), although this selectivity is not very stringent (10, 11). The core complex is thought to be a heteroheptamer of Sm proteins, and electron microscopic investigations of U snRNP core particles suggest that the Sm (12) and Lsm (13) cores are composed of a doughnut-shaped heteromer. The current paradigm is that seven Sm proteins (e.g., human B/B′, D1, D2, D3, E, F, and G) assemble stepwise into a heteroheptameric ring with snRNA through various intermediates, such as D1⋅D2 and E⋅F⋅G heteromers. This snRNP core complex is then imported to the nucleus for completion of spliceosome assembly. Functional complexes of RNA and homologous Sm protein septets, such as the Lsm 1–7 and Lsm 2–8 sets of yeast, are thought to assemble in a similar manner.
There are no atomic resolution structures of snRNP cores, although a recent electron cryomicroscopic study by Stark et al. (14) underscores the importance of Sm proteins in forming the core of the U1 snRNP. Crystal structure determinations of D1⋅D2 and D3⋅B heterodimers by Kambach et al. (15, 16) provide the only known Sm structures, and show that each monomer folds as a strongly bent, five-stranded antiparallel β-sheet. These monomers have nearly identical three-dimensional structures, as do the heterodimers. Kambach et al. used their heterodimer structures to model an Sm heteroheptamer with a positively charged central hole. We now report the 1.75-Å crystal structure of Sm-like archaeal protein (SmAP)—a heptameric Sm protein from the hyperthermophilic archaeon Pyrobaculum aerophilum—and describe the implications of this structure for the Sm core of eukaryotic snRNPs.
Materials and Methods
Protein Preparation and Crystallization.
P. aerophilum SmAP was cloned and over-expressed in Escherichia coli cells. Heat treatment of lysed cells (80°C) was followed by standard chromatographic steps and proteolytic removal of a C-terminal His6 tag. Final purification steps yielded full-length, wild-type SmAP with an appended glycine. Monoclinic crystals of SmAP (space group C2; a = 100.26 Å, b = 95.74 Å, c = 62.16 Å, β = 92.69°; VM = 2.33 Å3/Da for seven monomers per asymmetric unit) were used to solve the structure reported here by multiwavelength anomalous diffraction (MAD) phasing of an iridium derivative (Na3IrCl6).
Data Collection, Phasing, and Refinement.
Synchrotron data were collected at 105 K. Data were processed with denzo/scalepack (17), and MAD phasing proceeded by the usual methods of heavy atom location [shelxd (http://shelx.uni-ac.gwdg.de/SHELX/) and solve (18)], maximum likelihood phase refinement [mlphare (19)], and density modification [dm (20)]. Phase extension to 1.75 Å permitted automated model building for most of the protein with warp/arp (21). Model building and refinement were done in o (22) and cns (23), respectively. Averaging with sevenfold noncrystallographic symmetry led us to conclude that the only significant asymmetry within the heptameric ring is attributable to side-chain rotameric variation (each monomer of the final model was refined independently). Refinement rounds ended with inspection of the model and σA-weighted 2Fo−Fc, Fo−Fc, and simulated annealing omit maps (the latter only as necessary). The final model consists of a SmAP heptamer (seven chains, labeled A–G), 130 waters, and five glycerols. Each monomer is complete except for (i) the absence of 4 (monomer F) to 13 (monomer C) N-terminal residues per monomer, and (ii) an average of 3 residues per monomer that are truncated to either alanine or glycine. Ramachandran plots (procheck, ref. 24) and errat (25) were used for model validation. Structure factors and atomic coordinates have been deposited in the Protein Data Bank (ID code 1I8F).
Sequence and Structure Analyses.
PSI-blast (26) and clustalw (27) were used for database searches and multiple sequence alignments, respectively. Pairwise alignments were calculated by the Smith–Waterman algorithm. Similar protein structures [rms deviation (rmsd) < ≈2.5 Å, e.g., human D2, D3 proteins to SmAP] were easily superimposed with align (28), whereas more dissimilar structures (rmsd > ≈2.5 Å by align, e.g., human B, D1 proteins) were optimally aligned by combinatorial extension (29). Electrostatic and surface area calculations were performed with grasp (30). Figs. 2b, 2c, 3a, and 5 were produced with weblab viewerpro 3.7 (Molecular Simulations); Figs. 3 b and c were produced with setor (31, 32); and Fig. 4 was created in grasp.
Figure 2.

SmAP monomer and dimer structures. (a) A cartoon of the SmAP fold, along with the B4 and B5 strands of the two neighboring monomers. Dashed lines indicate side-chain interactions that supplement the backbone hydrogen bonding of the B4–B5 pairs. (b) A depth-cued illustration of the structure of one of the SmAP monomers (gray) is superimposed on the Cα traces of four aligned structures: human Sm D3 (violet, 1.0 Å rmsd), B (green, 1.1 Å rmsd), D2 (magenta, 1.2 Å rmsd), and D1 (orange, 1.7 Å rmsd). The extensive L4 loop of Sm B has been truncated for clarity, and the segments of strands B4 and B5 that bind adjacent monomers are colored as thick green and red lines as in a. This orientation illustrates the strongly bent five-stranded antiparallel β-sheet that forms the Sm structures. (c) The human D3⋅B heterodimer (cyan⋅orange) is superimposed on an SmAP homodimer (red⋅blue). Asterisks mark the conserved dimer interface, and colored balls give the positions of conserved residues shown in Fig. 1. Note that the archaeal homodimer, taken directly from the SmAP heptamer crystal structure, has essentially the same structure as the human heterodimer (1.4 Å rmsd over main-chain atoms).
Figure 3.

Structure of the SmAP heptamer and the cationic pore. (a) A ribbon diagram of the SmAP heptamer, viewed from the “bottom” face, parallel to the sevenfold axis. (b) The enlarged view of experimental 2Fo−Fc electron density for the Arg-29 side chains that line the pore (green, contoured at 1.2 σ). The anomalous difference density marks the sites of the eight bound iridium ions (indigo, contoured at 3.5 σ). (c) Illustration of the molecular surface for a sagittal section of the heptamer, with the sevenfold axis vertical. Arg-29 side chains are rendered as ball-and-stick models, and reveal that the most constricted section of the hourglass-shaped pore (traced in black) has a diameter of 8.8 Å [heavy, closed arrows ( ) are pore dimensions; light, open arrows (↑) are for the entire heptamer]. Horizontal bars indicate the concentric charged rings that are discussed in the text (red = anionic; blue = cationic).
Figure 4.

Electrostatic properties of the SmAP heptamer surface. The molecular surfaces of both faces of the SmAP heptamer are shown color-coded by electrostatic potential (red = −9.3 kT; blue = +10.9 kT). Note that there are two distinctive features: a ring of strong positive potential that lines the heptamer pore toward the bottom face, and a diffuse zone of intense negative potential across most of the top face. Such pronounced charge asymmetry gives the heptameric disk a large dipole moment (calculated to be 553 debye).
The SmAP⋅ssRNA (Single-Stranded RNA) Model.
The SmAP⋅ssRNA model was constructed by manual docking of an ssRNA containing an Sm consensus sequence (GAU4GA) through the pore of the refined SmAP heptamer, and did not require any alteration of the SmAP structure. Adjustment of only four phosphate backbone α torsion angles near the poly (U) tract sufficiently extended the RNA so that it was easily accommodated in the SmAP pore; moreover, the values were not adjusted drastically—from α ≈ −50° in A-form RNA to α ≈ −30° in the extended form shown in Fig. 5. The planes of the first and last uracil bases threaded through the pore are separated by roughly 17 Å.
Figure 5.

Model of a SmAP⋅ssRNA complex. Orthogonal views are shown for a model of SmAP bound to a hypothetical 20-nucleotide ssRNA (e.g., eukaryotic snRNA) that consists of three segments from 5′ to 3′: a random string of 6 nucleotides in the A-form conformation (ACGAUC), followed by a minimal consensus Sm binding site (GAU4GA), and ending with 6 more nucleotides in A-form geometry (ACGAUC). The SmAP heptamer is depicted as a ribbon diagram along with the solvent-accessible surface. The Arg-29 ring that forms the pore is colored by atom type and rendered in space-filling form, with the ssRNA drawn as a stick model. The steric and electrostatic environment of the pore is ideally suited to accommodate a single-stranded polypyrimidyl nucleic acid.
Results
A Family of SmAPs.
The occurrence of Sm and Sm-like proteins is not limited to eukaryotes. Along with P. aerophilum SmAP, we have uncovered several archaeal Sm sequences within the genomes of Pyrococcus abyssi, Aeropryum pernix, Thermoplasma acidophilum, and a halobacterium (Fig. 1). Added to the initial list of five SmAPs reported by Salgado-Garrido et al. in 1999 (5), these archaeal sequences clearly form a well defined protein family that may be ancestral to modern eukaryotic Sm proteins. On the basis of the structure described here, we propose that P. aerophilum SmAP is a representative member of such a family of Sm-like archaeal proteins (SmAPs).
Figure 1.

Sequence analysis of SmAPs. A multiple sequence alignment of SmAPs is shown, along with the sequence of the most similar known Sm structure (human D3) as a reference point (residue numbering is for the P. aerophilum sequence). Pairwise sequence similarity scores between SmAP and its homologs are provided in the last column. The top diagram depicts the SmAP monomer in terms of secondary structure elements. Glycine (yellow), acidic (red), and basic (blue) residues are highlighted in the consensus sequence and are referred to in the text and in Fig. 2c.
SmAP Monomer and Dimer Structures.
The crystal structure of SmAP was determined by MAD phasing (Table
1), and reveals that this archaeal
protein—like the human Sm B, D1,
D2, and D3 monomers—folds
as a strongly bent, five-stranded, antiparallel β-sheet capped by an
N-terminal α-helix (Fig. 2
a
and b). This protein fold strongly resembles members of the
oligonucleotide/oligosaccharide-binding fold family (33). The
β-strands B2, B3, and B4 create this
peculiar structure by twisting back upon themselves to form a U-shaped
core that is roughly elliptical in cross section. The amount of
curvature may be measured as the distance between the N and C termini
of a strand. As an example of the SmAP curvature, the distance between
the B2 strand termini (C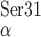 and
C
and
C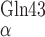 ) is 23.7 Å, whereas this distance would be
39.7 Å in an un-bent, fully-extended conformation. The structural
plasticity needed for such a high degree of β-strand curvature is
apparently provided by several strictly conserved glycines that occur
near the pivot points (Gly36, Gly64, Gly70 in the archael Sm sequences
shown in Fig. 1). The segment linking B4 and B5
lies at the top of the U-shaped trough to close the protein into a
β-barrel-like structure (Fig. 2b); it also positions
B5 for antiparallel hydrogen bonding to B1 within
the same monomer and the B4 strand of an adjacent
monomer (Fig. 2a).
) is 23.7 Å, whereas this distance would be
39.7 Å in an un-bent, fully-extended conformation. The structural
plasticity needed for such a high degree of β-strand curvature is
apparently provided by several strictly conserved glycines that occur
near the pivot points (Gly36, Gly64, Gly70 in the archael Sm sequences
shown in Fig. 1). The segment linking B4 and B5
lies at the top of the U-shaped trough to close the protein into a
β-barrel-like structure (Fig. 2b); it also positions
B5 for antiparallel hydrogen bonding to B1 within
the same monomer and the B4 strand of an adjacent
monomer (Fig. 2a).
Table 1.
Crystallographic statistics
| Data collection & MAD phasing
|
Model
refinement
|
|||||
|---|---|---|---|---|---|---|
| Native | Inflection | Peak | Low-λ remote | |||
| Wavelength, Å | 0.9794 | 1.1058 | 1.1055 | 1.0960 | Resolution range, Å | 20–1.75 |
| Resolution range, Å | 100–1.71 | 100–1.95 | 100–1.95 | 100–1.95 | No. of protein atoms | 3.796 |
| Completeness, % | 98.5 | 97.3 | 97.0 | 95.0 | No. of solvent molecules (H2O/glycerol) | 130/5 |
| I/σ(I) | 44.4 | 24.3 | 25.2 | 22.1 | Rcryst/Rfree, %§ | 23.5/26.6 |
| Rmerge, %* | 4.0 | 4.8 | 4.8 | 4.0 | 〈B-factor〉 protein, Å2 | 38.5 |
| # Ir sites/asymmetric unit | — | 8 | — | rmsds: bonds, Å | 0.018 | |
| Phasing resolution range, Å | 20–2.0 | 20–2.0 | 20–2.0 | angles, ° | 1.90 | |
| Rcullis†: acentric | — | 0.71/0.42 | 0.83/0.70 | |||
| centric | — | 0.64 | 0.82 | |||
| Figure of merit‡ | 0.65/0.82 | |||||
Rmerge(I) = Σhkl((Σi|Ihkl,i − 〈Ihkl〉|)/ΣiIhkl,i).
Rcullis = (Σhkl∥FPH ± FP| − FH,calc|)/Σhkl|FPH± FP|. Statistics for acentric reflections are given as isomorphous/anomalous.
Values are given before/after density modification.
Rcryst = Σhkl∥Fobs| − |Fcalc∥/Σhkl|Fobs|. Rfree was computed identically, except that 5% of the reflections were omitted as a test set.
This latter interaction—hydrogen bonding between strands B4i⋅⋅⋅B5i+1 for each monomer i—creates the seven dimer interfaces that orient the monomers head-to-tail around the ring. Specifically, each monomer contributes five residues from each B4 and B5 strand (Fig. 2a) to create an interface that occludes 1,682 ± 78 Å2 of surface area per dimer (Fig. 2c). The interfacial residues are predominantly apolar, and a few interesting side-chain interactions supplement the standard hydrogen bonds between the β-strand backbones. Such interactions include: (i) the guanidinium group of Arg-69B4 in most of the monomers engages in several hydrogen bonds with main-chain and side-chain atoms from the adjacent B5 strand, and (ii) van der Waals contact of the sulfur of Met-66B4 with the aromatic ring of Phe-75B5 suggests that a favorable S⋅⋅⋅π aromatic interaction (34, 35) may stabilize the interface.
The structural superimpositions shown in Fig. 2 illustrate the strong similarity between SmAP and human Sm monomers and dimers. SmAP aligns best with Sm D3 (31% sequence similarity, 1.0 Å rmsd over backbone atoms), and least well with D1 (40% sequence similarity, 1.7 Å rmsd). The twisted β-sheet core of each Sm monomer is nearly identical (Fig. 2b), the main difference being the shorter L4 loop of SmAP versus three of the human structures (B, D1, and D3). A similar trend toward shorter loops has also been found in other pairs of thermophilic-mesophilic proteins (see ref. 35 and references therein). Both D3⋅B and D1⋅D2 human heterodimer structures align very closely with a SmAP homodimer (1.4 Å and 1.6 Å rmsd, respectively). Superimposition of D3⋅B on SmAP shows that the dimer interface is conserved (Fig. 2c), although the amino acids in this region (B4 residues 65–69 and B5 residues 74–78) show greater phylogenetic variation than in the rest of the sequence (Fig. 1). This variation is explained by the fact that the dimer interface consists mainly of hydrogen bonds between the backbones of the β-strands. In addition to showing that human and archaeal Sm proteins belong to the same fold family, the SmAP structure is apparently an example of an ancestral homodimeric interface that evolved into several distinct, functional heterodimeric interfaces (D3⋅B, D1⋅D2, etc. interfaces in human).
The SmAP Heptamer.
The heptameric organization of SmAP shown in Fig. 3 was revealed by the binding of iridium ions (used for phasing purposes, Fig. 3b) and by in vitro biophysical characterization (C.M. and D.E., unpublished results). The refined crystallographic model reveals a disk-shaped homoheptameric ring that measures ≈65 Å in diameter and ≈38 Å in height (Fig. 3c), which is consistent with the dimensions from electron microscopy of human Sm cores (12). One of the most notable features of the heptamer is the propagation of the β-sheet core of individual monomers across the ring to give a circular, 35-stranded β-sheet. This extended β-sheet does not lie in a single plane because of the strong curvature of the constituent strands. Rather, the U-shaped monomers are arranged like the blades of a turbine, rotated by ≈45° out of the horizontal plane of the heptamer. In contrast to the model proposed for the human heptamer (15), the remarkable asymmetry in the electrostatic surface of the SmAP heptamer gives the disk a large dipole moment (Fig. 4). This feature may be of functional relevance because it imparts directionality to putative SmAP–RNA interactions.
Discussion
The Cationic Pore.
Interactions of SmAP with RNA (or DNA) are likely to occur within the shallow hourglass-shaped pore that runs along the sevenfold axis (Fig. 3 a and c). This cationic pore is ≈15 Å in depth and has a minimum diameter of 8.8 Å, the most constricted region being formed by the Arg-29 side chains of loop L2. The pore tapers steeply away on both sides of the narrow ring of arginines so that no other residues can be said to line the pore. Fig. 3c shows that four layers of charged heptagonal rings lie near to or within the pore. Starting at the bottom face of the heptamer, there is a negatively charged ring of glutamic acid residues (Glu-71, 18-Å diameter) followed by the narrow cationic ring of arginine residues (Arg-29, 8.8-Å diameter); a larger ring of aspartic acid residues (Asp-30, 11.5-Å diameter) is slightly above this; finally, there is a much wider ring of aspartic acid residues at the top face of the heptamer (Asp-57, 28.8-Å diameter). Note that unproductive SmAP⋅ssRNA bonding (i.e., interactions outside of the pore) would be diminished by such an arrangement of anionic rings.
Strict conservation of Arg-29 in archaeal Sm homologs (Fig. 1) suggests that a cationic pore with similar properties exists in all archaeal Sm heptamers (this arginine is also conserved in the human D3 protein). By superimposing the human heterodimers on the SmAP heptamer, we find that the cationic character of the SmAP pore is probably conserved in the human Sm heptamer. Several positively charged residues from the human proteins would lie near the SmAP pore (although not in exactly the same location as the Arg-29 ring), including Arg-51 and Lys-67 from D3, and Lys-54 from the B monomer.
The aforementioned residues that form the pore and charged heptagonal rings all lie in loops L2, L4, and L5. However, recent experiments by Urlaub et al. (37) demonstrate that residues from loop L3 in human Sm B and G proteins can crosslink with an RNA nonanucleotide containing the Sm binding site (AAU5GA). Assuming that snRNA binds in the pore in vivo, these results are not consistent with the relative locations of the pore and loop L3 in SmAP, and suggest that either (i) the human Sm monomers adopt altered conformations in the heteroheptamer to position the L3 loops more proximal to the pore, or (ii) the RNA nonanucleotide used by Urlaub et al. binds near the pore, but not within it.
Elucidation of Sm Binding Site Specificity.
Notably, the SmAP heptamer structure offers a simple explanation for
the specificity of the snRNA Sm binding site
(RAU4–6GR). The depth of the pore varies
depending on exactly how it is defined, and a lower bound estimate of
the pore depth (15 Å) is shown in Fig. 3c. More
importantly, the aperture of the cationic Arg-29 ring is clearly
defined in electron density (Fig. 3b) and gives the pore a
minimum diameter of 8.8 Å. Fig. 5 shows
a model of the SmAP heptamer complexed with a 20-nucleotide ssRNA that
contains the consensus Sm binding site. The minimal diameter of the
pore is ideally suited for binding of a single strand of polypyrimidyl
RNA, but is too narrow to accommodate a polypurine, thereby affording
steric discrimination between polypyrimidine and polypurine tracts. At
least four or five nucleotides are required to span the ≈16 Å length
of the pore, thus explaining the minimum requirement of four to five
uridines in the Sm binding site. A more difficult matter to explain is
the selection of poly(U) versus the other possible polypyrimidine,
poly(C). It is possible that the cationic pore favors poly(U) because
it is a better hydrogen-bond acceptor than the slightly more
electropositive poly(C). Most importantly, the eighth iridium peak in
anomalous difference Fourier maps is located in the pore, ≈3 Å below
the cationic Arg-29 plane (Fig. 3b). This peak proves that
the pore is accessible to polyvalent anions (e.g., octahedral
IrCl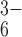 ) by an approach from the bottom face of the
heptamer.
) by an approach from the bottom face of the
heptamer.
The Sm Core of Eukaryotic snRNPs.
The crystal structure of SmAP and the model of it bound to an snRNA Sm binding site have several implications for the Sm core of eukaryotic snRNPs. We note that there are two possible paths by which SmAP may bind snRNA. Either the RNA is threaded through the pore (from the bottom face), or the heptamer assembles onto the snRNA binding site. The SmAP structure shows that the latter scenario is more plausible because purine bases cannot fit in the Sm pore. In vitro evidence for stable, subheptamers of human Sm proteins (e.g., an E⋅F⋅G heterotrimer, ref. 8), along with the fact that a minimal complex of five Sm proteins will bind RNA, lends credence to this stepwise core snRNP assembly pathway.
The regulation of such an assembly pathway could be achieved by noting the recent finding by Brahms et al. (32) that the C-terminal Arg-Gly dipeptides of human Sm proteins D1 and D3 are symmetrically dimethylated on the arginine η1 and η2 nitrogens in vivo. Fig. 1 shows that a C-terminal Arg-69–Gly-70 dipeptide of SmAP is strictly conserved among archaeal Sm proteins and aligns with one of the Arg-Gly dipeptides of human D3 protein. Inspection of the structural environment of Arg-69 in the human and SmAP structures shows that it lies at the Sm dimer interface, where it makes many contacts (Fig. 2c). Dimethylation of Arg-69 guanidinium groups by a protein arginine methyltransferase is predicted to interfere sterically with this interface, preventing Sm heptamer formation. Therefore, the structure of SmAP leads us to propose that regulation of the snRNP core assembly may be achieved by arginine dimethylation.
Because SmAP is a homoheptamer, our results support a mechanism of modern eukaryotic Sm protein evolution by early gene duplication events. Presumably, an archaeal SmAP gene was duplicated and gradually accrued neutral point mutations, with the structural restraint that the independently evolving monomers still form a heteroheptamer. Such a scheme would give rise to modified/modern Sm heteroheptamers, such as the human B⋅D1⋅D2⋅D3⋅E⋅F⋅G heptamer, with modified/modern biochemical activities, such as U snRNA binding. This model implies asymmetric heteroheptamers composed of paralogous Sm proteins, and explains the diversification of Sm protein function. Our preliminary crystallographic results with another archaeal Sm protein suggest that it also assembles into a homoheptamer, thereby supporting this model. Finally, we note that these results raise several interesting questions regarding the potential RNA that P. aerophilum SmAP interacts with in vivo, the cellular role of this putative RNA binding, and the possibility of snRNP-like particles in archaeal species.
Acknowledgments
We thank Dr. Kym Faull for help with mass spectrometry, and Adam Frankel, Linda Columbus, Drs. Dan Anderson, Doug Black, Jim Bowie, Guillaume Chanfreau, Sorel Fitz-Gibbon, Celia Goulding, and Ioannis Xenarios for useful discussions. We also thank Brookhaven National Lab for the use of beamline X8C of the National Synchrotron Light Source. We gratefully acknowledge financial support from the National Institutes of Health, the Department of Energy, and a National Science Foundation predoctoral fellowship (to C.M.).
Abbreviations
- snRNP
small nuclear ribonucleoprotein
- snRNA
small nuclear RNA
- Lsm
Sm-like
- SmAP
Sm-like archaeal protein
- MAD
multiwavelength anomalous diffraction
- ssRNA
single-stranded RNA
- rmsd
root mean square deviation
Footnotes
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code 1I8F).
References
- 1.Yu Y-T, Scharl E C, Smith C M, Steitz J A. In: The RNA World. Gesteland R F, Cech T R, Atkins J F, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1999. pp. 487–524. [Google Scholar]
- 2.Burge C B, Tuschl T, Sharp P A. In: The RNA World. Gesteland R F, Cech T R, Atkins J F, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1999. pp. 525–560. [Google Scholar]
- 3.Lerner M R, Steitz J A. Proc Natl Acad Sci USA. 1979;76:5495–5499. doi: 10.1073/pnas.76.11.5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seraphin B. EMBO J. 1995;14:2089–2098. doi: 10.1002/j.1460-2075.1995.tb07200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salgado-Garrido J, Bragado-Nilsson E, Kandels-Lewis S, Seraphin B. EMBO J. 1999;18:3451–3462. doi: 10.1093/emboj/18.12.3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bouveret E, Rigaut G, Shevchenko A, Wilm M, Seraphin B. EMBO J. 2000;19:1661–1671. doi: 10.1093/emboj/19.7.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He W, Parker R. Curr Opin Cell Biol. 2000;12:346–350. doi: 10.1016/s0955-0674(00)00098-3. [DOI] [PubMed] [Google Scholar]
- 8.Raker V A, Plessel G, Luhrmann R. EMBO J. 1996;15:2256–2269. [PMC free article] [PubMed] [Google Scholar]
- 9.Mattaj I W. Cell. 1986;46:905–911. doi: 10.1016/0092-8674(86)90072-3. [DOI] [PubMed] [Google Scholar]
- 10.Jones M H, Guthrie C. EMBO J. 1990;9:2555–2561. doi: 10.1002/j.1460-2075.1990.tb07436.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raker V A, Hartmuth K, Kastner B, Luhrmann R. Mol Cell Biol. 1999;19:6554–6565. doi: 10.1128/mcb.19.10.6554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kastner B, Bach M, Luhrmann R. Proc Natl Acad Sci USA. 1990;87:1710–1714. doi: 10.1073/pnas.87.5.1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Achsel T, Brahms H, Kastner B, Bachi A, Wilm M, Luhrmann R. EMBO J. 1999;18:5789–5802. doi: 10.1093/emboj/18.20.5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stark H, Dube P, Luhrmann R, Kastner B. Nature (London) 2001;409:539–542. doi: 10.1038/35054102. [DOI] [PubMed] [Google Scholar]
- 15.Kambach C, Walke S, Young R, Avis J M, de la Fortelle E, Raker V A, Luhrmann R, Li J, Nagai K. Cell. 1999;96:375–387. doi: 10.1016/s0092-8674(00)80550-4. [DOI] [PubMed] [Google Scholar]
- 16.Kambach C, Walke S, Nagai K. Curr Opin Struct Biol. 1999;9:222–230. doi: 10.1016/s0959-440x(99)80032-3. [DOI] [PubMed] [Google Scholar]
- 17.Otwinowski Z, Minor W. Methods Enzymol. 1997;276A:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 18.Terwilliger T C, Berendzen J. Acta Crystallogr D. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Collaborative Computational Project No. 4. Acta Crystallogr D. 1994;50:760–763. [Google Scholar]
- 20.Cowtan K, Main P. Acta Crystallogr D. 1998;54:487–493. doi: 10.1107/s0907444997011980. [DOI] [PubMed] [Google Scholar]
- 21.Perrakis A, Morris R, Lamzin V S. Nat Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 22.Jones T A, Zou J Y, Cowan S W, Kjeldgaard Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 23.Brunger A T, Adams P D, Clore G M, DeLano W L, Gros P, Grosse-Kunstleve R W, Jiang J S, Kuszewski J, Nilges M, Pannu N S, et al. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 24.Laskowski R A, Moss D S, Thornton J M. J Mol Biol. 1993;231:1049–1067. doi: 10.1006/jmbi.1993.1351. [DOI] [PubMed] [Google Scholar]
- 25.Colovos C, Yeates T O. Protein Sci. 1993;2:1511–1519. doi: 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Altschul S F, Madden T L, Schaffer A A, Zhang J, Zhang Z, Miller W, Lipman D J. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Higgins D G, Thompson J D, Gibson T J. Methods Enzymol. 1996;266:383–402. doi: 10.1016/s0076-6879(96)66024-8. [DOI] [PubMed] [Google Scholar]
- 28.Cohen G H. J Mol Biol. 1986;190:593–604. doi: 10.1016/0022-2836(86)90245-7. [DOI] [PubMed] [Google Scholar]
- 29.Shindyalov I N, Bourne P E. Protein Eng. 1998;11:739–747. doi: 10.1093/protein/11.9.739. [DOI] [PubMed] [Google Scholar]
- 30.Nicholls A, Sharp K A, Honig B. Proteins Struct Funct Genet. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 31.Evans S V. J Mol Graph. 1993;11:134–138. doi: 10.1016/0263-7855(93)87009-t. [DOI] [PubMed] [Google Scholar]
- 32.Brahms H, Raymackers J, Union A, de Keyser F, Meheus L, Luhrmann R. J Biol Chem. 2000;275:17122–17129. doi: 10.1074/jbc.M000300200. [DOI] [PubMed] [Google Scholar]
- 33.Murzin A G. EMBO J. 1993;12:861–867. doi: 10.1002/j.1460-2075.1993.tb05726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reid K S C, Lindley P F, Thornton J M. FEBS Lett. 1985;190:209–213. [Google Scholar]
- 35.Druhan L J, Swenson R P. Biochemistry. 1998;37:9668–9678. doi: 10.1021/bi980770b. [DOI] [PubMed] [Google Scholar]
- 36.Thompson M J, Eisenberg D. J Mol Biol. 1999;290:595–604. doi: 10.1006/jmbi.1999.2889. [DOI] [PubMed] [Google Scholar]
- 37.Urlaub H, Raker V A, Kostka S, Luhrmann R. EMBO J. 2001;20:187–196. doi: 10.1093/emboj/20.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]