

Abstract
DmmA is a haloalkane dehalogenase (HLD) identified and characterized from the metagenomic DNA of a marine microbial consortium. Dehalogenase activity was detected with 1,3-dibromopropane as substrate, with steady-state kinetic parameters typical of HLDs (Km = 0.24 ± 0.05 mM, kcat = 2.4 ± 0.1 s−1). The 2.2-Å crystal structure of DmmA revealed a fold and active site similar to other HLDs, but with a substantially larger active site binding pocket, suggestive of an ability to act on bulky substrates. This enhanced cavity was shown to accept a range of linear and cyclic substrates, suggesting that DmmA will contribute to the expanding industrial applications of HLDs.
Keywords: haloalkane dehalogenase, marine microbial consortium, α/β hydrolase, CurN, DmmA
Introduction
Haloalkane dehalogenases (HLDs) remove halogens from alkanes by hydrolysis, producing an alcohol, a halide ion, and a proton. These dehalogenating enzymes are of long-standing interest for industrial applications such as degradation of environmental pollutants1, 2 and biocatalysis.3 Newer applications include remediation of chemical weapons,4 biosensing,5, 6 and cellular imaging.7 The applications for HLDs are growing with knowledge of their functional and structural properties.
Each HLD has a unique substrate selectivity, which can include chlorinated, brominated, and iodinated alkanes of varying length. Some HLDs also act on ring-containing substrates, or those with alcohol, ether, nitrile, or alkene functional groups.8 Substrate selectivity is not easily predicted by sequence analysis. For HLDs of known structure, the size and shape of the active-site cavity are better predictors of substrate preference than is phylogenetic analysis.8 The natural substrates are, in general, unknown.
HLDs are members of the α/β hydrolase superfamily, with the active site located between a conserved core subdomain and a more variable lid structure. The HLD active site departs from the typical α/β hydrolase catalytic triad by the replacement of the common Ser nucleophile with an Asp and the addition of two halide-stabilizing residues (Trp and Trp/Asn) to form a “catalytic pentad.”9 The side chains of the catalytic pentad are identically positioned in the active site cleft of all HLDs of known structure, although some pentad residues vary in their locations in the primary sequence. This sequence-location difference of the catalytic pentad residues defines three evolutionary subfamilies of HLDs.10 Two structures are known from evolutionary subfamily I (DhlA11–13 and DppA14), and four from subfamily II (DhaA,15, 16 LinB,17–20 DbjA,21 and Rv257822). Several structures include bound substrates, products, or covalent intermediates, which together with many kinetic studies, led to a detailed reaction mechanism for this enzyme class.9
DmmA is a putative HLD of subfamily II.10 This protein was originally annotated as CurN, and presumed to be the final gene product of the curacin A biosynthetic gene cluster23 from the marine cyanobacterium Lyngbya majuscula (now designated Moorea producta24). However, resequencing revealed the authentic 3′ end of the gene cluster, which lacks curN,25 suggesting that dmmA was cloned as part of a chimeric cosmid from the original metagenomic DNA library.23 Herein, we present the crystal structure and HLD activity of DmmA.
Results
Biological source of DmmA (formerly CurN)
In the original curacin pathway sequencing,23 DNA encoding 64 amino acids at the CurN N-terminus overlapped with DNA encoding the CurM C-terminus in a different reading frame. This unusually long overlap and peculiarities of the CurM protein sequence led us to isolate a new cosmid, and to resequence the curM-curN region of the gene cluster, resulting in a corrected 3′ terminus that lacked curN.25 In addition, no CurN coding sequence was identified by amplification from M. producta (formerly L. majuscula) genomic DNA, nor was CurN located in the recently sequenced M. producta 3L genome.26 Furthermore, the G + C content of curN (61%) contrasts with the rest of the curacin biosynthetic gene cluster (45.5%). The scaffolds used to assemble the M. producta genome ranged from 37 to 66% G + C content, with the great majority falling between 40 and 45%.26 In addition, CurN (now referred to as DmmA (dehalogenase A from a marine microbe) in accord with other dehalogenases of unknown biological function8) has high similarity (∼50%) to HLDs from other marine bacteria, although none are from the cyanobacterial phyla. From these data, we conclude that curN (dmmA) is most likely a product of one of the organisms that grew in close association with the M. producta field isolate whose metagenomic DNA cosmid library was used to sequence the cur gene cluster. It is evident that the 3′ end of the curacin gene cluster concatenated with curN (dmmA) genetic material from one of the heterotroph bacteria that was growing on the M. producta filament at the time of cosmid preparation.
Because the biological source and the natural termini of DmmA are uncertain, we turned to sequence analysis to design dmmA constructs for protein expression. HLD homologs have C-termini of very similar length and sequence to the DmmA C-terminus, but the N-termini are poorly conserved and of variable length.10 In addition, DmmA amino acids 1-40 are predicted to be unstructured. Strong sequence conservation among subfamily-II HLDs begins at DmmA residue 48. This evidence could suggest that the N-terminus of DmmA is not in the original gene, however several considerations led us to consider the full open reading frame as the natural length of DmmA. First, the DNA sequence upstream of the putative translation start site contains sequence motifs and spacers similar to prokaryotic σ70 promoter regions and ribosome binding sequences (Supporting Information Fig. 1). In addition, several other annotated HLDs have N-termini of similar length to DmmA.10 Thus, we conclude that the natural protein likely includes all the residues encoded in the curN (dmmA) open reading frame. Two dmmA constructs were made, one encoding the full-length protein (DmmAlong, residues 1-341) and one with a 43-residue N-terminal truncation (DmmAshort, residues 44-341) to produce a potentially more structured N-terminus (Supporting Information Fig. 2). Both DmmAshort and DmmAlong were produced in high yield, as stable and soluble proteins in an E. coli expression system.
HLD activity
As for many other HLDs, the natural substrate for DmmA is unknown. Small di-halogenated alkanes are typical substrates to assess the activity of HLDs. We chose 1,3-dibromopropane, a standard substrate, for DmmA assays. HLD activity was tested in a colorimetric assay to detect the production of protons.27, 28 Both DmmAshort and DmmAlong were active in this assay, however DmmAshort exhibited tenfold higher activity than the longer form of the protein (Table I), suggesting that the N-terminus may interfere with catalysis. Steady-state kinetic constants were determined for DmmAshort (Table II), which revealed that these parameters are comparable to other HLDs.15, 27, 29 DhlA has threefold greater catalytic efficiency than DmmA for 1,3-dibromopropane (fourfold lower kcat, 12-fold lower Km),27 whereas DhaA has a nearly equal kcat and a 50-fold lower Km for 1,3-dibromopropane resulting in a 50-fold greater catalytic efficiency.29
Table I.
DmmA Activity Toward Halogenated Substrates
| Construct | Substrate | % Activitya |
|---|---|---|
| DmmAshort | 1,3-dibromopropane | 100 |
| DmmAshort | 1,6-dibromohexane | 3.5 ± 0.7 |
| DmmAshort | bromocyclohexane | 14.8 ± 0.6 |
| DmmAshort | 1,6-dichlorohexane | 3.7 ± 1.4 |
| DmmAshort | 1,6-diiodohexane | 3.2 ± 0.7 |
| DmmAlong | 1,3-dibromopropane | 10 ± 2 |
Normalized to activity of DmmAshort with 1 mM 1,3-dibromopropane.
Table II.
Steady State Kinetic Parameters of DmmAshort
| kcat (s−1) | Km (mM) | kcat/Km (M−1s−1) |
|---|---|---|
| 2.4 ± 0.1 | 0.24 ± 0.05 | 1.0 ± 0.3 × 104 |
Structure of DmmA
We solved crystal structures for both DmmAshort and DmmAlong (Table III). The first structure was determined from selenomethionyl DmmAshort (2.2 Å). DmmAlong (2.9 Å) was solved from the DmmAshort structure. The short and long variants crystallized under different conditions in different crystal forms, each with two polypeptides per asymmetric unit. The two structures thus provide four independent views of the DmmA dehalogenase.
Table III.
Crystallographic Summary
| DmmAshort (SeMet) | DmmAshort (SeMet) 1,5-dibromopentane soaked | DmmAlong | |
|---|---|---|---|
| Diffraction Data | |||
| Space group | P62 | P62 | P3121 |
| X-ray source | APS 23-ID-D | APS 23-ID-D | APS 23-ID-D |
| a, b, c (Å) | 99.8, 99.8, 122.0 | 100.3, 100.3, 121.4 | 144.7, 144.7, 105.0 |
| α, β, γ (°) | 90, 90, 120 | 90, 90, 120 | 90, 90, 120 |
| Wavelength (Å) | 0.97939 | 0.92003 | 0.97939 |
| dmin (Å) | 2.20 (2.28–2.2)a | 2.10 (2.18–2.10) | 2.50 (2.50–2.64) |
| Avg I/σI | 13.2 (2.9) | 16.7 (2.1) | 10.4 (3.5) |
| Rsymmb | 0.122 (0.549) | 0.118 (0.75) | 0.171 (0.607) |
| Completeness | 100.0 (99.9) | 99.7 (97.6) | 100 (100) |
| Avg. redundancy | 5.8 (5.2) | 10.5 (7.6) | 9.6 (8.6) |
| Unique reflections | 35,082 | 40,262 | 44,165 |
| Refinement | |||
| Data range (Å) | 86.39–2.20 | ||
| No. reflections | 34,919 | ||
| Rwork/Rfreec | 0.139/0.177 | ||
| RMS deviations | |||
| Bonds (Å) | 0.009 | ||
| Angles (°) | 1.195 | ||
| Avg B-factors (Å2) | |||
| Protein | 23.4 | ||
| Ligand/Ion | 53.3 | ||
| Water | 35.0 | ||
| Ramachandran | |||
| Allowed | 99.66% | ||
| Outliers | 0.34% | ||
| Number of Atoms | |||
| Protein | 4649 | ||
| Ligand/Ion | 18 | ||
| Water | 497 | ||
Outermost shell in parentheses.
Including anomalous differences.
The Rfree data set included a random 5% of reflections.
Due to the higher-resolution of DmmAshort, our efforts focused on analysis of that form of the protein. DmmA adopts the α/β hydrolase fold as expected [Fig. 1(A)], consisting of core and lid sub-domains with the active site at the top of the core, which is covered by the lid [Fig. 1(A,B)]. DmmAshort and DmmAlong are identical within experimental error (root-mean-square deviation (RMSD) = 0.32 Å for 298 Cα atoms). The DmmAshort structure is complete with no disordered residues. In DmmAlong, six additional N-terminal residues are visible compared with DmmAshort, but 37 residues at the N-terminus are disordered [Fig. 1(C)].
Figure 1.

Structure of DmmA. A: DmmA polypeptide. The stereo ribbon diagram is colored as a rainbow from blue at the N-terminus to red at the C-terminus with catalytic pentad residues in stick form with magenta C. B: Topology diagram. The α/β hydrolase core is conserved among all α/β hydrolases and the lid is conserved among haloalkane dehalogenases. Residues of the catalytic pentad are labeled. C: Superposition of DmmAlong (gray) and DmmAshort (rainbow main-chain, magenta active site). Six additional residues on the N-terminus of DmmAlong are visible. D: Protein-protein contact in the crystal structures. Only the left molecules of DmmAshort (rainbow main-chain, magenta active site) and DmmAlong (gray, purple N-terminal extension) are superimposed. The near superposition of the right molecules highlights the similar interfaces. Contacts of α10, β8, and N-terminal extension are labeled.
DmmA oligomeric state
A common protein-protein contact exists in the unrelated crystal forms of DmmAlong and DmmAshort. The contact has twofold (non-crystallographic) symmetry, suggesting that the protein may be dimeric [Fig. 1(D)]. The primary interaction occurs within the HLD core between α10 and β8, and is comprised of both hydrophobic and hydrophilic contacts and buries only four water molecules. The additional six ordered residues in DmmAlong (residues 38–43) associate with the partner subunit at this interface, resulting in a larger buried surface area in DmmAlong compared to DmmAshort (980 Å2 vs. 610 Å2) [Fig. 1(D)]. The small size of the interface led us to question whether it exists in solution. Other HLDs have been characterized as monomers, dimers, or dimers only under low-salt conditions. Among HLDs of known structure, only DbjA is dimeric in solution.36 The helix α10 is central to the subunit contacts in both DmmA and DbjA, but the interfaces otherwise differ (Supporting Information Fig. 3).
The quaternary structure in solution was probed by analytical gel filtration chromatography. Both DmmAshort and DmmAlong eluted with apparent molecular weights intermediate between calculated values for the monomer and the dimer (48 kDa observed vs. 34.6 kDa for a DmmAshort monomer, and 59 kDa vs. 38.8 kDa for DmmAlong) (Supporting Information Fig. 4). This result implies a dynamic equilibrium between monomeric and dimeric forms, however the retention volume did not change over a 20-fold concentration range of either DmmAshort or DmmAlong. In addition, the larger buried surface in the putative DmmAlong dimer compared to DmmAshort does not result in a more dimeric retention volume. Thus, we conclude that the interface observed in the crystal structures does not represent an interface in solution and that DmmA is monomeric. The aberrant elution volumes may result from the dynamic behavior of a monomeric DmmA.
DmmA active site
The active site has an intact and well ordered catalytic pentad, consisting of the nucleophile (Asp144), base (His315), acid (Glu168), and two halide-stabilizing residues (Trp145 and Asn78) [Fig. 2(A,B)]. The nucleophile (Asp144), base (His315), and Trp145 are conserved in all HLDs. Asn78 is conserved in subfamilies II and III, and Glu168 is present only in subfamily II. The catalytic pentad resides are in an otherwise hydrophobic chamber that is accessed through an entrance tunnel. This feature and the location of the pentad are well conserved in structures of all subfamily II HLDs [Fig. 2(C)]. Density resembling malonate, a component of the crystallization solution, was found in the active site cleft [Fig. 2(A), Supporting Information Fig. 5]. A product alcohol is bound at this position in structures of several other HLDs [Fig. 2(C)]. Difference density for a stronger scatterer than water (peak height 7.7 σ) appeared in the putative halide-binding site within hydrogen bonding distance of Trp145 and Asn78, the halide-stabilizing residues [Fig. 2(A), Supporting Information Fig. 5). A halide occupies this position in the structures of several other HLDs [Fig. 2(C)]. The halide-binding site was confirmed by Br anomalous scattering from a crystal of DmmAshort soaked in 1,5-dibromopentane prior to data collection at the energy of the bromine K absorption edge. Br− binding resulted in an anomalous difference peak of height 9 σ; no density was observed for the hydroxyl product (Supporting Information Fig. 6).
Figure 2.
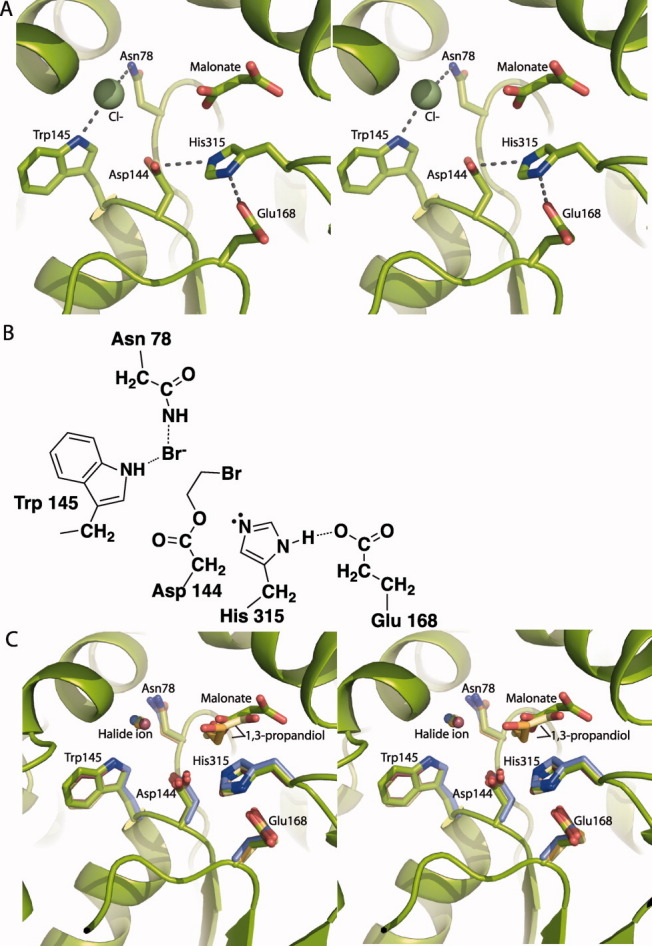
DmmA active site. A: DmmA active site showing catalytic pentad (sticks), Cl− and malonate. B: Schematic of the DmmA active site showing the acyl-enzyme intermediate. The halide-stabilizing residues (Asn78 and Trp 145) bind the halide released when generating the acyl enzyme intermediate. The acyl enzyme on Asp144 will subsequently be hydrolyzed by a water activated by His315 C. Active site of subfamily II haloalkane dehalogenases, based on superposition of core Cα atoms. The catalytic pentad is shown in sticks and bound halide ion as a sphere for DmmA (green C), LinB 1IZ819 (orange C), Rv2578 2O2I22 (yellow C), DbjA 3A2M21 (purple C), and DhaA 1CQW15 (mauve C). Bound malonate is shown for DmmA and bound product, 1,3-propandiol, for Rv2578 (yellow C) and LinB (orange C).
Comparison to other HLDs
The DmmA structure is similar to structures of other subfamily II HLDs (0.5–0.7 Å RMSD for 219–226 Cα atoms of proteins that are 38–48% identical overall to DmmA). The largest difference between the most similar structure, DhaA, and DmmA is in lid helix α4 and the core-to-lid linker from β6 to α4 [Fig. 3(A)]. As expected, DmmA is less similar to the subfamily I HLDs, DhlA (1.4 Å RMSD for 203 Cα) and DppA (1.9 Å for 209 Cα), which have lower overall sequence identity to DmmA.
Figure 3.
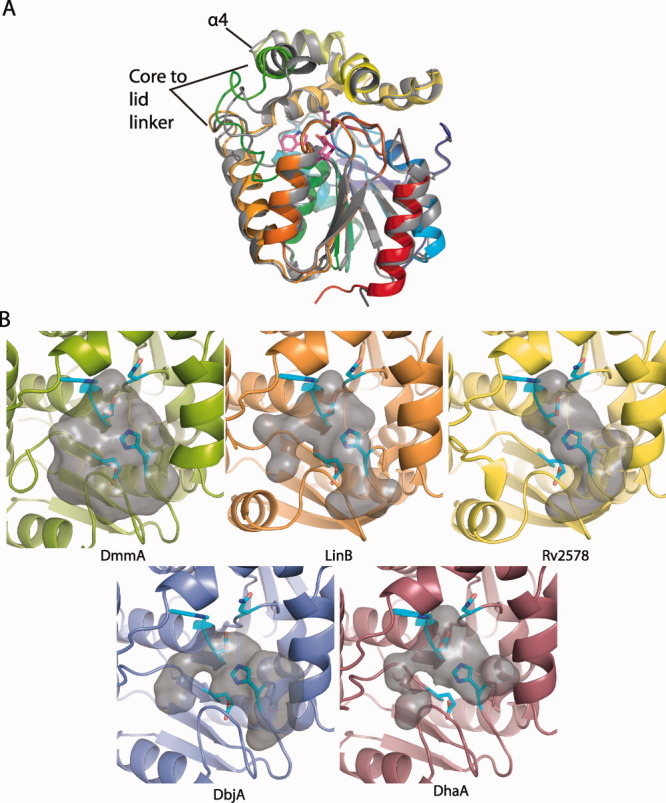
DmmA and other HLDs. A: Superposition of DmmA (rainbow main-chain, magenta active site) and the HLD DhaA (1BN715). Differences in the lid-to-core linker and α4 are labeled. B: Active site cavity of DmmA and other type II haloalkane dehalogenases: DmmA (green), LinB 1IZ819 (orange), Rv2578 2O2I22 (yellow), DbjA 3A2M21 (purple), and DhaA 1CQW15 (mauve). The gray surface represents the internal volume of the cavity. Residues that form the forward surface are omitted. Active site residues are shown in sticks (cyan C, blue N, red O).
Structure-function studies of other HLDs have demonstrated that the size of the active site cavity and entry tunnel play a role in substrate specificity8 and chiral selectivity.21 DmmA has a larger entry tunnel and wider substrate cleft than other HLDs [Fig. 3(B)]. This difference is due primarily to the position of lid helix α4 and the preceding loop, and to a lesser extent to amino acid substitutions within the cavity. The loop and helix α4 are the parts of the DmmA structure that differ most from other HLDs. Other HLDs with large active sites17, 21 have very broad substrate specificity and react well with larger substrates.8 DmmA, with an even larger active site, should accommodate substrates of greater size and shape diversity. To test this, we assayed DmmA for dehalogenase activity with several halogenated molecules larger than the 1,3-dibromopropane standard substrate (1,6-dibromohexane, 1,6-dichlorohexane, 1,6-diiodohexane, and bromocyclohexane) (Table I). In contrast to those tested previously,8 DmmA has greater activity for the bromocyclohexane compared to the larger straight-chain substrates. This observation is consistent with the wide cavity of DmmA and suggests that it has robust activity toward other bulky substrates.
Discussion
This work establishes that DmmA is a HLD. The kinetic properties of DmmA with a standard substrate, 1,3-dibromopropane, are within the range reported for other HLDs. However, as for most HLDs, the natural substrates are unknown. Of the two protein variants studied, DmmAshort was significantly faster than DmmAlong, exhibiting tenfold greater activity. Although most of the additional 43 residues on the N-terminus of DmmAlong are disordered, they may interfere with substrate access to the active site, resulting in the reduced activity observed for the longer form of the protein.
The DmmA structure has the same fold and active site position as other HLDs, as expected. Nevertheless, DmmA has unique structural features that set it apart from other enzymes within this class. The active site cavity of DmmA is significantly larger than the cavity of other homologs, which was not predictable from straightforward sequence comparisons to other HLDs of known structure. Instead, sequence differences led to subtle structural variation in the position of helix α4 and the core-to-lid linker, which resulted in a large cavity volume. The size of amino acid side chains lining the active site pocket makes a secondary contribution to the cavity size. The larger cavity volume may confer an ability to accept a wide range of large, bulky substrates compared to other HLDs. Among the larger substrates tested, DmmA had a preference for the bulky bromocyclohexane over corresponding linear molecules. This suggests that the natural DmmA substrate may bear a halogenated ring system. The large active site also suggests the value of DmmA as a potential chemoenzymatic tool for large substrates.
The ultimate biological source of DmmA is unknown, as attempts to amplify dmmA from M. producta (formerly L. majuscula) DNA or to find dmmA in the genome sequence have been unsuccessful. dmmA (originally annotated as curN23) was originally found in the cosmid library developed from an M. producta field isolate, which also contained bacterial symbionts or associants as a microbial consortium. To our knowledge, no other HLD has been found in a cyanobacterium, which also suggests an origin outside M. producta, in a symbiont or associated bacterium. Presumably, the natural function of DmmA is to dehalogenate molecules present in the native marine environment of M. producta. In this respect, M. producta has been a rich source of halogenated metabolites, including in this strain, barbamide, which possesses a trichloromethyl group,37 and in another strain, jamaicamide, which possesses vinyl chloride and alkynyl bromide groups.38 Despite its orphan status, the large active-site cavity poises DmmA for development as a new biotechnology tool.
Materials and Methods
Cloning, protein expression, and purification
Gene fragments encoding DmmA were generated from cosmid pLM17,23 inserted into the pET-24b vector, and verified by sequencing to form pDmmAlong (encoding residues 1-341) and pDmmAshort (encoding residues 44-341). E. coli strain BL21(DE3) was transformed with the expression plasmid, grown at 37°C in 1 L 2xYT to an OD600 of 0.5, cooled to 18°C, induced with IPTG (final concentration 0.4 mM) and grown for an additional 18 h. Selenomethionyl (SeMet) DmmAshort was produced in the same strain in SeMet minimal media.39
All purification steps were performed at 4°C. The cell pellet from 1 L of cell culture was resuspended in 40-mL Buffer A (20 mM Tris pH 7.9, 500 mM NaCl, 10% glycerol) plus 20-mM imidazole, lysed by sonication, and the soluble fraction loaded onto a 5-mL HisTrap Ni NTA column (GE Healthcare). DmmA was eluted with Buffer A with a linear gradient of 20–650 mM imidazole. DmmA was further purified by size exclusion chromatography with a HiPrep 16/60 Sephacryl S100 HR column (GE Healthcare) prequilibrated with Buffer A. DmmA was concentrated to 14 mg/mL, flash frozen in liquid N2, and stored at −80°C. SeMet DmmAshort was purified as the wild type with addition of 2 mM DDT to Buffer A during the size exclusion step. Yields per 1 L culture were 150 mg for DmmAshort, 80 mg for DmmAlong, and 15 mg for SeMet DmmAshort.
Crystallization
Crystals of DmmAshort (native and SeMet) grew by vapor diffusion at 20°C within 24 h from a 1:1 mix of protein stock (6 mg/mL DmmAshort in Buffer A) and well solution (2.2 M sodium malonate pH 7.0, 5% glycerol). Crystals of DmmAlong formed at 20°C within 24–48 h from a 1:1 mix of protein stock (14 mg/mL DmmAlong in Buffer A) and well solution (0.6 M lithium sulfate, 27.5% PEG3350, and 0.1 M BisTris pH 5.5). Crystals of DmmAlong were cyroprotected by a 5–10 s transfer to a mixture of well solution also containing 25% PEG400.
Data collection and structure determination
Data were collected at the GM/CA-CAT beamline 23ID-D at the advanced photon source (APS) at Argonne National Lab (Argonne, IL). A 2.2 Å single-wavelength anomalous diffraction dataset was collected at the wavelength of peak absorption at the selenium edge from a DmmAshort SeMet crystal. A 2.9 Å dataset was collected from a DmmAlong crystal. All data were processed using the HKL2000 suite.40 Determination of selenium atomic positions, experimental phasing, density modification phase refinement, and initial model building were performed using the programs SOLVE and RESOLVE.41, 42 And 22 of the 24 expected selenium positions were identified. The model was finished manually in COOT.43 REFMAC530 from the CCP4 suite31 was used for refinement. The DmmAlong structure was solved by molecular replacement in Phaser32 using DmmAshort as a search model. Model quality was evaluated with MolProbity.33 Figures were made using PyMOL34 and hollow.35 Electron density was complete throughout the polypeptide chain for both polypeptides for DmmAshort (residues 44-341) and DmmAlong (residues 38–341). No density was observed for the C-terminal His tag of either DmmA variant or for the first 37 residues of DmmAlong.
Enzyme assay
Activity was measured using a pH indicator dye-based colorimetric method.27, 28 Substrate [0.1–6.0 mM 1,3-dibromopropane (Aldrich)] was prepared in an indicator solution (20 μg/mL phenol red, 20 mM Na2SO4, 1 mM EDTA, 1 mM Hepes pH 8.2). Reactions were initiated by addition of DmmAshort (1 μM final concentration, 100-μL final volume). Absorbance at 550 nm was monitored at 5-second intervals during the reaction to detect the decrease in pH generated by the release of H+. Parallel negative controls lacking the enzyme or substrate were used to correct for nonenzymatic dehalogenation or pH change. A standard curve relating absorbance at 550 nm to [H+] was used to convert absorbance to [H+]. The initial rates were fit using KaleidaGraph to the Michaelis-Menten equation (vinitial = ([enzyme]*[substrate]*kcat)/(Km+[substrate]) to obtain steady state kinetic constants. Comparison with other substrates (1,6-dibromohexane (Acros), 1,6-dichlorohexane (Aldrich), 1,6-diiodohexane (Aldrich), bromocyclohexane (Aldrich)) were performed at 1 mM substrate concentration in duplicate and normalized to the percent of 1,3-dibromopropane activity.
PDB Coordinates
Coordinate and structure factors for DmmAshort have been deposited in the Protein Data Bank with accession number 3U1T.
Acknowledgments
Beamline 23ID-D is supported by the National Institutes of Health, National Institute of General Medical Sciences Grant Y1-GM-1104 and National Cancer Institute Grant Y1-CO-1020 through the GM/CA Collaborative Access Team at the APS, which is supported by the United States Department of Energy.
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Stucki G, Thuer M. Experiences of a large-scale application of 1,2-dichloroethane degrading microorganisms for groundwater treatment. Environ Sci Technol. 1995;29:2339–2345. doi: 10.1021/es00009a028. [DOI] [PubMed] [Google Scholar]
- 2.Alcalde M, Ferrer M, Plou FJ, Ballesteros A. Environmental biocatalysis: from remediation with enzymes to novel green processes. Trends Biotechnol. 2006;24:281–287. doi: 10.1016/j.tibtech.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 3.Swanson PE. Dehalogenases applied to industrial-scale biocatalysis. Curr Opin Biotechnol. 1999;10:365–369. doi: 10.1016/S0958-1669(99)80066-4. [DOI] [PubMed] [Google Scholar]
- 4.Prokop Z, Oplustil F, DeFrank J, Damborsky J. Enzymes fight chemical weapons. Biotechnol J. 2006;1:1370–1380. doi: 10.1002/biot.200600166. [DOI] [PubMed] [Google Scholar]
- 5.Campbell DW, Muller C, Reardon KF. Development of a fiber optic enzymatic biosensor for 1,2-dichloroethane. Biotechnol Lett. 2006;28:883–887. doi: 10.1007/s10529-006-9014-x. [DOI] [PubMed] [Google Scholar]
- 6.Bidmanova S, Chaloupkova R, Damborsky J, Prokop Z. Development of an enzymatic fiber-optic biosensor for detection of halogenated hydrocarbons. Anal Bioanal Chem. 2010;398:1891–1898. doi: 10.1007/s00216-010-4083-z. [DOI] [PubMed] [Google Scholar]
- 7.Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, Simpson D, Mendez J, Zimmerman K, Otto P, Vidugiris G, Zhu J, Darzins A, Klaubert DH, Bulleit RF, Wood KV. HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol. 2008;3:373–382. doi: 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- 8.Koudelakova T, Chovancova E, Brezovsky J, Monincova M, Fortova A, Jarkovsky J, Damborsky J. Substrate specificity of haloalkane dehalogenases. Biochem J. 2011;435:345–354. doi: 10.1042/BJ20101405. [DOI] [PubMed] [Google Scholar]
- 9.Janssen DB. Evolving haloalkane dehalogenases. Curr Opin Chem Biol. 2004;8:150–159. doi: 10.1016/j.cbpa.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 10.Chovancova E, Kosinski J, Bujnicki JM, Damborsky J. Phylogenetic analysis of haloalkane dehalogenases. Proteins. 2007;67:305–316. doi: 10.1002/prot.21313. [DOI] [PubMed] [Google Scholar]
- 11.Verschueren KH, Kingma J, Rozeboom HJ, Kalk KH, Janssen DB, Dijkstra BW. Crystallographic and fluorescence studies of the interaction of haloalkane dehalogenase with halide ions. Studies with halide compounds reveal a halide binding site in the active site. Biochemistry. 1993;32:9031–9037. doi: 10.1021/bi00086a008. [DOI] [PubMed] [Google Scholar]
- 12.Verschueren KH, Seljee F, Rozeboom HJ, Kalk KH, Dijkstra BW. Crystallographic analysis of the catalytic mechanism of haloalkane dehalogenase. Nature. 1993;363:693–698. doi: 10.1038/363693a0. [DOI] [PubMed] [Google Scholar]
- 13.Pikkemaat MG, Ridder IS, Rozeboom HJ, Kalk KH, Dijkstra BW, Janssen DB. Crystallographic and kinetic evidence of a collision complex formed during halide import in haloalkane dehalogenase. Biochemistry. 1999;38:12052–12061. doi: 10.1021/bi990849w. [DOI] [PubMed] [Google Scholar]
- 14.Hesseler M, Bogdanovic X, Hidalgo A, Berenguer J, Palm GJ, Hinrichs W, Bornscheuer UT. Cloning, functional expression, biochemical characterization, and structural analysis of a haloalkane dehalogenase from Plesiocystis pacifica SIR-1. Appl Microbiol Biotechnol. 2011;91:1049–1060. doi: 10.1007/s00253-011-3328-x. [DOI] [PubMed] [Google Scholar]
- 15.Newman J, Peat TS, Richard R, Kan L, Swanson PE, Affholter JA, Holmes IH, Schindler JF, Unkefer CJ, Terwilliger TC. Haloalkane dehalogenases: structure of a Rhodococcus enzyme. Biochemistry. 1999;38:16105–16114. doi: 10.1021/bi9913855. [DOI] [PubMed] [Google Scholar]
- 16.Stsiapanava A, Dohnalek J, Gavira JA, Kuty M, Koudelakova T, Damborsky J, Kuta Smatanova I. Atomic resolution studies of haloalkane dehalogenases DhaA04, DhaA14 and DhaA15 with engineered access tunnels. Acta Crystallogr D Biol Crystallogr. 2010;66:962–969. doi: 10.1107/S0907444910027101. [DOI] [PubMed] [Google Scholar]
- 17.Marek J, Vevodova J, Smatanova IK, Nagata Y, Svensson LA, Newman J, Takagi M, Damborsky J. Crystal structure of the haloalkane dehalogenase from Sphingomonas paucimobilis UT26. Biochemistry. 2000;39:14082–14086. doi: 10.1021/bi001539c. [DOI] [PubMed] [Google Scholar]
- 18.Oakley AJ, Prokop Z, Bohac M, Kmunicek J, Jedlicka T, Monincova M, Kuta-Smatanova I, Nagata Y, Damborsky J, Wilce MC. Exploring the structure and activity of haloalkane dehalogenase from Sphingomonas paucimobilis UT26: evidence for product- and water-mediated inhibition. Biochemistry. 2002;41:4847–4855. doi: 10.1021/bi015734i. [DOI] [PubMed] [Google Scholar]
- 19.Streltsov VA, Prokop Z, Damborsky J, Nagata Y, Oakley A, Wilce MC. Haloalkane dehalogenase LinB from Sphingomonas paucimobilis UT26: X-ray crystallographic studies of dehalogenation of brominated substrates. Biochemistry. 2003;42:10104–10112. doi: 10.1021/bi027280a. [DOI] [PubMed] [Google Scholar]
- 20.Oakley AJ, Klvana M, Otyepka M, Nagata Y, Wilce MC, Damborsky J. Crystal structure of haloalkane dehalogenase LinB from Sphingomonas paucimobilis UT26 at 0.95 A resolution: dynamics of catalytic residues. Biochemistry. 2004;43:870–878. doi: 10.1021/bi034748g. [DOI] [PubMed] [Google Scholar]
- 21.Prokop Z, Sato Y, Brezovsky J, Mozga T, Chaloupkova R, Koudelakova T, Jerabek P, Stepankova V, Natsume R, van Leeuwen JG, Janssen DB, Florian J, Nagata Y, Senda T, Damborsky J. Enantioselectivity of haloalkane dehalogenases and its modulation by surface loop engineering. Angew Chem Int Ed Engl. 2010;49:6111–6115. doi: 10.1002/anie.201001753. [DOI] [PubMed] [Google Scholar]
- 22.Mazumdar PA, Hulecki JC, Cherney MM, Garen CR, James MN. X-ray crystal structure of Mycobacterium tuberculosis haloalkane dehalogenase Rv2579. Biochim Biophys Acta. 2008;1784:351–362. doi: 10.1016/j.bbapap.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 23.Chang Z, Sitachitta N, Rossi JV, Roberts MA, Flatt PM, Jia J, Sherman DH, Gerwick WH. Biosynthetic pathway and gene cluster analysis of curacin A, an antitubulin natural product from the tropical marine cyanobacterium Lyngbya majuscula. J Nat Prod. 2004;67:1356–1367. doi: 10.1021/np0499261. [DOI] [PubMed] [Google Scholar]
- 24.Engene N, Rottacker EC, Kastovsky J, Byrum T, Choi H, Ellisman MH, Komarek J, Gerwick WH. Moorea producta gen. nov., sp. nov. and Moorea bouillonii comb. nov., tropical marine cyanobacteria rich in bioactive secondary metabolites. Int J Syst Evol Microbiol. doi: 10.1099/ijs.0.033761-0. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gu L, Wang B, Kulkarni A, Gehret JJ, Lloyd KR, Gerwick L, Gerwick WH, Wipf P, Hakansson K, Smith JL, Sherman DH. Polyketide decarboxylative chain termination preceded by o-sulfonation in curacin a biosynthesis. J Am Chem Soc. 2009;131:16033–16035. doi: 10.1021/ja9071578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones AC, Monroe EA, Podell S, Hess WR, Klages S, Esquenazi E, Niessen S, Hoover H, Rothmann M, Lasken RS, Yates JR, III, Reinhardt R, Kube M, Burkart MD, Allen EE, Dorrestein PC, Gerwick WH, Gerwick L. Genomic insights into the physiology and ecology of the marine filamentous cyanobacterium Lyngbya majuscula. Proc Natl Acad Sci U S A. 2011;108:8815–8820. doi: 10.1073/pnas.1101137108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schindler JF, Naranjo PA, Honaberger DA, Chang CH, Brainard JR, Vanderberg LA, Unkefer CJ. Haloalkane dehalogenases: steady-state kinetics and halide inhibition. Biochemistry. 1999;38:5772–5778. doi: 10.1021/bi982853y. [DOI] [PubMed] [Google Scholar]
- 28.Marvanova S, Nagata Y, Wimmerova M, Sykorova J, Hynkova K, Damborsky J. Biochemical characterization of broad-specificity enzymes using multivariate experimental design and a colorimetric microplate assay: characterization of the haloalkane dehalogenase mutants. J Microbiol Methods. 2001;44:149–157. doi: 10.1016/s0167-7012(00)00250-5. [DOI] [PubMed] [Google Scholar]
- 29.Bosma T, Pikkemaat MG, Kingma J, Dijk J, Janssen DB. Steady-state and pre-steady-state kinetic analysis of halopropane conversion by a rhodococcus haloalkane dehalogenase. Biochemistry. 2003;42:8047–8053. doi: 10.1021/bi026907m. [DOI] [PubMed] [Google Scholar]
- 30.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 31.CCP4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 32.Zwart PH, Afonine PV, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, McKee E, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Storoni LC, Terwilliger TC, Adams PD. Automated structure solution with the PHENIX suite. Methods Mol Biol. 2008;426:419–435. doi: 10.1007/978-1-60327-058-8_28. [DOI] [PubMed] [Google Scholar]
- 33.Chen VB, Arendall WB, III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DeLano WL. The PyMOL Molecular graphics system, Version 1.3. Palo Alto, CA: DeLano Scientific; 2002. Schrödinger, LLC. [Google Scholar]
- 35.Ho BK, Gruswitz F. HOLLOW: generating accurate representations of channel and interior surfaces in molecular structures. BMC Struct Biol. 2008;8:49. doi: 10.1186/1472-6807-8-49. –54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hasan K, Fortova A, Koudelakova T, Chaloupkova R, Ishitsuka M, Nagata Y, Damborsky J, Prokop Z. Biochemical characteristics of the novel haloalkane dehalogenase DatA, isolated from the plant pathogen Agrobacterium tumefaciens C58. Appl Environ Microbiol. 2011;77:1881–1884. doi: 10.1128/AEM.02109-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chang Z, Flatt P, Gerwick WH, Nguyen VA, Willis CL, Sherman DH. The barbamide biosynthetic gene cluster: a novel marine cyanobacterial system of mixed polyketide synthase (PKS)-non-ribosomal peptide synthetase (NRPS) origin involving an unusual trichloroleucyl starter unit. Gene. 2002;296:235–247. doi: 10.1016/s0378-1119(02)00860-0. [DOI] [PubMed] [Google Scholar]
- 38.Edwards DJ, Marquez BL, Nogle LM, McPhail K, Goeger DE, Roberts MA, Gerwick WH. Structure and biosynthesis of the jamaicamides, new mixed polyketide-peptide neurotoxins from the marine cyanobacterium Lyngbya majuscula. Chem Biol. 2004;11:817–833. doi: 10.1016/j.chembiol.2004.03.030. [DOI] [PubMed] [Google Scholar]
- 39.Guerrero SA, Hecht HJ, Hofmann B, Biebl H, Singh M. Production of selenomethionine-labelled proteins using simplified culture conditions and generally applicable host/vector systems. Appl Microbiol Biotechnol. 2001;56:718–723. doi: 10.1007/s002530100690. [DOI] [PubMed] [Google Scholar]
- 40.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode methods in enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 41.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr D Biol Crystallogr. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Terwilliger TC. Automated main-chain model building by template matching and iterative fragment extension. Acta Crystallogr D Biol Crystallogr. 2003;59:38–44. doi: 10.1107/S0907444902018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.