

Abstract
AAA+ proteases employ a hexameric ring that harnesses the energy of ATP binding and hydrolysis to unfold native substrates and translocate the unfolded polypeptide into an interior compartment for degradation. What determines the ability of different AAA+ enzymes to unfold and thus degrade different native protein substrates is currently uncertain. Here, we explore the ability of the E. coli Lon protease to unfold and degrade model protein substrates beginning at N-terminal, C-terminal, or internal degrons. Lon has historically been viewed as a weak unfoldase, but we demonstrate robust and processive unfolding/degradation of some substrates with very stable protein domains, including mDHFR and titinI27. For some native substrates, Lon is a more active unfoldase than related AAA+ proteases, including ClpXP and ClpAP. For other substrates, this relationship is reversed. Thus, unfolding activity does not appear to be an intrinsic enzymatic property. Instead, it depends on the specific protease and substrate, suggesting that evolution has diversified rather than optimized the protein unfolding activities of different AAA+ proteases.
Keywords: protein unfolding, AAA+ proteolytic machine, ATP-dependent denaturation, ranking unfolding power
Introduction
Intracellular protein degradation is used to eliminate damaged proteins, to sculpt the detailed composition of the proteome, and to regulate processes such as cell division, DNA replication, gene expression, and metabolite synthesis.1–4 Because proteolysis is irreversible, it must be carefully regulated to avoid random destruction of cellular proteins. Indeed, AAA+ proteases, the enzymes responsible for most nonlysosomal protein degradation within cells, are designed to carry out highly selective proteolysis in protein-rich environments. This selectivity depends on sequestering the proteolytic active sites in a chamber with small entry portals that exclude native proteins.4 As a consequence, AAA+ proteases must bind a protein substrate, unfold any native tertiary structure, and then translocate the denatured polypeptide into the proteolytic chamber. Unfolding is therefore an integral step in protein degradation by AAA+ proteases.
Escherichia coli utilizes five different AAA+ proteases: Lon, ClpXP, ClpAP, HslUV, and FtsH.1 Related enzymes are widespread throughout the bacterial kingdom, in archaea, and in the organelles of eukaryotes. The eukaryotic, archaeal, and mycobacterial proteasomes are also AAA+ proteases. Substantial variation in the protein-unfolding activities of AAA+ proteases has been observed, suggesting that certain AAA+ enzymes are intrinsically more powerful than others and thus that unfolding activity may play an important role in substrate selection.5 In each AAA+ protease, a hexameric ring recognizes a substrate, unfolds the protein, and translocates the denatured polypeptide through a central axial pore and into the proteolytic chamber.4 Protein unfolding is initiated when loops that line the narrow axial pore of the AAA+ ring bind an unstructured segment of a protein substrate, often called a degradation tag or degron. Repeated cycles of ATP binding and hydrolysis then power conformational changes that pull the tag through the pore and eventually tug the native portion of the substrate against the AAA+ ring, creating an unfolding force. Depending on the native substrate and enzyme, successful unfolding can require anywhere from a few to many hundreds of cycles of ATP hydrolysis.6–8 Moreover, when an unfolding attempt fails, two outcomes are possible. The enzyme can maintain its grip on the substrate and simply try again.8 Alternatively, the substrate can slip or dissociate from the enzyme's grasp, requiring rebinding before further unfolding attempts.9–11 If a partially degraded substrate dissociates from the protease, rebinding is only possible if a functional degradation tag is still present. Otherwise, partially degraded substrate fragments accumulate as proteolytic products, which is often observed during degradation in vitro of multi-domain substrates containing very stable interior domains.5, 9, 10, 12, 13
Following attachment of suitable degrons, mouse dihydrofolate reductase (mDHFR) is degraded by many AAA+ proteases in vitro.5, 9 Importantly, however, degradation can be blocked by the inhibitor methotrexate (MTX), which binds mDHFR tightly, increases its stability, and prevents enzymatic unfolding. Koodathingal et al. designed substrates in which AAA+ proteases encounter the mDHFR domain only after degrading a recognition tag and intervening protein domains.5 Under these circumstances, the protease partitions between mDHFR unfolding/degradation (kdeg) and substrate release (krel), which is generally irreversible. Because MTX completely blocks degradation, an unfolding processivity ratio (U = kdeg/krel) can be calculated from the intensities of partially degraded products observed by SDS-PAGE in the presence (I+MTX) or absence (I–MTX) of MTX [U = (I+MTX/I–MTX) – 1)].5 For Lon degradation, the processivity ratio was ∼0.1, indicating that Lon dissociates approximately 10-fold more frequently than it unfolds and degrades mDHFR. By contrast, substantially higher processivity ratios were observed for ClpXP, ClpAP, and other AAA+ proteases, suggesting that these enzymes are more powerful protein unfoldases than Lon. At one level, it might not seem surprising that Lon appears to have a weak protein-unfolding activity, as Lon plays a prominent role in degradation of unfolded and misfolded proteins.14 However, Lon also degrades native proteins, such as SulA and UmuD.15, 16 Moreover, we previously reported that the Lon enzymes from E. coli and Mesoplasma florum were capable of degrading model substrates containing stable titinI27 domains.7, 17, 18
Here, we probe the unfolding and degradation activity of E. coli Lon using multi-domain substrates containing mDHFR, titinI27, or green fluorescent protein (GFP). Each of these proteins displays significant thermodynamic and/or kinetic stability.6, 19–22 The titinI27, and GFP proteins are also quite stable to mechanical denaturation in atomic-force microscopy experiments,23, 24 and all three proteins have been used as model substrates for degradation by ClpXP and ClpAP.6, 13, 20, 25–29 In comparison with other AAA+ proteases, we find that Lon can be either a stronger or a weaker unfoldase, depending on the substrate and position of the degradation tag. For example, Lon is more active than ClpXP and ClpAP in unfolding some native substrates but less active for other substrates. We conclude that unfolding activity depends upon the specific protease and target substrate. Our results also demonstrate that Lon can initiate degradation from an internal degron flanked by native protein domains and can engage and degrade a very poor substrate, if it is tethered to Lon via a domain with a good Lon degron. We discuss the implications of these results for physiology and enzyme/substrate coevolution.
Results
Lon degrades tagged mDHFR efficiently
To examine Lon degradation of mDHFR, we initially fused this protein to the sul20C tag, which consists of the 20 C-terminal amino acids of E. coli SulA and serves as an efficient recognition signal for Lon degradation.7 As assayed by SDS-PAGE, E. coli Lon degraded untagged mDHFR very slowly (Fig. 1, top panel). When MTX was present, Lon also failed to degrade detectable amounts of the tagged mDHFR-sul20C protein (Fig. 1, middle panel). In the absence of MTX, however, Lon (0.3 μM hexamer) degraded mDHFR-sul20C (5 μM) almost completely in 10 min [Fig. 1(A), bottom panel], corresponding to an average degradation rate of ∼1.6 substrates min−1 enz−1. Insolubility of mDHFR-sul20C at higher concentrations precluded determination of Vmax for degradation, which could be substantially higher than the observed degradation rate given the KM values for other sul20C-tagged substrates.7 Irrespective of the exact value of Vmax, mDHFR does not appear to be inherently resistant to unfolding or to other steps in Lon-mediated proteolysis. Moreover, the observed rate of ∼1.6 substrates min−1 enz−1 for Lon degradation of DHFR-sul20C is similar to the maximal rates at which many protein substrates are degraded by the AAA+ ClpXP and ClpAP proteases (see Discussion).
Figure 1.
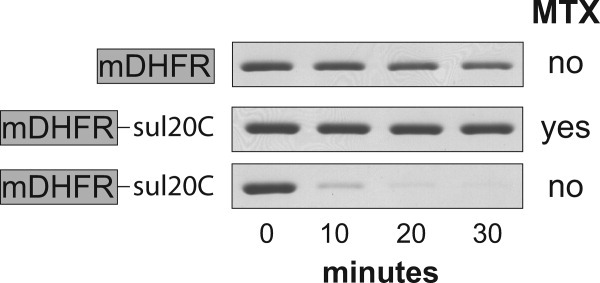
Degradation of mDHFR variants by Lon. Protein samples (5 μM) were incubated with Lon protease (0.3 μM hexamer) and ATP (2 mM; plus a regeneration system) at 37°C in the presence or absence of methotrexate (MTX; 10 μM). At different times, aliquots were removed, quenched, and then analyzed by SDS-PAGE and staining with Coomassie blue. Untagged mDHFR was a poor substrate (upper panel) as was mDHFR-sul20C in the presence of MTX (middle panel). In the absence of MTX, mDHFR-sul20C was rapidly degraded (lower panel).
Degradation of mDHFR fusion proteins
Next, we constructed and purified an mDHFR-titinI27-sul20C fusion protein. In this construct, Lon should unfold and degrade most of the titin domain before engaging the mDHFR domain. Indeed, following degradation by Lon, a partially degraded fragment slightly larger than mDHFR was observed by SDS-PAGE when MTX was present [Fig. 2(A), left lanes], but was not detected when MTX was absent [Fig. 2(A), right lanes]. Mass spectrometry indicated that this partially degraded fragment contained mDHFR plus a “tail” (SGLIEVEKPLYGVEVFVGET AHFEIEL) from the titin portion of the substrate. The calculated MR of the mDHFR-tail fragment (26,059.9 Da) was essentially identical to the experimental value (26,060.0 Da), and the C-terminal leucine of the mDHFR-tail fragment is consistent with the specificity of Lon peptide-bond cleavage.30 In the absence of MTX, the mDHFR-tail fragment was not detected and complete degradation of the substrate occurred [Fig. 2(A), right lanes]. This result differs from previous studies in which Lon showed little ability to processively degrade mDHFR-fusion proteins.5
Figure 2.
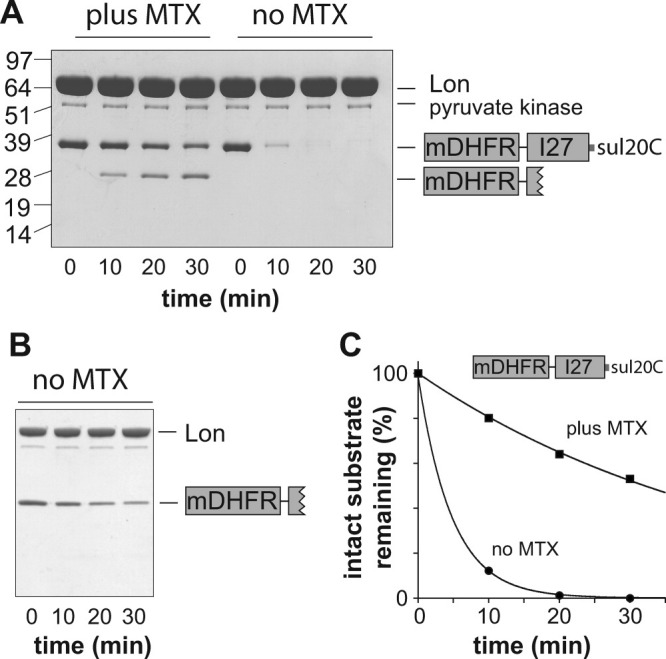
Degradation of C-tagged mDHFR-fusion proteins. A: Degradation of mDHFR-titinI27-sul20C (5 μM) by Lon (2 μM hexamer) in the presence or absence of MTX (10 μM). Pyruvate kinase is part of the ATP-regeneration system. The numbers on the left side of the gel are MR values (kDa) of protein standards. Other conditions were the same as in Figure 1. B: Degradation of the purified mDHFR-tail protein (5 μM) by Lon (0.3 μM hexamer) in the absence of MTX. C: The intensities of the full-length substrate bands in panel A were determined by densitometry, plotted as a function of time, and fit to single-exponential functions (R > 0.999; amplitude 100%) with rate constants of 2.1 × 10−1 min−1 (no MTX) and 2.1 × 10−2 min−1 (plus MTX).
We considered the possibility that Lon, upon encountering the mDHFR domain, dissociates but can then rebind the mDHFR-tail fragment for repeated unfolding attempts (i.e., that the tail sequence serves as a Lon degron). To test the feasibility of this model, we constructed and purified a protein corresponding to mDHFR-tail and found that ∼75% of this substrate (5 μM initial concentration) was degraded by Lon (0.3 μM) over the course of 30 min [Fig. 2(B)]. Thus, if Lon did dissociate while attempting to unfold the mDHFR domain, it could rebind and resume attempts at unfolding/degradation. However, several observations are inconsistent with a model in which most mDHFR-titinI27-sul20C degradation occurs by dissociation and rebinding. First, a degradation intermediate was not detected in the experiment without MTX [Fig. 2(A)]. Second, when MTX was present to block degradation of the mDHFR domain, the full-length substrate disappeared and the intermediate appeared at rates ∼10-fold slower than the overall rate of degradation in the no-MTX experiment [Fig. 2(A,C)]. Why is the titin portion of the fusion protein degraded faster in the presence than absence of MTX? We tested the possibility that MTX directly affected Lon degradation of titinI27-sul20C but found no difference in proteolysis rates in the presence and absence of MTX (not shown). Although other mechanisms may be possible, we were able to model these results by setting the rate constant for degradation of the mDHFR domain (no MTX) to a value at least 10-fold larger than the rate constant for dissociation of the partially degraded mDHFR-tail fragment from Lon. The unfolding processivity value (U) is defined as the ratio of the rate constant for mDHFR degradation divided by the rate constant for mDHFR release and thus would be ≥10 in this experiment. In other words, >90% of the mDHFR-titinI27-sul20C substrate would be degraded without Lon dissociation. In their experiments, by contrast, Koodathingal et al. observed a ∼100-fold lower U value (∼0.1), corresponding to ∼90% dissociation, for C-terminal unfolding of the mDHFR domain by Lon.5
To test mDHFR unfolding under conditions in which Lon degradation initiates at the N terminus, we constructed a β20-H6-titinI27-mDHFR fusion protein. The β20 degron can function at either terminus or at an internal position in a substrate.17 Lon proteolysis of this N-tagged substrate resulted in loss of the full-length substrate at roughly comparable rates with and without MTX [Fig. 3(A,B)]. Moreover, a partially degraded mDHFR fragment was produced in the presence and absence of MTX [Fig. 3(A,C)], although at substantially higher levels with MTX. Based on I+MTX and I–MTX values obtained from the fits of the Figure 3C data, the unfolding processivity value is ∼3, which is ∼30-fold larger than the value obtained by Koodathingal et al.5 In our studies, Lon was somewhat more efficient at processively unfolding mDHFR from the C terminus (U ≥ 10) than from the N terminus (U ≍ 3). This difference could arise because the N-terminal element of mDHFR secondary structure is a β strand that is completely buried in the protein core and thus is difficult to dislodge, whereas the C-terminal element is a β strand on the periphery of the protein, which might be displaced without global unfolding. Alternatively, differences in the degrons employed or Lon's grip on the substrates might be responsible for the more processive unfolding/degradation from the C terminus.
Figure 3.
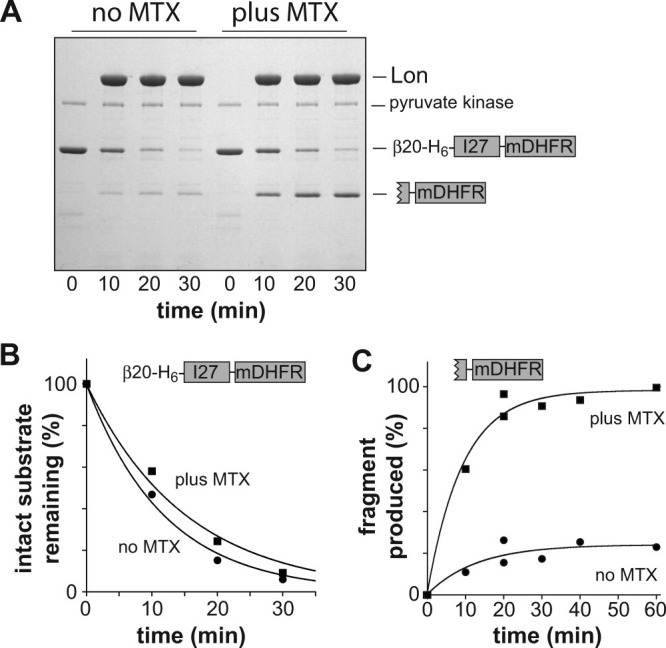
Degradation of N-tagged mDHFR-fusion proteins. A: Degradation of β20-H6-mDHFR (5 μM) by Lon (0.3 μM hexamer) in the presence or absence of MTX (10 μM). Other conditions as in Fig. 1. B: The intensities of the full-length substrate bands in panel A were determined by densitometry, plotted, and fit to single-exponential functions (R > 0.993; amplitude 100%) with rate constants of 8.5 × 10−2 min−1 (no MTX) and 6.5 × 10−2 min−1 (plus MTX). C: The intensities of the tail-mDHFR fragment bands in panel A and in a second experiment with time points of 0, 20, 40, and 60 min (not shown) were determined by densitometry, plotted, and fit to single-exponential functions (R > 0.999). No MTX: rate constant, 8.8 × 10−2 min−1; amplitude, 18%. Plus MTX: rate constant 1.0 × 10−1 min−1; amplitude, 96%.
Directional effects on Lon degradation of titinI27 domains
Is Lon a better unfoldase from the C terminus or does the substrate dictate in which direction degradation is more efficient? To direct Lon degradation of a titinI27 domain, we fused either the N or C terminus of this protein to amino acids 3–93 of E. coli β-galactosidase,17 an unstructured sequence that contains the β20 degron. The 3–93 sequence was used rather than a β20 degron to allow detection of potential degradation intermediates by SDS-PAGE. Lon degraded the β-gal3-93-titinI27 substrate rapidly and without detectable intermediates [Fig. 4(A), left lanes]. By contrast, Lon degraded the titinI27-β-gal3-93 substrate more slowly and a partially degraded fragment was observed [Fig. 4(A), right lanes]. Mass spectrometry indicated that this fragment (calculated MR 14,521.3 Da; experimental MR 14,521.8 Da) consisted of the complete titinI27 domain followed by 23 N-terminal residues (MITDSLAVVLQRRDWE NPGVTQL) from the β-gal3-93 tag. An experiment using higher concentrations of Lon relative to substrate revealed that this fragment formed transiently and was eventually degraded [Fig. 4(B)]. These observations suggest that Lon-degradation proceeds rapidly through the unfolded C-terminal portion of the titinI27-β-gal3-93 substrate but slows when the enzyme attempts to unfold titinI27 by pulling on its C terminus. By contrast, Lon degradation of titinI27 from the N terminus proceeded more rapidly and without intermediates. These results differ from those using mDHFR, suggesting that the properties of the substrate have a major influence on unfolding from the N or the C terminus.
Figure 4.
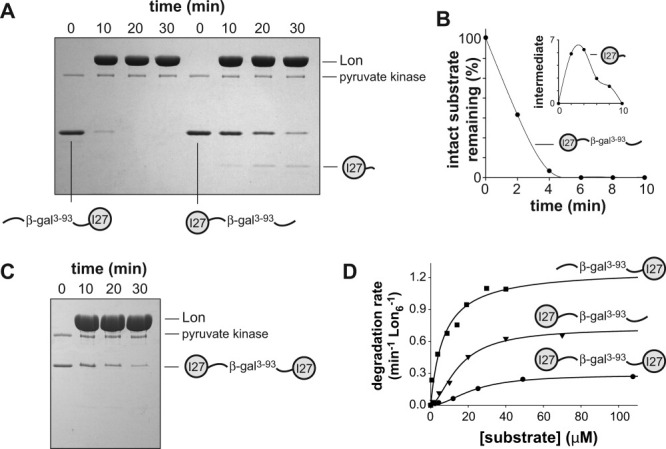
Degradation of titinI27-fusion proteins. A: The four left lanes show degradation of β-gal3-93-titinI27 (5 μM) by Lon (2 μM hexamer). The four right lanes show degradation of titinI27-β-gal3-93 (5 μM) by Lon (2 μM hexamer). Other conditions were the same as in Figure 1. B: Degradation of titinI27-β-gal3-93 (3 μM) by Lon (6 μM hexamer). The inset shows that the partially degraded fragment initially accumulates and is then degraded. C: Degradation of titinI27-β-gal3-93-titinI27 (3 μM) by Lon (6 μM hexamer). D: Steady-state rates of degradation of different concentrations of 35S-labeled titinI27-fusion variants by Lon (0.1 μM hexamer) were determined by release of acid-soluble peptides. The solid lines are fits to the Hill equation (rate = Vmax × [S]n/(KMn+[S]n). Kinetic parameters are listed in Table I.
To determine if Lon degradation could proceed from a degradation tag flanked by native proteins, we placed the β-gal3-93 recognition sequence between two titinI27 domains. As assayed by SDS-PAGE, Lon degraded this titinI27-β-gal3-93-titinI27 substrate without detectable intermediates, albeit slowly [Fig. 4(C)]. We conclude that Lon can initiate degradation from an internal degron between folded domains.
For studies of steady-state degradation kinetics, we purified 35S-labeled variants of β-gal3-93-titinI27, titinI27-β-gal3-93-titinI27, and titinI27-β-gal3-93 and determined KM and Vmax for Lon proteolysis [Fig. 4(D); Table I]. The substrate with titin domains at both termini was degraded at the slowest maximal rate, the substrate with the degradation tag at the C terminus was degraded at an intermediate rate, and the substrate with the tag at the N terminus was degraded at the fastest maximal rate. These results are consistent with the SDS-PAGE assays and confirm that the rate at which Lon can unfold and degrade titinI27 depends on the location of the degradation tag and thus the direction of unfolding/degradation. The doubly capped substrate is probably degraded at the slowest rate because two polypeptide chains need to be concurrently translocated through Lon and both titin domains need to be denatured.
Table I.
Steady-State Parameters for Lon Degradation of TitinI27-Fusion Substrates
| KM (μM) | Vmax (min−1 Lon6−1) | Hill coefficient | |
|---|---|---|---|
| β-gal3-93-titinI27 | 7 ± 1.9 | 1.3 ± 0.1 | 1.0 ± 0.3 |
| titinI27-β-gal3-93 | 15 ± 2.0 | 0.7 ± 0.05 | 1.7 ± 0.3 |
| titinI27-β-gal3-93- titinI27 | 23 ± 1.9 | 0.28 ± 0.01 | 1.9 ± 0.2 |
GFP-fusion proteins resist Lon degradation from the N-terminus
Previous studies have shown that variants of GFP with C-terminal degradation tags are degraded very slowly by Lon.17, 31 We constructed GFP-fusion proteins with a β20-tagged titinI27 domain at the N terminus or a titinI27 domain with a sul20C tag at the C terminus. As assayed by SDS-PAGE, Lon degraded most of the titinI27 portion of both fusion substrates but left partially degraded fragments larger than GFP [Fig. 5(A)]. When degradation initiated at the N terminus, the full-length substrate disappeared ∼10-fold more rapidly than when degradation initiated at the C terminus [Fig. 5(A,B)].
Figure 5.
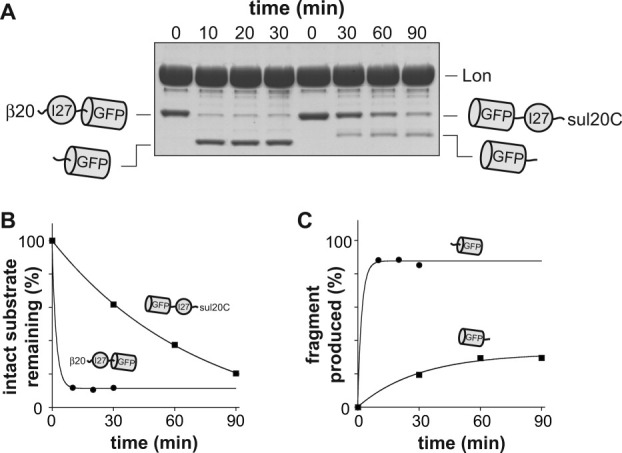
Degradation of GFP-fusion proteins by Lon. A: The four left lanes show degradation of β20-titinI27-GFP-H6 (5 μM) by Lon (2 μM hexamer). The four right lanes show degradation of H6-GFP-titinI27-sul20C (5 μM) by Lon (2 μM hexamer). B: Intensities of the full-length substrate bands from panel A were fit to single-exponential functions. For the β20-tagged substrate, the amplitude of the fit was 88% (∼12% of substrates were nondegradable, possibly because they lost the degradation tag) and the rate constant was 0.49 min−1 (R > 0.999). For the sul20C-tagged substrate, the amplitude of the fit was 100% and the rate constant was 0.017 min−1 (R > 0.999). C: The intensities of the partially degraded GFP fragments in panel A were fit to single-exponential functions. For the sul20C-tagged substrate, the amplitude of the fit was 32% and the rate constant was 0.033 min−1 (R > 0.995). For the β20-tagged substrate, the amplitude of the fit was 87.5% and the rate constant was 0.5 min−1 (R > 0.999).
In the experiment with β20-titinI27-GFP, the steady-state intensity of the partially degraded fragment was ∼85% of the intensity of the initial substrate, suggesting that little if any of the GFP domain was degraded [Fig. 5(A,C)]. With the GFP-titinI27-sul20C substrate, by contrast, the steady-state intensity of the partially degraded fragment was ∼30% of the initial substrate [Fig. 5(A,C)], indicating that more than half of the GFP domain had been degraded. These results suggest that Lon unfolds/degrades GFP slightly more frequently than it dissociates when approaching from the C terminus but largely fails when it attempts to denature GFP by pulling on its N-terminal residues. As discussed above, a preference for C-terminal unfolding was also observed for mDHFR substrates, whereas a preference for N-terminal unfolding was observed for titinI27 substrates. It seems likely, therefore, that the physical properties of the substrate largely determined these preferences.
Tag-dependent degradation of a disulfide-linked polypeptide
Current and previous results suggest that Lon can initiate degradation at many positions along a polypeptide chain.17 We sought to test if a protein that was normally a poor substrate could be degraded by Lon if it was covalently attached to a substrate that could be bound efficiently, but not degraded, by the protease. For these experiments, we constructed a sul20C-tagged variant of mDHFR with a cysteine between mDHFR and the sul20C tag (mDHFR-cys-sul20C). To allow disulfide crosslinking, we also constructed a titinI27 variant in which the two wild-type cysteines were replaced with aspartic acids and a new cysteine was introduced at the C terminus (titinI27-CD-cys). The Cys→Asp substitutions in the hydrophobic core of native titinI27 result in an unfolded protein.17 By activating the cysteine in titinI27-CD-cys by reaction with DTNB and then adding mDHFR-cys-sul20C, efficient disulfide crosslinking of mDHFR-cys-sul20C to titinI27-CD-cys was observed [Fig. 6(A)].
Figure 6.
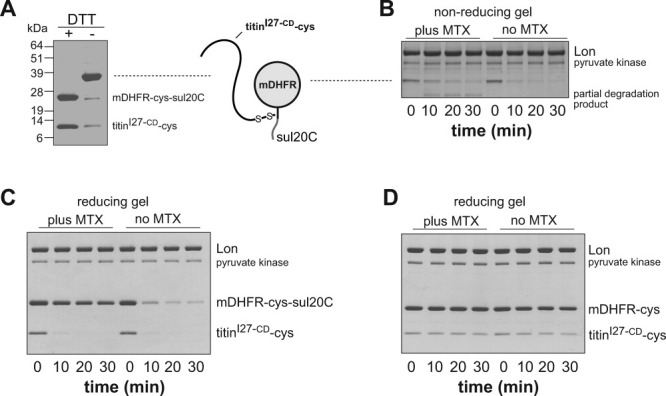
Lon degradation of a disulfide-bonded substrate. A: Nonreducing SDS-PAGE analysis of a protein in which native mDHFR-cys-sul20C was disulfide bonded to unfolded titinI27-CD-cys. The left lane shows the sample after reduction with DTT. The right lane shows the unreduced sample. The numbers on the left side of the gel are MR values (kDa) of protein standards. B: Nonreducing SDS-PAGE of Lon (0.3 μM hexamer) degradation of the disulfide-linked substrate (5 μM) in the presence and absence of MTX. Other conditions as in Figure 1. C: Reducing SDS-PAGE of Lon (0.3 μM hexamer) degradation of the disulfide-linked substrate (5 μM) in the presence and absence of MTX. D: Reducing SDS-PAGE of Lon (0.3 μM hexamer) degradation of a control disulfide-linked substrate lacking the sul20C degradation tag (5 μM) in the presence and absence of MTX.
As assayed by nonreducing SDS-PAGE, the intact crosslinked substrate was degraded by Lon in the presence and absence of MTX but a partial degradation product accumulated when MTX was present [Fig. 6(B)]. Analysis by reducing SDS-PAGE showed that Lon degraded both the titinI27-CD-cys and mDHFR-cys-sul20C portions of the disulfide-bonded substrate in the absence of MTX [Fig. 6(C), right lanes]. With MTX present, Lon degraded the titinI27-CD-cys portion of the crosslinked substrate efficiently but degraded little of the mDHFR-cys-sul20C moiety [Fig. 6(C), left lanes]. Without the sul20C tag, neither the mDHFR-cys nor the titinI27-CD-cys portions of the disulfide-linked substrate were degraded efficiently by Lon [Fig. 6(D]. When we assayed degradation of titinI27-CD-cys in the absence of disulfide crosslinking to mDHFR-cys-sul20C, Lon degraded the free titin protein at a rate ∼sixfold slower than the titin portion of the linked construct (data not shown). In combination, these results indicate that the sul20C tag does not need to be proteolyzed to direct Lon degradation of nearby unstructured sequences that would normally be poor Lon substrates.
Discussion
Several of our results suggest that Lon can translocate two polypeptide strands during protein degradation. For example, complete degradation of the disulfide-bonded protein consisting of titinI27-CD-cys and mDHFR-cys-sul20C would require passage of two polypeptides through the Lon pore. Similarly, initiation of degradation of the titinI27-β-gal3-93-titinI27 substrate from the interior degradation tag would require concurrent translocation of two polypeptide chains. In this regard, Lon resembles ClpXP, HslUV, and the 26S proteasome, each of which has been shown to degrade disulfide-bond substrates and/or to initiate from interior degrons.9,12,32–36 For ClpXP, HslUV, and the proteasome, the subunits of the AAA+ ring interact noncovalently with the subunits of the peptidase ring. For Lon, by contrast, the AAA+ domain is covalently fused to the peptidase domain. The ability of AAA+ proteases to concurrently translocate multiple polypeptide chains appears therefore to be a conserved property of the AAA+ ring.
We also found that tethering a poor substrate (titinI27-CD-cys) to Lon via a sul20C-tagged mDHFR domain allowed degradation even when MTX prevented degradation of the mDHFR-sul20C portion of the disulfide-linked protein. This result demonstrates that the sul20C degron can direct degradation, without itself being engaged and degraded by Lon. We imagine that tethering, in this case, increases the effective concentration of the unfolded titinI27-CD-cys polypeptide with respect to the engagement and translocation machinery of Lon, allowing degradation of an otherwise poor substrate. The same principle allows adaptor proteins to deliver “poor” substrates to other AAA+ proteases.2, 4 Moreover, polyubiquitin chains tether protein substrates to the 26S proteasome, but efficient degradation also requires engagement of an unfolded region of polypeptide.3, 35
Prior to translocation, Lon must unfold native substrates. We find that the unfolding activity of E. coli Lon depends on the specific protein domain and the direction of unfolding/degradation, which is determined by the position of the degradation tag. For example, Lon degrades mDHFR very efficiently from the C-terminus and reasonably efficiently from the N-terminus. For the titinI27 domain, Lon degradation was also efficient but unfolding from the N-terminus resulted in faster degradation than unfolding from the C-terminus. For GFP, no Lon degradation was detected from the N-terminus and slow degradation was observed from the C-terminus.
How does the unfolding/degradation of protein substrates by Lon compare with these activities for other AAA+ proteases? One way to rank the activities of different enzymes is to compare Vmax, the rate of degradation at substrate saturation using proteins for which unfolding is rate limiting. Table II lists Vmax values for Lon, ClpXP, and ClpAP degradation of titinI27 and GFP substrates with recognition tags at the C-terminus or the N-terminus. For C-tagged titinI27 substrates, Lon displayed the highest Vmax, ClpXP was next highest, and ClpAP had no detectable activity (Table II). For N-tagged titinI27 proteins, ClpAP had the highest Vmax, and Lon and ClpXP were about 10-fold less active. For C-tagged GFP, the order of unfolding/degradation activities was ClpAP>ClpXP>Lon. For N-tagged GFP, Lon and ClpXP were essentially inactive, whereas ClpAP was very active. These comparisons indicate that the ranking of enzyme unfolding activity changes as a function of the substrate and position of the degradation tag. Moreover, we previously found that Vmax for degradation of titinI27 from the C-terminus varied ∼sixfold, depending on the degron employed. Thus, although Lon appears to be a better unfoldase for C-tagged titinI27 substrates than ClpXP and substantially more active than ClpAP (Table II), these rankings might be degron dependent.
Table II.
Activities of AAA+ Proteases Against Different Substrates
| E. coli ClpXP | E. coli ClpAP | E. coli Lon | ||
|---|---|---|---|---|
| Vmax (min−1 enz−1) | Vmax (min−1 enz−1) | Vmax (min−1 enz−1) | ||
| C-tagged titin | 0.25a | <0.05b | 0.4–2.5c,d,e | |
| N-tagged titin | 1.4b | 8–13e | 1.3d | |
| C-tagged GFP | 0.94–1.2g | 4.9g | ∼0.05h | |
| N-tagged GFP | <0.05b | 1.2–1.4i | <0.05d | |
| E. coli ClpXP processivity ratio (U) | E. coli ClpAP processivity ratio (U) | E. coli Lon processivity ratio (U) | E. coli Lon processivity ratio (U) | |
| C-tagged mDHFR | 1.7 ± 0.2j | >9k | ≥10d | 0.1 ± 0.05j |
| N-tagged mDHFR | not determined | >9k | 3d | <0.1j |
Koodathingal et al. defined unfolding activity in terms of a processivity ratio that relates the rate with which a AAA+ protease unfolds a domain encountered in the midst of a multi-domain substrate to the rate at which it dissociates from the substrate.5 In their studies, E. coli Lon had a processivity ratio of 0.1 or less for unfolding of mDHFR from the C terminus or from the N terminus (Table II), whereas we found values ≥10 for C-terminal unfolding and ∼3 for N-terminal unfolding. Thus, processivity ratios for Lon unfolding of the same domain can vary substantially. We suspect that the difference between our results and those of Koodathingal et al. result from their use of much lower substrate concentrations, produced by translation in vitro. We previously found that Lon is an allosteric enzyme for which positively cooperative degron binding could shift the equilibrium between enzyme conformations with different proteolytic activities.7 Thus, very low substrate concentrations might be unable to stabilize a Lon conformation that can processively degrade mDHFR. Alternatively, the use of different degrons in the two sets of experiments could account for our finding that Lon can unfold very stable model proteins.
Our results do reinforce many of the major conclusions of Koodathingal et al.5 Namely, individual AAA+ proteases can differ substantially in their ability to unfold native proteins, and the direction of degradation can have a large influence on unfolding activity. Presumably, the degradation tags of natural substrates that are difficult to unfold have evolved to optimize targeting to AAA+ proteases that can best unfold these domains. Different AAA+ proteases have also apparently evolved to allow more efficient degradation of certain types of protein domains. For example, an N-tagged or a C-tagged protein similar to GFP would be a very poor Lon substrate but a good ClpAP substrate. By contrast, a C-tagged protein with properties similar to titinI27 would be expected to be a good Lon substrate but a very poor ClpAP substrate.
What structural factors in a substrate influence enzymatic unfolding? Global stability plays some role, as metastable domains will be easier to unfold than domains with greater thermodynamic and/or kinetic stabilities. This principle affects substrate choice by the FtsH protease, which only appears to be capable of unfolding and degrading protein substrates with low intrinsic stabilities.37 Importantly, however, Lee et al. first showed that other structural factors must also be important, as the degradation rates of stable model substrates by AAA+ proteases correlated poorly with their overall thermodynamic or kinetic stabilities.9 They proposed that the local stability of the element of secondary structure immediately adjacent to the degradation tag of the substrate was important in determining resistance to enzymatic unfolding,9 a model supported by subsequent studies.6, 38 In principle, this model could account for the observation that a specific protein domain can often be unfolded/degraded at different rates depending on the N-terminal or C-terminal placement of the degradation tag. However, if the local stability of terminal substrate structure was the dominant factor in determining enzymatic unfolding, then a protein domain that is degraded faster from one end by one AAA+ protease should also show the same trend with other AAA+ proteases. Although this result is generally observed, exceptions are evident. For example, the HslUV protease unfolds E. coli DHFR ∼100-fold more efficiently from the N terminus than the C terminus,5 whereas ClpXP and Lon unfold this domain two to threefold more efficiently when degradation initiates at the C terminus rather than the N terminus (Table II). Moreover, even when the direction-of-unfolding trend holds, quantitative correspondence can be poor. For example, ClpAP degraded N-tagged titinI27 substrates >100-fold faster than C-tagged titinI27 substrates (Ref.29; J. Kenniston, personal communication), whereas we found that Lon degraded N-tagged titinI27 only twice as fast as the corresponding C-tagged variant (Table I). Thus, some AAA+ proteases appear to be better than others at unfolding certain protein domains from one direction.
What properties of a AAA+ protease influence the rate at which it can unfold a specific protein domain? The rate of ATP hydrolysis, which sets the rate at which enzymatic pulling events can be repeated, may be important. For example, ClpXP unfolding of C-tagged GFP has been shown to depend on a sufficiently rapid rate of ATP hydrolysis.13, 39 In this case, an initial ClpXP power stroke extracts the terminal strand from the GFP β-barrel and rapid additional power strokes are needed to translocate this strand and unfold the remaining portion of the barrel before the native structure reforms. Other protease properties that probably influence unfolding activity include the enzyme's grip on the substrate during pulling and the mechanical force it can generate per power stroke.
Lon's ability to unfold stable protein domains probably plays an important role in protein-quality control following heat shock or other unfolding stresses. Lon degrons are over-represented in protein sequences that are normally buried in the three-dimensional native structure,17 and unfolding would reveal these degrons and allow Lon to initiate degradation. However, in a multi-domain protein, some domains could remain intact and Lon's ability to unfold these structures would, in many cases, allow processive degradation. In instances in which Lon did dissociate before completing degradation, the partially degraded substrates would contain ∼25 residue unstructured tails that could allow Lon or another AAA+ protease to rebind and resume unfolding/degradation. We also note that the model domains used in our studies represent very stable proteins chosen to test the limits of enzymatic unfolding. In the cell, proteases and their substrates co-evolve, and thus natural selection should ensure that proteins that need to be degraded will be good substrates for one or more AAA+ proteases.
Materials and Methods
Protein purification and modification
Purification of E. coli Lon, N-terminal H6-tagged variants of human titinI27, 35S-labeled titinI27 substrates, and H6-tagged GFP variants were performed as described.17 H6-tagged mDHFR variants were expressed from a T7 promoter in 1-L cultures. Harvested cells were resuspended in 25 mL of Tris-HCl [pH 8.0] (T25), 500 mM NaCl, 20 mM imidazole, and 0.02 mg/mL lysozyme and were lysed with a French pressure cell press or by sonication. The supernatants were mixed in a 50 mL tube with 2 mL Ni-NTA resin (Qiagen), prewashed with the same buffer, and incubated at 4°C for 15 min. The resin was washed three times with 20 mL of the same buffer, resuspended in 5 mL of buffer and transferred to a gravity column, and washed once with 10 mL of buffer. Proteins were eluted with 3 mL of T25, 500 mM NaCl, and 250 mM imidazole and loaded on a gel-filtration column (GE Healthcare 16/60 Superdex-75), pre-equilibrated with T25, 300 mM NaCl. Following purification, glycerol was added to a final concentration of 10% and the protein solutions were stored at –80°C.
For purification of the cysteine-crosslinked variant, mDHFR-cys-sul20C was purified and then loaded on an agarose-methotrexate column (1 mL) pre-equilibrated with T25, 300 mM NaCl. The bound protein was washed with 10 mL of the same buffer. In parallel, H6-tagged-titinI27-CD-cys was purified by Ni-NTA affinity, as described above, activated by reaction with 5,5'-dithiobis-(2-nitrobenzoic acid (DTNB) and loaded on the mDHFR-bound agarose-methotrexate column. Following a wash with 10 mL of T25, 300 mM NaCl, the cross-linked protein was eluted with 3 mL buffer containing 2 mM folic acid, and was then loaded on a gel-filtration column (GE Healthcare 16/60 Superdex-75) pre-equilibrated with T25, 300 mM NaCl. The eluted protein was concentrated, glycerol was added to a final concentration of 10%, and the protein solution was stored frozen at –80°C.
Protein concentrations were determined by UV absorbance using extinction coefficients calculated from the amino-acid sequence. The gene and protein sequences of protein substrates are available upon request.
Degradation assays
Lon degradation assays were performed at 37°C as described.17 Briefly, the degradation buffer contained T25, 100 mM KCl, 10 mM MgCl2, 1 mM DTT, 2 mM ATP, and an ATP regeneration system consisting of pyruvate kinase (10 U/mL) and phosphoenolpyruvate (up to 60 mM). Following SDS-PAGE and staining with Coomassie blue, U values were calculated as described,5 by quantifying the intensities of the full-length substrate band and the intermediate band using ImageJ software (http://rsbweb.nih.gov/ij/index.html). Intensities were normalized to the intensity of the substrate at time = 0 (defined as 100%).
Acknowledgments
The authors thank T. Baker, E. Kloss, A. Matouschek, and M. Wohlever for helpful discussions.
References
- 1.Gottesman S. Proteolysis in bacterial regulatory circuits. Annu Rev Cell Dev Biol. 2003;19:565–587. doi: 10.1146/annurev.cellbio.19.110701.153228. [DOI] [PubMed] [Google Scholar]
- 2.Baker TA, Sauer RT. ATP-dependent proteases of bacteria: recognition logic and operating principles. Trends Biochem Sci. 2006;31:647–653. doi: 10.1016/j.tibs.2006.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schrader EK, Harstad KG, Matouschek A. Targeting proteins for degradation. Nat Chem Biol. 2009;5:815–822. doi: 10.1038/nchembio.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sauer RT, Baker TA. AAA+ proteases: ATP-fueled machines of destruction. Annu Rev Biochem. 2011;80:587–612. doi: 10.1146/annurev-biochem-060408-172623. [DOI] [PubMed] [Google Scholar]
- 5.Koodathingal P, Jaffe NE, Kraut DA, Prakash S, Fishbain S, Herman C, Matouschek A. ATP-dependent proteases differ substantially in their ability to unfold globular proteins. J Biol Chem. 2009;284:18674–18684. doi: 10.1074/jbc.M900783200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kenniston JA, Baker TA, Fernandez JM, Sauer RT. Linkage between ATP consumption and mechanical unfolding during the protein processing reactions of an AAA+ degradation machine. Cell. 2003;114:511–520. doi: 10.1016/s0092-8674(03)00612-3. [DOI] [PubMed] [Google Scholar]
- 7.Gur E, Sauer RT. Degrons in protein substrates program the speed and operating efficiency of the AAA+ Lon proteolytic machine. Proc Natl Acad Sci USA. 2009;106:18503–18508. doi: 10.1073/pnas.0910392106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aubin-Tam ME, Olivares AO, Sauer RT, Baker TA, Lang MJ. Single-molecule protein unfolding and translocation by an ATP-fueled proteolytic machine. Cell. 2011;145:257–267. doi: 10.1016/j.cell.2011.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee C, Schwartz MP, Prakash S, Iwakura M, Matouschek A. ATP-dependent proteases degrade their substrates by processively unraveling them from the degradation signal. Mol Cell. 2001;7:627–637. doi: 10.1016/s1097-2765(01)00209-x. [DOI] [PubMed] [Google Scholar]
- 10.Kenniston JA, Baker TA, Sauer RT. Partitioning between unfolding and release of native domains during ClpXP degradation determines substrate selectivity and partial processing. Proc Natl Acad Sci USA. 2005;102:1390–1395. doi: 10.1073/pnas.0409634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tian L, Holmgren RA, Matouschek A. A conserved processing mechanism regulates the activity of transcription factors Cubitus interruptus and NF-kappaB. Nat Struct Mol Biol. 2005;12:1045–1053. doi: 10.1038/nsmb1018. [DOI] [PubMed] [Google Scholar]
- 12.Hoskins JR, Yanagihara K, Mizuuchi K, Wickner S. ClpAP and ClpXP degrade proteins with tags located in the interior of the primary sequence. Proc Natl Acad Sci USA. 2002;99:11037–11042. doi: 10.1073/pnas.172378899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martin A, Baker TA, Sauer RT. Protein unfolding by a AAA+ protease: critical dependence on ATP-hydrolysis rates and energy landscapes. Nat Struct Mol Biol. 2008;15:139–145. doi: 10.1038/nsmb.1380. [DOI] [PubMed] [Google Scholar]
- 14.Kowit JD, Goldberg AL. Intermediate steps in the degradation of a specific abnormal protein in Escherichia coli. J Biol Chem. 1977;252:8350–8357. [PubMed] [Google Scholar]
- 15.Sonezaki S, Ishii Y, Okita K, Sugino T, Kondo A, Kato Y. Overproduction and purification of SulA fusion protein in Escherichia coli and its degradation by Lon protease in vitro. Appl Microbiol Biotechnol. 1995;43:304–309. doi: 10.1007/BF00172829. [DOI] [PubMed] [Google Scholar]
- 16.Gonzalez M, Frank EG, Levine AS, Woodgate R. Lon-mediated proteolysis of the Escherichia coli UmuD mutagenesis protein: in vitro degradation and identification of residues required for proteolysis. Genes Dev. 1998;12:3889–3899. doi: 10.1101/gad.12.24.3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gur E, Sauer RT. Recognition of misfolded proteins by Lon a AAA+ protease. Genes Dev. 2008;22:2267–2277. doi: 10.1101/gad.1670908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gur E, Sauer RT. Reductive evolution of mycoplasma forced a proteolytic specificity switch in degradation of ssrA-tagged proteins. Proc Natl Acad Sci USA. 2008;105:16113–16118. doi: 10.1073/pnas.0808802105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clark AC, Frieden C. Native Escherichia coli and murine dihydrofolate reductases contain late-folding non-native structures. J Mol Biol. 1999;285:1765–1776. doi: 10.1006/jmbi.1998.2402. [DOI] [PubMed] [Google Scholar]
- 20.Kim YI, Burton RE, Burton BM, Sauer RT, Baker TA. Dynamics of substrate denaturation and translocation by the ClpXP degradation machine. Mol Cell. 2000;5:639–648. doi: 10.1016/s1097-2765(00)80243-9. [DOI] [PubMed] [Google Scholar]
- 21.Fukuda H, Arai M, Kuwajima K. Folding of green fluorescent protein and the cycle3 mutant. Biochemistry. 2000;39:12025–12032. doi: 10.1021/bi000543l. [DOI] [PubMed] [Google Scholar]
- 22.Blois TM, Hong H, Kim TH, Bowie JU. Protein unfolding with a steric trap. J Am Chem Soc. 2009;131:13914–13915. doi: 10.1021/ja905725n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carrion-Vazquez M, Oberhauser AF, Fowler SB, Marszalek PE, Broedel SE, Clarke J, Fernandez JM. Mechanical and chemical unfolding of a single protein: a comparison. Proc Natl Acad Sci USA. 1999;96:3694–3699. doi: 10.1073/pnas.96.7.3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dietz H, Rief M. Exploring the energy landscape of GFP by single-molecule mechanical experiments. Proc Natl Acad Sci USA. 2004;101:16192–16197. doi: 10.1073/pnas.0404549101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weber-Ban EU, Reid BG, Miranker AD, Horwich AL. Global unfolding of a substrate protein by the Hsp100 chaperone ClpA. Nature. 1999;401:90–93. doi: 10.1038/43481. [DOI] [PubMed] [Google Scholar]
- 26.Singh SK, Grimaud R, Hoskins JR, Wickner S, Maurizi MR. Unfolding and internalization of proteins by the ATP-dependent proteases ClpXP and ClpAP. Proc Natl Acad Sci USA. 2000;97:8898–8903. doi: 10.1073/pnas.97.16.8898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flynn JM, Levchenko I, Seidel M, Wickner SH, Sauer RT, Baker TA. Overlapping recognition determinants within the ssrA degradation tag allow modulation of proteolysis. Proc Natl Acad Sci USA. 2001;11:10584–10589. doi: 10.1073/pnas.191375298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang KH, Sauer RT, Baker TA. ClpS modulates but is not essential for bacterial N-end rule degradation. Genes Dev. 2007;21:403–408. doi: 10.1101/gad.1511907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang KH, Román-Hernández G, Grant RA, Sauer RT, Baker TA. The molecular basis of N-end rule recognition. Mol Cell. 2008;32:406–414. doi: 10.1016/j.molcel.2008.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Melderen L, Thi MH, Lecchi P, Gottesman S, Couturier M, Maurizi MR. ATP-dependent degradation of CcdA by Lon protease: Effects of secondary structure and heterologous subunit interactions. J Biol Chem. 1996;271:27730–27738. doi: 10.1074/jbc.271.44.27730. [DOI] [PubMed] [Google Scholar]
- 31.Choy JS, Aung LL, Karzai AW. Lon protease degrades transfer-messenger RNA-tagged proteins. J Bacteriol. 2007;189:6564–6571. doi: 10.1128/JB.00860-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bolon DN, Grant RA, Baker TA, Sauer RT. Nucleotide-dependent substrate handoff from the SspB adaptor to the AAA+ ClpXP protease. Mol Cell. 2004;16:343–350. doi: 10.1016/j.molcel.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 33.Kwon AR, Trame CB, McKay DB. Kinetics of protein substrate degradation by HslUV. J Struct Biol. 2004;146:141–147. doi: 10.1016/j.jsb.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 34.Lee C, Prakash S, Matouschek A. Concurrent translocation of multiple polypeptide chains through the proteasomal degradation channel. J Biol Chem. 2002;277:34760–34765. doi: 10.1074/jbc.M204750200. [DOI] [PubMed] [Google Scholar]
- 35.Prakash S, Tian L, Ratliff KS, Lehotzky RE, Matouschek A. An unstructured initiation site is required for efficient proteasome-mediated degradation. Nat Struct Mol Biol. 2004;11:830–837. doi: 10.1038/nsmb814. [DOI] [PubMed] [Google Scholar]
- 36.Fishbain S, Prakash S, Herrig A, Elsasser S, Matouschek A. Rad23 escapes degradation because it lacks a proteasome initiation region. Nat Commun. 2011;2:192. doi: 10.1038/ncomms1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Herman C, Prakash S, Lu CZ, Matouschek A, Gross CA. Lack of a robust unfoldase activity confers a unique level of substrate specificity to the universal AAA protease FtsH. Mol Cell. 2003;11:659–669. doi: 10.1016/s1097-2765(03)00068-6. [DOI] [PubMed] [Google Scholar]
- 38.Kenniston JA, Burton RE, Siddiqui SM, Baker TA, Sauer RT. Effects of local protein stability and the geometric position of the substrate degradation tag on the efficiency of ClpXP denaturation and degradation. J Struct Biol. 2004;146:130–140. doi: 10.1016/j.jsb.2003.10.023. [DOI] [PubMed] [Google Scholar]
- 39.Nager AR, Baker TA, Sauer RT. Stepwise unfolding of a β-barrel protein by the AAA+ ClpXP protease. J Mol Biol. 2011;413:4–16. doi: 10.1016/j.jmb.2011.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]