

Abstract
In the AAA+ HslUV protease, substrates are bound and unfolded by a ring hexamer of HslU, before translocation through an axial pore and into the HslV degradation chamber. Here, we show that the N-terminal residues of an Arc substrate initially bind in the HslU axial pore, with key contacts mediated by a pore loop that is highly conserved in all AAA+ unfoldases. Disordered loops from the six intermediate domains of the HslU hexamer project into a funnel-shaped cavity above the pore and are positioned to contact protein substrates. Mutations in these I-domain loops increase KM and decrease Vmax for degradation, increase the mobility of bound substrates, and prevent substrate stimulation of ATP hydrolysis. HslU-ΔI has negligible ATPase activity. Thus, the I domain plays an active role in coordinating substrate binding, ATP hydrolysis, and protein degradation by the HslUV proteolytic machine.
Keywords: AAA+ machine, ATP-dependent degradation, HslUV, substrate binding
Introduction
Energy-dependent proteolysis is a key process in sculpting the proteomes of cells from all kingdoms of life. Degradation can clear damaged or misfolded proteins, remove superfluous or unneeded proteins following a shift in growth conditions or developmental programs, and play important roles in regulatory circuits that drive the cell cycle or mediate transcriptional responses to environmental stress.1, 2 The ATP-fueled proteases that execute these processes must be highly specific to avoid degradation of essential proteins. They also provide paradigms for a wide range of molecular machines that perform mechanical tasks in intracellular settings.
Escherichia coli contains five ATP-dependent proteases: HslUV, ClpXP, ClpAP, Lon, and FtsH.2 Related proteases, including the 26S proteasome, are found in most eubacteria and archaebacteria, in mitochondria and chloroplasts, and in the cytoplasm of eukaryotic cells.3 Each of these multi-subunit proteases contains a hexameric AAA+ enzyme, which functions to recognize, unfold, and translocate specific target proteins into the degradation chamber of a self-compartmentalized peptidase. In HslUV, for example, the HslU hexamer serves as the AAA+ protein unfoldase/translocase, whereas HslV forms a double-ring dodecamer that encloses the proteolytic compartment. Like its AAA+ relatives, HslU contains large and small AAA+ domains that couple ATP binding and hydrolysis to the conformational changes that drive substrate unfolding and translocation. These mechanical processes are thought to occur via ATP-powered movements of loops that project into the axial pore of the hexamer. In HslU, ClpX, and ClpA, these loops contain a highly conserved GYVG sequence, which appears to contact some substrates and to play roles in translocation and unfolding.4–8
Substrates are typically targeted to specific AAA+ proteases by peptide sequences.2 For example, the ssrA-tag sequence binds in the axial pores of ClpX and ClpA, resulting in degradation of ssrA-tagged substrates by ClpXP or ClpAP.4, 6, 7–9 Other peptide sequences tether substrates or adaptor proteins to family specific auxiliary domains in the AAA+ hexamer. For instance, the N domain of ClpX binds to a tethering sequence in the UmuD/D′ protein, helping to mediate ClpXP degradation.10 Our understanding of substrate recognition by HslUV is rudimentary. Studies of a handful of natural or model HslUV substrates show that peptide sequences are important determinants of targeting for degradation,5, 11–16 but how any protein substrate interacts with HslU is poorly understood.
Numerous crystal structures of HslU and HslUV have been solved, including the first views of a AAA+ ring unfoldase in complex with its self-compartmentalized peptidase.12, 17–20 Packing between highly conserved large and small AAA+ domains of neighboring subunits stabilizes the HslU hexameric ring. In addition, an intermediate (I) domain, which is only found in the HslU family, is inserted between two neighboring helices of the large AAA+ domain. One of these helices follows the GYVG loop and the other precedes the Walker-B motif, which plays important roles in ATP hydrolysis. The I domains project upward from the top surface of the HslU ring, forming a funnel-shaped cavity above the axial pore. However, the role of the I domain in HslU function is currently unclear.
One of the few well-characterized HslUV substrates is Arc repressor, a dimeric protein with disordered N-terminal residues that target it to HslU.13, 14, 16, 21, 22 Here, we probe the interaction of Arc substrates with HslU variants bearing mutations in the GYVG pore loop or the I domain. Our results support a model in which N-terminal residues of Arc initially interact with the GYVG loop in the axial pore of HslU, while other portions of Arc contact disordered I-domain loops (residues 175–209) that project into the substrate-binding funnel above the pore. The I-domain interactions constrain the mobility of enzyme-bound Arc, facilitate efficient degradation, and are required for substrate stimulation of ATP hydrolysis. Surprisingly, we discovered that deleting the I domain reduced the basal level of ATP hydrolysis ∼50-fold. In combination, these results indicate that the I domain plays an active role in coordinating substrate binding, protein degradation, and ATP hydrolysis.
Results
Effects of HslU pore-loop mutations on substrate binding and degradation
ATP-dependent unfoldases contain highly conserved axial-pore loops, which have been implicated in substrate binding, unfolding, and translocation.7, 8, 23 In previous studies of the HslU GYVG pore loop,5 the Y91F mutation (GFVG) was shown to slow degradation of an MBP-SulA fusion protein as assayed by SDS-PAGE, whereas the Y91A mutation (GAVG) prevented degradation; both mutants hydrolyzed ATP and stimulated HslV peptidase activity. Using 35S-labeled Arc-st11-ssrA as a substrate,16 we monitored degradation by wild-type HslUV, Y91F HslUV, and Y91A HslUV [Fig. 1(A); Table I]. At each substrate concentration tested, degradation by wild-type HslUV was slightly faster than degradation by Y91F HslUV. Vmax was 11 ± 0.8 min−1 enz−1 for the wild-type enzyme and 8.7 ± 0.1 min−1 enz−1 for Y91F; KM values for the wild-type and Y91F enzymes were 20 ± 2 μM and 26 ± 1 μM, respectively (Table I). Y91A HslUV was largely inactive in degradation [Fig. 1(A)], with a second-order rate constant ∼280-fold smaller than for wild-type HslUV degradation. Y91A and Y91F HslU hydrolyzed ATP at rates somewhat faster than wild-type HslU and stimulated HslV to comparable levels of peptidase activity (Table I). Thus, as expected,5 the degradation defects of the HslU pore mutants are not caused by their inability to form hexamers, hydrolyze ATP, or interact with HslV.
Figure 1.
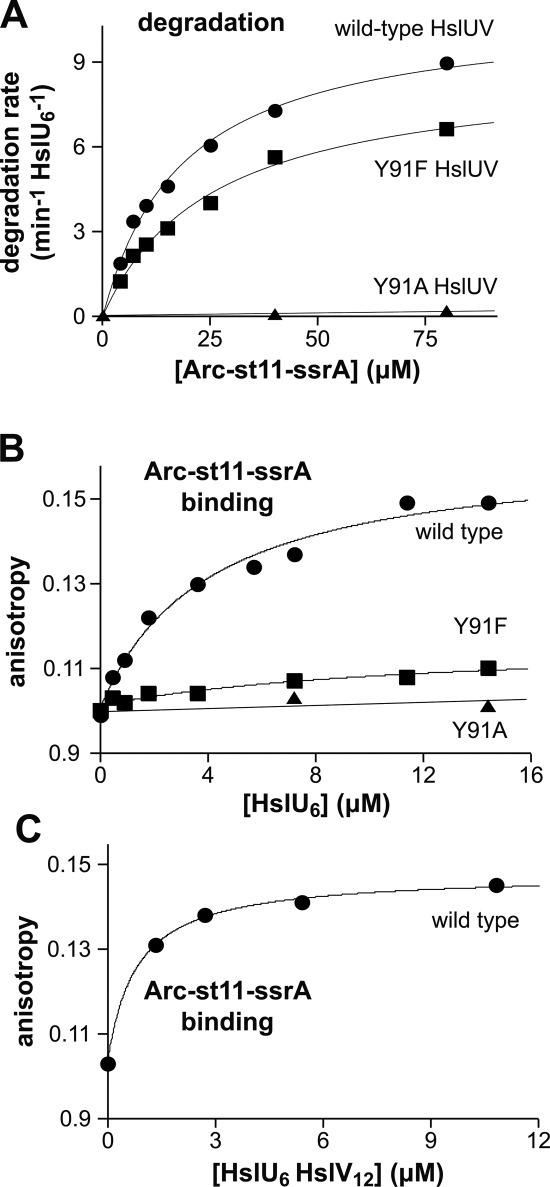
Functional properties of HslU pore-loop mutants. A: Steady-state kinetics of Arc-st11-ssrA degradation by wild-type (WT), Y91F, and Y91A HslUV (100 nM HslU6; 300 nM HslV12). For WT and Y91F, the lines are fits to the Michaelis–Menten equation: KM = 18.7 μM, Vmax = 10.8 min−1 enz−1 (WT); KM = 25.8 μM, Vmax = 8.8 min−1 enz−1 (Y91F). Average values from multiple experiments are listed in Table I. For Y91A, the line is a linear fit. B: Binding of wild-type, Y91F, and Y91A HslU to fluorescent Arc-st11-ssrA (0.8 μM) in the presence of 3 mM ATPγS. For WT and Y91F, the lines are fits to the equation b + a*(1+KD/[HslU6])−1: b = 0.10, a = 0.061; KD = 4.0 μM (WT); b = 0.10, a = 0.015; KD = 12 μM (Y91F). Average KD values from multiple experiments are listed in Table I. For Y91A, the line is a linear fit. C: Increasing wild-type HslU6 was titrated against fluorescent Arc-st11-ssrA (0.8 μM) in the presence of 10.8 μM HslV12 and 3 mM ATPγS. The line is a fit to a quadratic form of the equation in panel B to account for near stoichiometric binding: b = 0.10, a = 0.045; KD = 0.46 μM.
Table I.
Functional Parameters
| HslUV degradation of Arc-st11-ssrA | Arc-st11-ssrA binding | gt1 peptide binding | Basal ATP hydrolysis | Activation of HslV peptidase activity | ||
|---|---|---|---|---|---|---|
| HslU variant |
Vmax (min−1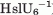 ) ) |
KM (μM) | KD (μM) | KD (μM) | min−1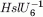
|
min−1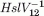
|
| wild type | 11 ± 0.8 | 20 ± 2.2 | 3.8 ± 1.2 | 0.91 ± 0.29 | 130 ± 23 | 120 ± 20 |
| Y91F | 8.7 ± 0.1 | 26 ± 0.0 | 10 ± 5 | 5.7 ± 1.7 | 270 ± 25 | 87 ± 18 |
| Y91A | Very slow degradation | Very slow degradation | Very weak binding | Very weak binding | 200 ± 22 | 100 ± 26 |
| Δ175–209linker | 5.5 ± 1.3 | 125 ± 41 | 3.5 ± 1.9 | 1.1 ± 0.12 | 67 ± 10 | 100 ± 31 |
| Δ175–209GG | Not tested | Not tested | Not tested | 1.3 ± 0.20 | 14 ± 3.9 | 120 ± 39 |
| Δ108–243GG | a | No degradation | Not tested | Not tested | 3.8 ± 2.5 | Not tested |
Errors were calculated as SQRT((n − 1)−1 × ∑(value − mean)2) where n is the number of independent experiments, typically 2–4.
No degradation observed for (M1L)-Arc-ssrA or Arc-ssrA(ΔLAA) substrates, which are described in Ref.16.
To determine whether the Y91F and Y91A mutations affect substrate binding, we labeled amino groups in Arc-st11-ssrA with a fluorescent dye and used changes in fluorescence anisotropy to assay binding in the presence of ATPγS, an ATP analog that HslU does not hydrolyze [Fig. 1(B); Table I]. Wild-type HslU bound this fluorescent substrate with an average affinity of 3.8 ± 1.2 μM, whereas the Y91A variant showed no detectable binding. The average KD for Y91F was 10 ± 5 μM, although this binding reaction resulted in a very small change in anisotropy compared to wild type [Fig. 1(B)], suggesting that the Y91F-bound substrate has greater mobility than in the wild-type complex. We conclude that the wild-type Tyr91 side chain plays an important role in allowing HslU to bind Arc-st11-ssrA.
For wild-type and Y91F HslU, the KD values for binding fluorescent Arc-st11-ssrA were 2- to 5-fold tighter than the KM values for ATP-dependent HslUV degradation of the unmodified substrate (Table I). This difference was not caused by the absence of HslV in the binding reactions, as titrations with wild-type HslUV resulted in even stronger binding [KD = 0.46 μM; Fig. 1(C)]. It is possible that the fluorescent modifications strengthen binding or that the KD/KM discrepancy reflects occupancy differences that result from using ATPγS for binding versus ATP for degradation (see Discussion).
The N-terminal residues of Arc play an important role in HslU recognition.14 Indeed, when we deleted the seven N-terminal amino acids of Arc-st11-ssrA, the purified Δ7N variant was not degraded by HslUV and did not bind to HslU following fluorescent labeling (data not shown). Moreover, the unlabeled Δ7N-Arc-st11-ssrA variant did not compete well for binding of HslU to fluorescent Arc-st11-ssrA [Fig. 2(A)]. The small amount of competition observed in this experiment probably reflects a small decrease in the affinity of HslU for an Arc heterodimer containing one wild-type subunit and one Δ7N subunit. The gt1 peptide (MRYFFKKKLRFY) was designed as a mimic of the N-terminal residues of Arc.14 We assayed binding of fluorescein-labeled gt1 to determine whether the GYVG mutations altered recognition. Wild-type HslU bound gt1 with an average KD of 0.91 ± 0.29 μM, Y91F HslU bound with an average KD of 5.7 ± 1.7 μM, and Y91A HslU did not show detectable binding [Fig. 2(B); Table I]. A model in which the N-terminal residues of Arc bind in the axial pore of HslU with major contacts made by the aromatic ring of Tyr91 and minor contacts made by the side chain –OH group would explain the failure of Y91A HslU to bind to Arc-st11-ssrA or the gt1 peptide, the reduced affinity of Y91F for Arc-st11-ssrA and gt1, and the failure of wild-type HslU to bind the Δ7N variant of Arc.
Figure 2.
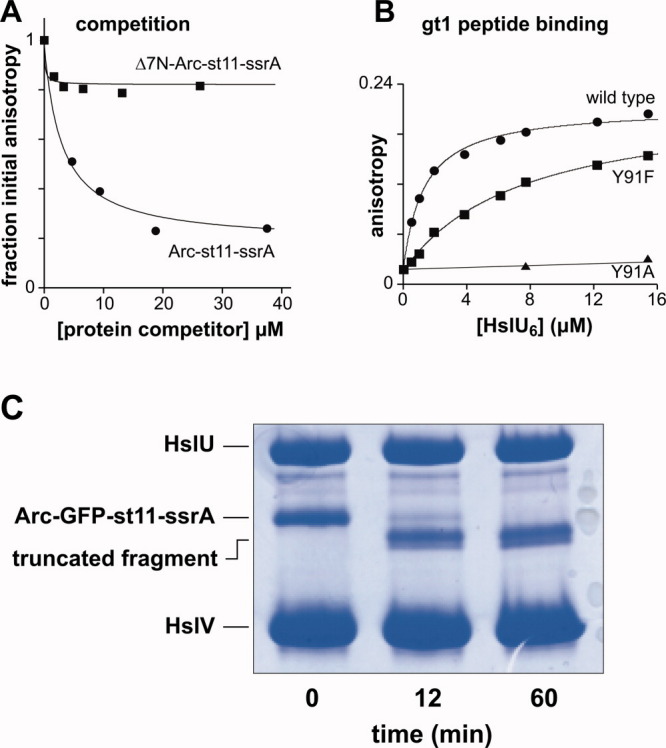
Competition, peptide binding, and fusion-protein degradation. A: Wild-type HslU6 (15 μM) was mixed with fluorescent Arc-st11-ssrA (0.4 μM), different amounts of unlabeled Arc-st11-ssrA or Δ7N-Arc-st11-ssrA were added as competitor, and binding was measured by anisotropy. Fits are to the hyperbolic equation used in panel 1B. B: Binding of wild-type, Y91F, and Y91A HslU to fluorescent gt1 peptide (200 nM) in the presence of 3 mM ATPγS. For WT and Y91F, the fits are to the hyperbolic equation used in panel 1B: b = 0.020; a = 0.19; KD = 1.2 μM (WT); b = 0.016; a = 0.20; KD = 7.0 μM (Y91F). Average KD values from multiple experiments are listed in Table I. For Y91A, the line is a linear fit. C: Degradation of Arc-GFP-st11-ssrA (4 μM) by HslUV (1 μM HslU6; 3 μM HslV12) was monitored by SDS-PAGE and staining with Coomassie blue. The bands corresponding to the truncated products were excised together, digested with trypsin, and analyzed by LC-MS/MS. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Degradation of an Arc fusion protein starts at the N-terminus
If the N terminal residues of Arc initially bind in the axial pore of HslU, then unfolding and translocation should begin at the N-terminus and proceed towards the C-terminus. To test this model, we constructed a substrate with an N-terminal Arc domain, followed by GFP, followed by the st11-ssrA sequence (Arc-GFP-st11-ssrA). As assayed by SDS-PAGE, HslUV degradation of this substrate resulted in accumulation of two truncated products of similar molecular weight that were slightly smaller than the full-length fusion substrate [Fig. 2(C)]. We excised the truncated products from the gel, digested them with trypsin, and characterized the resulting peptides by LC-MS/MS. Arc-GFP-st11-ssrA contains 314 residues total and tryptic peptides corresponding to residues 17–23 (Arc), 32–40 (Arc), 59–81 (GFP), 82–95 (GFP), 129–134 (GFP), 135–140 (GFP), 141–151 (GFP), 152–162 (GFP), 182–195 (GFP), 196–211 (GFP), 214–221 (GFP), and 300–314 (st11-ssrA) were recovered. The identity of all these peptides was confirmed by MS/MS sequencing. Importantly, the last peptide was one of the most abundant and its sequence (NQHDAANDENYALAA) corresponded to the 15 C-terminal residues of the Arc-GFP-st11-ssrA substrate. No peptides from the N-terminal 16 residues of the fusion substrate were recovered. Moreover, both truncated products cross-reacted with an anti-H6 antibody and bound to Ni++-NTA resin (data not shown), demonstrating that they contain the H6 sequence in the st11 tag near the C-terminus of Arc-GFP-st11-ssrA. Over the time period that the truncated degradation products were formed, we observed no change in native GFP fluorescence, showing that the GFP domain was not unfolded or degraded. In combination, these results demonstrate that most HslUV degradation of Arc-GFP-st11-ssrA begins at the N-terminus but stalls when it encounters GFP.
The intermediate domain of HslU is required for robust ATP hydrolysis
Previous studies have shown that the I domain of HslU (residues 108–243) is required for degradation of some native substrates.12, 19 To probe the function of this domain in greater depth, we constructed and purified E. coli HslU variants in which the entire I domain was replaced with a GG dipeptide (Δ108–243GG) or the I domain 175–209 loop, which is disordered in all HslU and HslUV crystal structures, was replaced either with a GG dipeptide (Δ175–209GG; an enzyme characterized in Ref.19) or with a longer SGAGGTSGEGGS linker (Δ175–209linker).
As anticipated from prior studies,12, 19 each I-domain variant purified as a hexamer and all those that were tested stimulated HslV peptidase activity normally (Table I). When we measured the rate of hydrolysis of 2.5 mM ATP by 0.3 μM enzyme, each I-domain mutant was less active than wild type and the variant with the full I-domain deletion had almost no activity [Fig. 3(A); Table I]. Importantly, however, the ATP-hydrolysis rates of each of the I-domain mutants scaled linearly with enzyme concentration [Fig. 3(B)], showing that hexamer destabilization is not responsible for reduced ATPase activity. Based on the slopes of the Figure 3(B) fits, the mutant ATPase activities as a percentage of the wild-type value were: Δ108–243GG (∼2%), Δ175–209GG (∼12%), and Δ175–209linker (∼43%). Adding HslV stimulated the ATPase activity of the Δ175–209linker mutant to a level similar to wild-type HslUV but the activity of the Δ175–209GG variant remained at ∼10% of the level of wild-type HslUV [Fig. 3(C)]. In contrast to our results, Song et al. reported 80–100% of wild-type ATPase activity for the Δ175–209GG mutant and for a variant missing the entire I domain, although they did not show data or specify experimental details.19 To ensure that these differences were not caused by our use of a sub-saturating nucleotide concentration, we assayed the dependence of turnover on ATP concentration for wild-type and Δ175–209GG HslU and found that Vmax for the mutant was ∼10-fold lower than for wild-type HslU [Fig. 3(D)]. In combination, our results indicate that the I domain of HslU is required for robust ATP hydrolysis and that the 175–209 loop plays a role in modulating hydrolysis.
Figure 3.
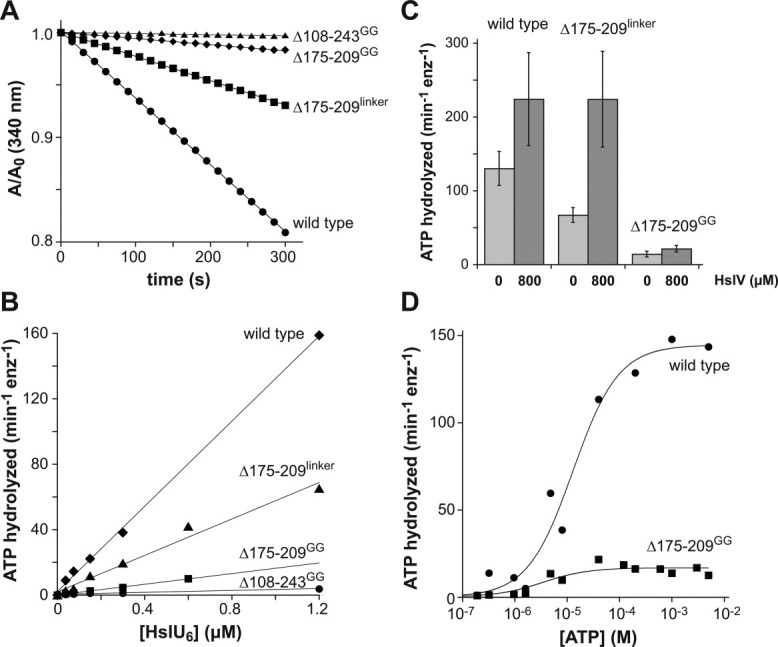
ATP hydrolysis. A: The rate of hydrolysis of ATP (2.5 mM) by wild-type HslU or I-domain mutants (0.3 μM hexamer) was measured using a continuous assay in which production of ADP is linked to oxidation of NADH, which is monitored by a decrease in absorbance at 340 nm.29 The lines are linear fits, corresponding to ATP hydrolysis rates of 137, 67, 14, and 3 min−1 HslU6−1 for wild type, Δ175-209linker, Δ175-209GG, and Δ108-243GG, respectively. B: Experiments like those shown in panel A were performed using different concentrations of wild-type HslU and I-domain mutants. The lines are linear fits with slopes corresponding to ATP hydrolysis rates of 130 ± 23, 67 ± 10, 14 ± 3.9, and 3.8 ± 2.5 min−1 HslU6−1 for wild type, Δ175–209linker, Δ175–209GG, and Δ108–243GG, respectively. C: ATP hydrolysis rates by 0.3 μM wild-type HslU6, Δ175–209linker HslU6, and Δ175–209GG HslU6 in the absence (light gray bars) or presence of 0.8 μM HslV12 (dark gray bars). D: Rates of ATP hydrolysis by 0.3 μM wild-type or Δ175–209GG HslU6 were determined at different ATP concentrations and fitted to the Hill form of the Michaelis–Menten equation, rate = Vmax/(1+(KM/[ATP])n). Based on multiple experiments (n = 3), KM for ATP hydrolysis was 8.9 ± 3.8 μM for wild type HslU and 5.5 ± 0.9 for Δ175–209GG.
Functional roles of the 175–209 I-domain loop
We chose Δ175–209linker HslU for studies of degradation, because this I-domain mutant had the highest rate of ATP hydrolysis. In assays monitored by SDS-PAGE, Δ175–209linker HslUV degraded Arc-st11-ssrA much more slowly than wild-type HslUV [Fig. 4(A), inset]. To determine the mechanistic basis for slower proteolysis, we used steady-state kinetics to determine KM (average 125 ± 41 μM) and Vmax (average 5.5 ± 1.3 min−1 enz−1) for Δ175–209linker HslUV degradation of Arc-st11-ssrA [Fig. 4(A); Table I]. Thus, in comparison with wild-type HslUV, the Δ175–209linker mutation weakened KM ∼6-fold and slowed the maximal rate of Arc-st11-ssrA degradation ∼2-fold (Table I).
Figure 4.
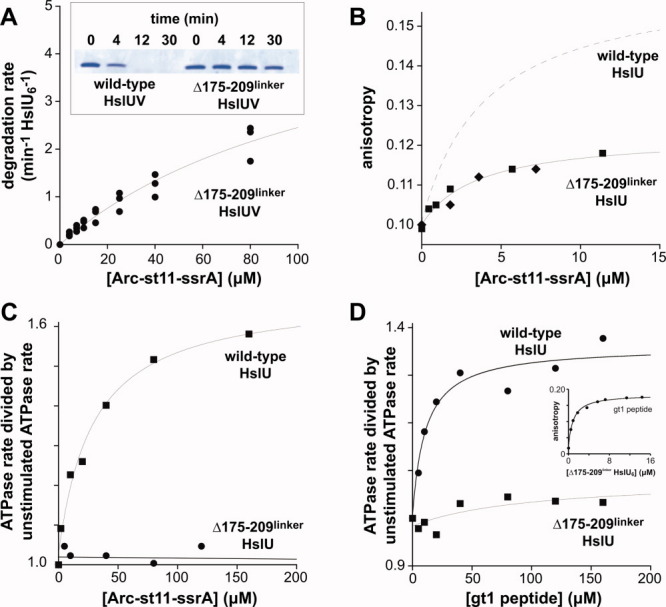
Properties of Δ175-209linker HslU. A: The inset shows SDS-PAGE analysis of HslUV degradation (0.3 μM HslU6; 0.8 μM HslV12) of Arc-st11-ssrA (10 μM) by the wild-type and Δ175–209linker proteases. The main plot shows the rate of degradation of different concentrations of Arc-st11-ssrA by 0.3 μM Δ175–209linker HslU6 and 0.8 μM HslV12. Data from three independent experiments are plotted. The solid line is a fit of the combined data to the Michaelis–Menten equation (KM = 125 ± 41 μM; Vmax = 5.5 ± 1.3 min−1 enz−1). B: Binding of Δ175-209linker HslU6 to fluorescent Arc-st11-ssrA (0.8 μM) in the presence of 3 mM ATPγS. Data from two independent experiments are plotted. A fit of the combined data to the equation b + a*(1+KD/[HslU6])−1 gave b = 0.10 ± 0.001, a = 0.022 ± 0.003, and KD = 3.5 ± 1.3 μM. The dashed line shows the binding curve for wild-type HslU from panel 1B. C: ATP hydrolysis by wild-type HslU6 (0.1 μM) was stimulated by addition of Arc-st11-ssrA, but hydrolysis by Δ175–209linker HslU6 (0.1 μM) was not stimulated. For wild type, the line is a fit to b + a*(1+Kapp/[HslU6])−1 with b = 1.03 ± 0.02, a = 0.65 ± 0.046, and Kapp = 29 ± 7.1 μM. For Δ175–209linker, the line is a linear fit. D: The gt1 peptide strongly stimulates ATP hydrolysis by wild-type HslU6 (0.1 μM) but weakly stimulates hydrolysis by Δ175–209linker HslU6 (0.1 μM). The lines are fits to the equation used in panel C with b = 0.99 ± 0.03, a = 0.36 ± 0.04, and Kapp = 10.5 ± 3.9 μM for wild type HslU. For Δ175–209linker HslU, values for a and Kapp were unreliable as the associated errors were similar to or larger than the values themselves. The inset shows binding of Δ175–209linker HslU to the fluorescent gt1 peptide (0.2 μM). The fitted line corresponds to a KD of 1.1 ± 0.12 μM. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Despite its weakened KM, Δ175–209linker HslU bound fluorescent Arc-st11-ssrA with an average KD of 3.5 ± 1.9 μM [Fig. 4(B); Table I], a value within error of the KD for wild-type HslU [Fig. 1(B); Table I]. However, the maximum anisotropy for the substrate–enzyme complex was ∼0.12 for Δ175–209linker HslU [Fig. 4(B)] compared to ∼0.15 for wild-type HslU [Fig. 1(D)]. This difference indicates that the bound substrate is more mobile in the complex with Δ175–209linker HslU. Thus, contacts made by the 175–209 loop appear to decrease substrate mobility in the wild-type complex. The weak KM but normal KD observed for Δ175–209linker HslUV compared with wild type are consistent with a model in which contacts made by the 175–209 loop reduce the rate of Arc-st11-ssrA dissociation in a reaction that only occurs after ATP hydrolysis (see Discussion).
To test for potential alterations in ATP hydrolysis during protein degradation, we assayed hydrolysis of 2.5 mM ATP by wild-type and Δ175–209linker HslU in the presence of increasing concentrations of Arc-st11-ssrA [Fig. 4(C)]. This substrate half-maximally stimulated ATP hydrolysis by wild-type HslU at a concentration of 25 ± 6 μM, a value similar to the KM for degradation of Arc-st11-ssrA. Surprisingly, however, Arc-st11-ssrA did not stimulate ATP hydrolysis by Δ175–209linker HslU to any significant degree [Fig. 4(C)]. Thus, our results show that Arc-st11-ssrA binds with normal affinity to Δ175–209linker HslU but fails to stimulate ATP turnover. The gt1 peptide also stimulated ATP hydrolysis by Δ175–209linker HslU to a far lower degree than wild-type HslU [Fig. 4(D)], even though this peptide bound Δ175–209linker HslU with an average affinity (1.1 ± 0.12 μM) within error of the wild-type value [Fig. 4(D), inset]. Thus, the 175–209 loop in the I domain of wild-type HslU plays an important role in allowing bound peptides and protein substrates to modulate the rate of ATP hydrolysis.
Discussion
Our results support a degradation model in which the N-terminal residues of Arc substrates initially bind in the axial pore of the HslU hexamer [Fig. 5(A)]. Specifically, we find that the Y91F mutation, which changes the highly conserved GYVG motif in the axial pore of HslU to GFVG, increases KD for binding Arc-st11-ssrA and the gt1 peptide (which mimics interactions made by the N-terminus of Arc). Thus, the side-chain hydroxyl group of Tyr91 in wild-type HslU plays a role in recognition of Arc substrates. The aromatic ring of Tyr91 is even more important, as the alanine-substitution mutation abolishes detectable binding of Arc-st11-ssrA and gt1. Previous studies showed that the Y91G and Y91A mutations prevented HslUV degradation of SulA and MBP-SulA but did not distinguish between defects caused by diminished binding versus poor unfolding/translocation.5, 19 The AAA+ ClpX and ClpA protein unfoldases also have GYVG pore loops but have different substrate specificities than HslU. Thus, the GYVG loop must contribute to binding but does not determine substrate interaction specificity. Indeed, the GYVG loop of ClpX collaborates with two additional pore loops to bind substrates.7, 24
Figure 5.
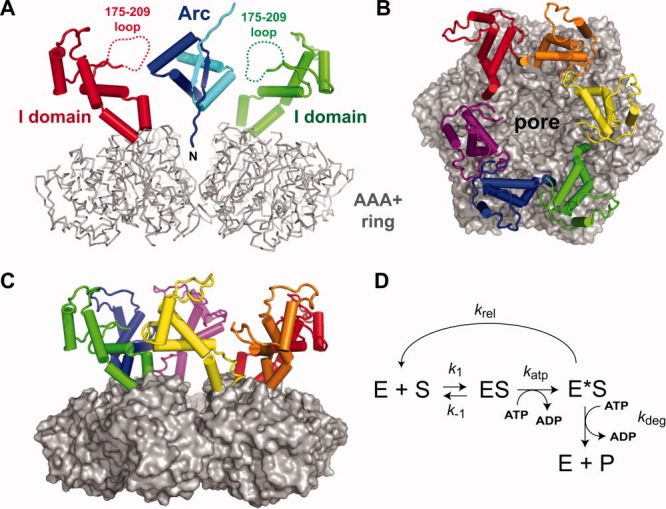
Structure and mechanism. A: Side view of the HslU hexamer with an Arc dimer modeled to allow one N-terminal arm to bind in the HslU pore. The AAA+ ring of HslU is colored gray and shown in backbone representation. Two I domains of HslU, colored red and green, are shown in cartoon representation; dashed lines indicate the disordered 175–209 loops. Arc is shown in cartoon representation, with one subunit colored blue and one colored cyan. The coordinates for HslU and Arc were taken from PDB files 1DO0 and 1PAR, respectively.17, 21 B: Substrate-binding view of an HslU hexamer with the AAA+ ring (gray) shown in surface representation and the I domains (colored individually) shown in cartoon representation. C: Side view of the hexamer with the same colors and structural representations as in panel B. D: Model of substrate binding and release. The k1 and k-1 steps model substrate association with and dissociation from the HslU hexamer that would occur in the presence of ATP or ATPγS. ATP hydrolysis converts ES to E*S with the rate constant katp. Substrate can be released from E*S (krel) or be degraded in additional steps that require ATP hydrolysis. For simplicity, these steps are combined into a single step with the rate constant kdeg.
If the N-terminal residues of Arc substrates bind in the axial pore, then degradation of these proteins should begin at the N-terminus. We confirmed this prediction by showing that HslUV degradation of an Arc-GFP-st11-ssrA fusion protein generates truncated products in which GFP and the C-terminal portion of the substrate remained intact. Previous studies showed that HslUV degraded most of the C-terminal SulA portion of a GFP-SulA fusion protein but failed to degrade the GFP portion.13 Thus, the β-barrel architecture of GFP resists HslUV unfolding from both the C-terminal and N-terminal ends.
The intermediate domains of HslU emerge from the top surface of the AAA+ hexameric ring, forming a funnel-shaped cavity that leads to the axial pore [Fig. 5(A–C)]. The 175-209 loop is disordered in all HslU and HslUV crystal structures but would project into the funnel [Fig. 5(A)], allowing multiple loops to interact with a substrate as it approached or was bound in the pore. Deletion of the I domain or 175–209 loop was previously shown to prevent or greatly diminish degradation of SulA or Arc-SulA.13, 19 Importantly, the crystal structure of a hexamer of Haemophilus influenzae HslU-ΔI (residues 108–243 replaced with a GG linker) bound to HslV12, revealed normal interactions with the peptidase and an essentially wild-type structure of the AAA+ ring.13
When we replaced the 175–209 loop of E. coli HslU with a GG linker, the protein displayed a ∼10-fold decrease in the steady-state rate of ATP hydrolysis compared to wild-type HslU. Replacing this loop with a longer linker increased ATPase activity to roughly half of the wild-type value for HslU alone and close to the wild-type value when HslV was present. Strikingly, however, even this protein (Δ175–209linker HslU) had major functional defects. For example, Δ175–209linker HslUV showed poor degradation of Arc-st11-ssrA, with a ∼6-fold weaker KM and ∼2-fold decrease in Vmax when compared with the wild-type enzyme. Moreover, the Arc-st11-ssrA substrate stimulated the ATPase activity of wild-type HslU ∼1.6-fold but did not stimulate hydrolysis by Δ175–209linker HslU.
Interestingly, Δ175–209linker HslU bound fluorescent Arc-st11-ssrA with a KD within error of the wild-type value in the presence of ATPγS, suggesting that the 6-fold KM defect arises from a reaction that occurs as a consequence of ATP hydrolysis. However, ATP-dependent degradation occurs more slowly for the Δ175–209linker variant than for wild-type HslUV and thus cannot explain the increase in KM. It is likely, therefore, that the intact substrate dissociates more rapidly from Δ175–209linker HslU than from wild-type HslU following ATP hydrolysis. Indeed, ClpXP has been shown to release bound substrates when ATP-dependent unfolding fails.25 Substrate binding to ATP-loaded HslU will be governed by rate constants for association (k1) and dissociation (k-1), with the ratio of these constants (KD = k−1/k1) defining the affinity measured in the presence of ATPγS. In the model of Figure 5(D), the ES complex is converted to an E*S complex by ATP hydrolysis (katp), and the substrate can then be released (krel) or be degraded in subsequent steps that require ATP hydrolysis but are combined into a single proteolysis step (kdeg) in the model. Mutations that increase krel should weaken KM and decrease Vmax. For example, simulating these reactions using k1 = 1 μM−1 min−1, k−1 = 4 min−1, katp = 250 min−1, krel = 12 min−1, and kdeg = 12 min−1 gave KD = 4 μM, KM = 24 μM, and Vmax = 11.0 min−1 enz−1, which are similar to the values we measure for binding and degradation of Arc-st11-ssrA by wild-type HslUV. Changing just the krel value to 250 min−1 in the simulation gave KD = 4 μM, KM = 123 μM, and Vmax = 5.7 min−1 enz−1, which are similar to the values measured for binding and degradation of Arc-st11-ssrA by Δ175–209linker HslUV. Thus, the Figure 5(D) model can account for the observed properties of the wild-type enzyme and the Δ175–209linker mutant.
Protein unfolding by AAA+ enzymes can require many cycles of ATP hydrolysis.26 We propose that the 175–209 loops in the I domains of an HslU hexamer help to keep a native protein substrate bound as the AAA+ ring of HslU hydrolyzes ATP in an unfolding attempt. Following ATP hydrolysis, the GYVG loops in the translocation pore may bind the N-terminal residues of the substrate less tightly and contacts with the 175–209 loops could minimize the chance of dissociation. Our finding that Arc-st11-ssrA is more mobile when bound to Δ175–209linker HslU than to wild-type HslU supports this model. Despite being disordered, the 175–209 loop contains a GVEIMAPPGMEEMTSQLQSMF sequence (residues 183–203 in E. coli HslU) that is ∼90% conserved among diverse species of γ-proteobacteria. We interpret this conservation as evidence that contacts between side chains of the loop and bound substrates are functionally important. All six 175–209 loops in the HslU hexamer would be positioned to contact a protein substrate in the funnel [Fig. 5(A–C)], and thus loop–substrate interactions could be relatively nonspecific, depending on adventitious polar or hydrophobic interactions. It is worth noting that six disordered 175–209 loops correspond to ∼200 amino acids or roughly twice the size of the Arc dimer depicted in Figure 5(A). Thus, the funnel region would be crowded and gel-like, and substrate binding might force the I domains outward, transmitting a mechanical signal to the AAA+ ring of HslU. Conversely, ATP hydrolysis by the AAA+ ring might force the I domains and 175–209 loops inward, contributing to substrate unfolding.
We were surprised to find that the I domain of HslU is required for robust ATP hydrolysis. For example, compared with wild type, we found that deletion of the I-domain reduced the rate of ATP hydrolysis ∼50-fold and replacing the 175–209 loop with a GG linker decreased ATPase activity ∼10-fold. By contrast, Song et al. reported that the same or very similar deletions caused almost no change in ATP hydrolysis but did not report assay conditions or experimental data.19 Why our results differ from their results is unclear. They seem to have used an end-point assay and may have used a time well beyond the linear range, thereby overestimating mutant activities relative to wild type. Nevertheless, our results unambiguously demonstrate that the I domain and the 175–209 loop play important roles in allowing HslU to hydrolyze ATP rapidly. In the HslU sequence, the I domain is followed by the Walker-B motif, which coordinates Mg++ binding to the β and γ phosphates of ATP and activates a water molecule for nucleophilic attack on the terminal phosphate.17, 18, 27 Changes in I-domain conformation could affect these active-site residues. Alternatively, I-domain structure could be linked to conformational changes in the hexameric ring of HslU, which limit the overall rate of ATP turnover. In addition to an overall role for the I domain in allowing robust ATP hydrolysis, the 175–209 loop also appears to be important for substrate stimulation of ATP hydrolysis, as Δ175–209linker HslU showed no stimulation with the Arc-st11-ssrA substrate and a very low level of stimulation with the gt1 peptide.
Among AAA+ enzymes, HslU is unique in having an I domain. The Tip48/49 family of snoRNA remodeling enzymes also has a family-specific insertion in a similar region of the large AAA+ domain, as does the LonB family of archaeal AAA+ proteases.28 In LonB, this insertion forms a pair of membrane helices that tether each subunit in the hexameric ring to the inner surface of the cytoplasmic membrane. It will be important to determine whether these membrane-binding domains, like the I domain of HslU, play roles in substrate binding and regulation of ATP hydrolysis by LonB proteases.
Materials and Methods
Proteins and peptides
E. coli HslU variants were constructed by PCR in a pET12b-H6-HslU plasmid background. E. coli HslU, E. coli HslU mutants, E. coli HslV, and unlabeled and 35S-labeled Arc-st11-ssrA were expressed and purified as described.16 To engineer a gene encoding Arc-GFP-st11-ssrA, unique NheI (GCTAGC) and SalI (GTCGAC) sites were introduced by PCR at the Arc/st11 junction in pET21b-Arc-st11-ssrA, unique NheI and SalI sites were introduced flanking the N- and C-terminal GFP coding sequence in a PCR fragment, these molecule were cut with NheI and SalI and appropriate fragments were ligated to generate the expression plasmid. Arc-GFP-st11-ssrA was purified using the Arc-st11-ssrA procedure,16 except a HiLoad 16/60 Superdex-200 column was used for the final gel-filtration step.
Labeling of purified Arc-st11-ssrA with FL-BODIPY-CASE and labeling of the synthetic gt1 peptide with fluorescein were performed as described.14 Peptides were purified by HPLC, and their molecular weights were confirmed by MALDI mass spectrometry prior to use. Protein concentrations were determined from A280, using extinction coefficients calculated from the amino-acid sequence. Peptide concentrations were determined using an extinction coefficient of 70,000 M−1 cm−1 for fluorescein at A492.
Biochemical assays
HslU or variants were incubated at hexamer concentrations of 150, 300, 600, or 1200 nM in 25 μL PD buffer (25 mM HEPES (pH 7.6), 5 mM KCl, 5 mM MgCl2, 0.032% (vol/vol) Igepal CA-630 (NP-40), and 10% (vol/vol) glycerol) at 37°C. We then added 25 μL of a buffer-matched solution containing 5 mM ATP, 2 mM NADH, 4 mM phosphoenolpyruvate, 6 U/mL lactate dehydrogenase, and 6 U/mL pyruvate kinase, and monitored NADH oxidation, which occurred in a coupled reaction as ADP was generated by ATP hydrolysis, by decreased absorbance at 340 nm.29 ATPase activities in units of mM ATP hydrolyzed min−1 were calculated as ΔA340/(Δtime × pathlength × 6.23 mM−1 cm−1), plotted against HslU concentration (mM), and specific activities in units of ATPs hydrolyzed HslU−1 min−1 were determined from a linear fit of this curve. Reactions were carried out in a 96-well plate, with a path length of 0.17 cm for the 50 μL volume used. To determine the ability of the peptidase to stimulate ATPase activity, we incubated HslU (300 nM) and HslV (800 nM) together before beginning the reaction with the addition of ATP. To determine the nucleotide dependence of hydrolysis activity, we fixed the final HslU concentration (100 nM) and varied the ATP concentration. To determine whether substrates or gt1 peptide altered ATP-hydrolysis rates, we fixed the final HslU concentration (100 nM) and varied the concentration of peptide/protein that was included in the 25 μL mix with ATP and the regeneration system.
Assays probing the ability of HslU variants to bind HslV and activate cleavage of 20 μM Gly-Gly-Leu-7-amino-4-methylcoumarin (GGL-AMC) were performed in PD buffer at 37°C and monitored by increased fluorescence at 440 nm using a SoftMax Pro5 fluorescence plate reader (Molecular Devices). Reactions contained ATPγS (2 mM), HslV12 (100 nM), and HslU6 (500 nM).
Degradation assays were performed at 37°C in PD buffer supplemented with ATP (5 mM), 16 mM creatine phosphate, and 10 μg/mL creatine kinase. Proteolysis of 35S-Arc-st11-ssrA by 100 nM HslU6, 300 nM HslV12 was assayed by the production of TCA-soluble radioactive peptides.16 For assays monitored by SDS-PAGE, reactions contained 10 μM Arc-st11-ssrA (300 nM HslU6, 800 nM HslV12) or 4 μM Arc-GFP-st11-ssrA (1 μM HslU6, 3 μM HslV12) and were quenched at different times by addition of SDS loading buffer (125 mM bis-Tris pH 6.8, 5% glycerol, 1% SDS, 0.5 mM EDTA, 100 mM DTT, 0.015% Coomassie Blue G-250).
The binding of HslU or variants to gt1 peptide or Arc-st11-ssrA was assayed at 37°C in PD buffer by monitoring increases in fluorescence anisotropy (excitation 492 nm; emission 520 nm) using the SoftMax Pro5 fluorescence plate reader (Molecular Devices) for gt1 binding or PTI QM-2000-4SE spectrofluorimeter for Arc-st11-ssrA. Samples (20 μL) contained 200 nM fluorescein-labeled gt1 or 800 nM fluorescein-labeled Arc-st11-ssrA, 3 mM ATPγS, varying concentrations of HslU, and if appropriate, 10.8 μM HslV12. Readings were corrected for scattering, and G-factors were re-calculated before each set of measurements and used in the analysis.
LC-MS/MS analysis and sequencing of tryptic peptides from the truncated products produced by HslUV degradation of Arc-GFP-st11-ssrA was performed by the MIT Biopolymers and Proteomics Facility.
Acknowledgments
Authors thank V. Baytshok, J. Chen, S. Glynn, I. Levchenko, S. Kim, I. Papayannopoulos, A. Nager, A. Olivares, and R. Mauldin, and for assistance and helpful discussions. T.A.B. is a Howard Hughes Medical Institute employee.
References
- 1.Gottesman S. Proteolysis in bacterial regulatory circuits. Annu Rev Cell Dev Biol. 2003;19:565–587. doi: 10.1146/annurev.cellbio.19.110701.153228. [DOI] [PubMed] [Google Scholar]
- 2.Baker TA, Sauer RT. ATP-dependent proteases of bacteria: recognition logic and operating principles. Trends Biochem Sci. 2006;31:647–653. doi: 10.1016/j.tibs.2006.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Striebel F, Kress W, Weber-Ban E. Controlled destruction: AAA+ ATPases in protein degradation from bacteria to eukaryotes. Curr Opin Struct Biol. 2009;19:209–217. doi: 10.1016/j.sbi.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 4.Siddiqui SM, Sauer RT, Baker TA. Role of the protein-processing pore of ClpX, an AAA+ ATPase, in recognition and engagement of specific protein substrates. Genes Dev. 2004;18:369–374. doi: 10.1101/gad.1170304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park E, Rho YM, Koh OJ, Ahn SW, Seong IS, Song JJ, Bang O, Seol JH, Wang J, Eom SH, Chung CH. Role of the GYVG pore motif of HslU ATPase in protein unfolding and translocation for degradation by HslV peptidase. J Biol Chem. 2005;280:22892–22898. doi: 10.1074/jbc.M500035200. [DOI] [PubMed] [Google Scholar]
- 6.Hinnerwisch J, Fenton WA, Furtak KJ, Farr GW, Horwich AL. Loops in the central channel of ClpA chaperone mediate protein binding, unfolding, and translocation. Cell. 2005;121:1029–1041. doi: 10.1016/j.cell.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 7.Martin A, Baker TA, Sauer RT. Diverse pore loops of the AAA+ ClpX machine mediate unassisted and adaptor-dependent recognition of ssrA-tagged substrates. Mol Cell. 2008;29:441–450. doi: 10.1016/j.molcel.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martin A, Baker TA, Sauer RT. Pore loops of the AAA+ ClpX machine grip substrates to drive translocation and unfolding. Nat Struct Mol Biol. 2008;15:1147–1151. doi: 10.1038/nsmb.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gottesman S, Roche E, Zhou Y, Sauer RT. The ClpXP and ClpAP proteases degrade proteins with carboxy-terminal peptide tails added by the SsrA-tagging system. Genes Dev. 1998;12:1338–1347. doi: 10.1101/gad.12.9.1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neher SB, Sauer RT, Baker TA. Distinct peptide signals in the UmuD and UmuD′ subunits of UmuD/D′ mediate tethering and substrate-processing by the ClpXP protease. Proc Natl Acad Sci USA. 2003;100:13219–13224. doi: 10.1073/pnas.2235804100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seong IS, Oh JY, Yoo SJ, Seol JH, Chung CH. ATP-dependent degradation of SulA, a cell division inhibitor, by the HslUV protease in Escherichia coli. FEBS Lett. 1999;456:211–214. doi: 10.1016/s0014-5793(99)00935-7. [DOI] [PubMed] [Google Scholar]
- 12.Kwon AR, Trame CB, McKay DB. Kinetics of protein substrate degradation by HslUV. J Struct Biol. 2004;146:141–147. doi: 10.1016/j.jsb.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 13.Kwon AR, Kessler BM, Overkleeft HS, McKay DB. Structure and reactivity of an asymmetric complex between HslV and I-domain deleted HslU, a prokaryotic homolog of the eukaryotic proteasome. J Mol Biol. 2003;330:185–195. doi: 10.1016/s0022-2836(03)00580-1. [DOI] [PubMed] [Google Scholar]
- 14.Burton RE, Baker TA, Sauer RT. Nucleotide-dependent substrate recognition by the AAA+ HslUV protease. Nat Struct Mol Biol. 2005;12:245–251. doi: 10.1038/nsmb898. [DOI] [PubMed] [Google Scholar]
- 15.Koodathingal P, Jaffe NE, Kraut DA, Prakash S, Fishbain S, Herman C, Matouschek A. ATP-dependent proteases differ substantially in their ability to unfold globular proteins. J Biol Chem. 2009;284:18674–18684. doi: 10.1074/jbc.M900783200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sundar S, McGinness KE, Baker TA, Sauer RT. Multiple sequence signals direct recognition and degradation of protein substrates by the AAA+ protease HslUV. J Mol Biol. 2010;403:420–429. doi: 10.1016/j.jmb.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bochtler M, Hartmann C, Song HK, Bourenkov GP, Bartunik HD, Huber R. The structures of HsIU and the ATP-dependent protease HsIU-HsIV. Nature. 2000;403:800–805. doi: 10.1038/35001629. [DOI] [PubMed] [Google Scholar]
- 18.Sousa MC, Trame CB, Tsuruta H, Wilbanks SM, Reddy VS, McKay DB. Crystal and solution structures of an HslUV protease-chaperone complex. Cell. 2000;103:633–643. doi: 10.1016/s0092-8674(00)00166-5. [DOI] [PubMed] [Google Scholar]
- 19.Song HK, Hartmann C, Ramachandran R, Bochtler M, Behrendt R, Moroder L, Huber R. Mutational studies on HslU and its docking mode with HslV. Proc Natl Acad Sci USA. 2000;97:14103–14108. doi: 10.1073/pnas.250491797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang J, Song JJ, Seong IS, Franklin MC, Kamtekar S, Eom SH, Chung CH. Nucleotide-dependent conformational changes in a protease-associated ATPase HslU. Structure. 2001;9:1107–1116. doi: 10.1016/s0969-2126(01)00670-0. [DOI] [PubMed] [Google Scholar]
- 21.Breg JN, van Opheusden JH, Burgering MJ, Boelens R, Kaptein R. Structure of Arc repressor in solution: evidence for a family of β-sheet DNA-binding proteins. Nature. 1990;346:586–589. doi: 10.1038/346586a0. [DOI] [PubMed] [Google Scholar]
- 22.Raumann BE, Rould MA, Pabo CO, Sauer RT. DNA recognition by beta-sheets in the Arc repressor-operator crystal structure. Nature. 1994;367:754–757. doi: 10.1038/367754a0. [DOI] [PubMed] [Google Scholar]
- 23.Sauer RT, Baker TA. AAA+ Proteases: ATP-fueled machines of protein destruction. Annu Rev Biochem. 2011;80:587–612. doi: 10.1146/annurev-biochem-060408-172623. [DOI] [PubMed] [Google Scholar]
- 24.Farrell CM, Baker TA, Sauer RT. Altered specificity of a AAA+ protease. Mol Cell. 2007;25:161–166. doi: 10.1016/j.molcel.2006.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kenniston JA, Baker TA, Sauer RT. Partitioning between unfolding and release of native domains during ClpXP degradation determines substrate selectivity and partial processing. Proc Natl Acad Sci USA. 2005;102:1390–1395. doi: 10.1073/pnas.0409634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kenniston JA, Baker TA, Fernandez JM, Sauer RT. Linkage between ATP consumption and mechanical unfolding during the protein processing reactions of an AAA+ degradation machine. Cell. 2003;114:511–520. doi: 10.1016/s0092-8674(03)00612-3. [DOI] [PubMed] [Google Scholar]
- 27.Zhang X, Wigley DB. The ‘glutamate switch’ provides a link between ATPase activity and ligand binding in AAA+ proteins. Nat Struct Mol Biol. 2008;15:1223–1227. doi: 10.1038/nsmb.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iyer LM, Leipe DD, Koonin EV, Aravind L. Evolutionary history and higher order classification of AAA+ ATPases. J Struct Biol. 2004;146:11–31. doi: 10.1016/j.jsb.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 29.Nørby JG. Coupled assay of Na+,K+-ATPase activity. Methods Enzymol. 1988;156:116–119. doi: 10.1016/0076-6879(88)56014-7. [DOI] [PubMed] [Google Scholar]