

Abstract
Repetitive DNA sequences are abundant in a broad range of species, from bacteria to mammals, and they cover nearly half of the human genome. Repeats have always presented technical challenges for sequence alignment and assembly programs. Next-generation sequencing projects, with their short read lengths and high data volumes, have made these challenges more difficult. From a computational perspective, repeats create ambiguities in alignment and assembly, which, in turn, can produce biases and errors when interpreting results. Simply ignoring repeats is not an option, as this creates problems of its own and may mean that important biological phenomena are missed. We discuss the computational problems surrounding repeats and describe strategies used by current bioinformatics systems to solve them.
DNA sequencing efficiency has increased by approximately 100,000-fold in the decade since sequencing of the human genome was completed. Next-generation sequencing (NGS) machines can now sequence the entire human genome in a few days, and this capability has inspired a flood of new projects that are aimed at sequencing the genomes of thousands of individual humans and a broad swath of animal and plant species1-3. New methods, such as whole-transcriptome sequencing (also called RNA sequencing (RNA-seq))4-7, chromatin immunoprecipitation followed by sequencing (ChIP–seq)8-11 and sequencing to identify methylated DNA (methyl-seq)12,13, are transforming our ability to capture an accurate picture of the molecular processes within the cell, which, in turn, is leading to a better understanding of human diseases14. Whole-genome resequencing combined with new, highly efficient alignment software is being used to discover large numbers of SNPs and structural variants in previously sequenced genomes15. In response to this influx of new laboratory methods, many novel computational tools have been developed to map NGS reads to genomes and to reconstruct genomes and transcriptomes11,16-22. Current NGS platforms produce shorter reads than Sanger sequencing (NGS reads are 50–150 bp), but with vastly greater numbers of reads, as many as 6 billion per run. By contrast, the original human genome project generated approximately 30 million reads using Sanger sequencing.
Some of the biggest technical challenges that are associated with these new methods are caused by repetitive DNA23: that is, sequences that are similar or identical to sequences elsewhere in the genome. Most large genomes are filled with repetitive sequences; for example, nearly half of the human genome is covered by repeats, many of which have been known about for decades24,25. Although some repeats appear to be nonfunctional, others have played a part in human evolution26,27, at times creating novel functions, but also acting as independent, ‘selfish’ sequence elements28,29. Repeats arise from a variety of biological mechanisms that result in extra copies of a sequence being produced and inserted into the genome. Repeats come in all shapes and sizes: they can be widely interspersed repeats, tandem repeats or nested repeats, they may comprise just two copies or millions of copies, and they can range in size from 1–2 bases (mono- and dinucleotide repeats) to millions of bases. Well-characterized repeats in the human genome (BOX 1) are sometimes separated into two classes: short tandem repeats (also called microsatellites) and longer interspersed repeats (called short interspersed nuclear elements (SINEs) and long interspersed nuclear elements (LINEs)). The most well-documented example of interspersed repeats in the human genome is the class of Alu repeat elements, which cover approximately 11% of the genome25. Repeats can also take the form of large-scale segmental duplications, such as those found on some human chromosomes30 and even whole-genome duplication, such as the duplication of the Arabidopsis thaliana genome31. High levels of repetitiveness are found across all kingdoms of life, and plant genomes contain particularly high proportions of repeats: for example, transposable elements cover >80% of the maize genome32. A recent study reported that the short-lived fish Nothobranchius furzeri has 21% of its genome occupied by tandem repeats, suggesting a possible role for tandem repeats in the ageing process33. Even bacterial genomes can exhibit repeat content up to 40%, as demonstrated by Orientia tsutsugamushi34.
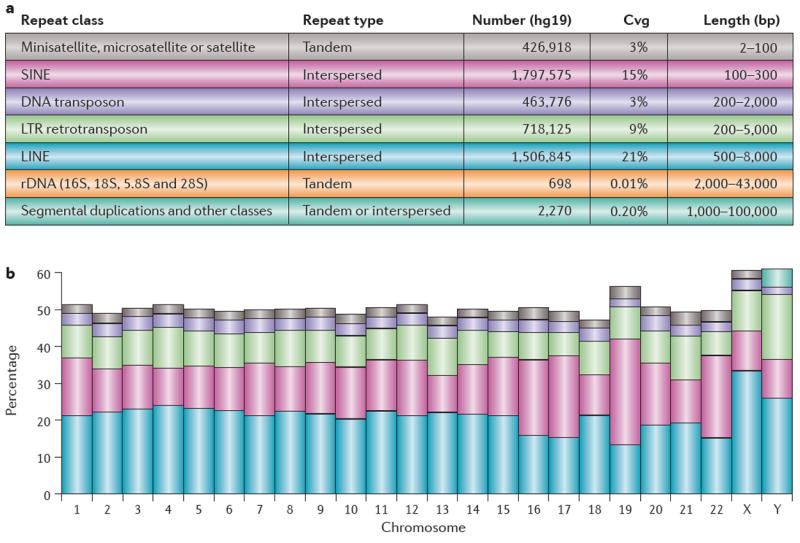
Box 1. Repetitive DNA in the human genome.
Approximately 50% of the human genome is comprised of repeats. The table in panel a shows various named classes of repeat in the human genome, along with their pattern of occurrence (shown as ‘repeat type’ in the table; this is taken from the RepeatMasker annotation). The number of repeats for each class found in the human genome, along with the percentage of the genome that is covered by the repeat class (Cvg) and the approximate upper and lower bounds on the repeat length (bp). The graph in panel b shows the percentage of each chromosome, based on release hg19 of the genome, covered by repetitive DNA as reported by RepeatMasker. The colours of the graph in panel b correspond to the colours of the repeat class in the table in panel a. Microsatellites constitute a class of repetitive DNA comprising tandem repeats that are 2–10 bp in length, whereas minisatellites are 10–60 bp in length, and satellites are up to 100 bp in length and are often associated with centromeric or pericentromeric regions of the genome. DNA transposons are full-length autonomous elements that encode a protein, transposase, by which an element can be removed from one position and inserted at another. Transposons typically have short inverted repeats at each end. Long terminal repeat (LTR) elements (which are often referred to as retrovirus-like elements) are characterized by the LTRs (200–5000 bp) that are harboured at each end of the retrotransposon. LINE, long interspersed nuclear element; rDNA, ribosomal DNA; SINE, short interspersed nuclear element.
Figure 5.
From a computational perspective, repeats create ambiguities in alignment and in genome assembly, which, in turn, can produce errors when interpreting results. Repeats that are sufficiently divergent do not present problems, so for the remaining discussion in this Review, we define a repeat as a sequence that is at least 100 bp in length, that occurs two or more times in the genome and that exhibits >97% identity to at least one other copy of itself. This definition excludes many repetitive sequences, but it includes those that present the principal computational challenges.
In this Review, we consider the challenges that are posed by repeats for genome resequencing projects, de novo genome assembly and RNA-seq analysis. We focus on two classes of computational tools: software for the alignment of NGS reads and software for the assembly of genomes and transcriptomes. Some of the more widely used programs in both categories are shown in TABLES 1,2, which illustrates the breadth of tools available. Rather than describing the algorithmic details of these programs, we will discuss their shared strategies for solving repeat-induced analysis problems in each situation and address some of their limitations.
Table 1.
Overview of current computational tools for next-generation sequencing genome alignment and assembly
| Scope | Program | Repeat-relevant parameters | Website | Refs |
|---|---|---|---|---|
| SV or CNV detection | BreakDancer | Specify the mapping quality threshold for ambiguous reads: -q | http://sourceforge.net/projects/breakdancer | |
| CNVnator | None available or none required | http://sv.gersteinlab.org/cnvnator/ | ||
| He et al. (2011) | Algorithm only, able to estimate CNV counts in repeat-rich regions | None | 47 | |
| PEMer | Maximum alignments per multi-read: --max_duplicates_per_score | http://sv.gersteinlab.org/pemer | ||
| VariationHunter | None available or none required | http://compbio.cs.sfu.ca/strvar.htm | ||
| SNP detection | GATK | None available or none required | http://www.broadinstitute.org/gsa/wiki/index.php/Downloading the GATK | |
| SAMtools | In repetitive regions, avoid calling ‘A’: -avcf ref.fa aln.bam | http://samtools.sourceforge.net | ||
| SOAPsnp | None required; multi-reads supported by read aligner parameters | http://soap.genomics.org.cn/soapsnp.html | ||
| Sniper | Read mapping policy: --all, --uniq, --best | http://kim.bio.upenn.edu/software/sniper.shtml | ||
| VarScan | None available or none required | http://varscan.sourceforge.net | ||
| Short-read alignment | Bowtie | Randomly distribute reads across repeats: --best –M 1 -strata | http://bowtie-bio.sourceforge.net | 16 |
| BFAST | Reports all locations by default | http://bfast.sourceforge.net | 69 | |
| Burrows–Wheeler Aligner (BWA) | Report one random hit for repetitive reads: -n 1 | http://bio-bwa.sourceforge.net | 70 | |
| mrFAST | Reports all locations by default, for best match: --best | http://mrfast.sourceforge.net | 71 | |
| SOAPAligner | Report all locations: -r 2 | http://soap.genomics.org.cn/soapaligner.html | 72 | |
| De novo assembly | Allpaths-LG | None required: incorporated into library insert size recipe | http://www.broadinstitute.org/software/allpaths-lg/blog/?page id=12 | 20 |
| CABOG | Re-assemble misclassified non-unique unitigs: doToggle=1 | http://wgs-assembler.sf.net | 73 | |
| SGA | Resolve small repeats at end of reads: -r 20 | http://github.com/jts/sga | ||
| SOAPdenovo | Use reads to solve small repeats: -R | http://soap.genomics.org.cn/soapdenovo.html | 17 | |
| Velvet | Use long reads to resolve repeats: -long, -exp cov auto | http://www.ebi.ac.uk/~zerbino/velvet | 74,75 |
The ‘Program’ column contains the name of program or algorithm. The ‘Repeat-relevant parameters’ column is a list of parameters that adjust how repeats are treated. The programs have many other parameters, but more careful treatment of repeats would start with modification of these. CNV, copy number variant; SV, structural variant.
Table 2.
Overview of current computational tools for next-generation sequencing transcriptome analysis
| Scope | Program | Repeat-relevant parameters | Website | Refs |
|---|---|---|---|---|
| Spliced read alignment | GSNAP | http://share.gene.com/gmap | ||
| MapSplice | http://www.netlab.uky.edu/p/bioinfo/MapSplice | |||
| RUM | http://www.cbil.upenn.edu/RUM | |||
| SpliceMap | http://www.stanford.edu/group/wonglab/SpliceMap | |||
| TopHat | http://tophat.cbcb.umd.edu | |||
| Reference- guided transcript assembly | Cufflinks | Improve repeat read mapping estimate: --multi-read-correct | http://cufflinks.cbcb.umd.edu | 18,19 |
| ERANGE | Use multi-read fractions: --withmultifraction | http://woldlab.caltech.edu/rnaseq | 5 | |
| G-Mo.R-Se | None required; multi-reads supported by read aligner parameters | http://www.genoscope.cns.fr/externe/gmorse | ||
| Myrna | None required; multi-reads supported by read aligner parameters | http://bowtie-bio.sourceforge.net/myrna | 46 | |
| Scripture | None required; multi-reads supported by read aligner parameters | http://www.broadinstitute.org/software/scripture | ||
| De novo transcript assembly | Multiple-k | None required or none available | http://www.surget-groba.ch/downloads | |
| Rnnotator | None required or none available | None | ||
| Trinity | Separate transcripts derived from paralogues: --run butterfly | http://trinityrnaseq.sourceforge.net | 21 | |
| Trans-ABySS | None required or none available through command line | http://www.bcgsc.ca/platform/bioinfo/software/trans-abyss | 76 | |
| Velvet-Oases | Use long reads to resolve repeats: -long, -exp cov auto | http://www.ebi.ac.uk/~zerbino/oases | 77 |
The ‘Program’ column contains the name of program or algorithm. The ‘Repeat-relevant parameters’ column is a list of parameters that adjust how repeats are treated. The programs have many other parameters, but more careful treatment of repeats would start with modification of these.
Genome resequencing projects
Genome resequencing allows researchers to study genetic variation by analysing many genomes from the same or from closely related species23,35-37. The primary requirement is for a high-quality reference genome onto which all of the short NGS reads can be mapped. After sequencing a sample to deep coverage, it is possible to detect SNPs, copy number variants (CNVs) and other types of sequence variation without the need for de novo assembly. The computational task involves aligning millions or billions of reads back to the reference genome using one of several short-read alignment programs (TABLE 1). The two most efficient of these aligners, Bowtie and the Burrows–Wheeler Aligner (BWA), achieve throughputs of 10–40 million reads per hour on a single computer processor. In spite of this recent progress, a major challenge remains when trying to decide what to do with reads that map to multiple locations (that is, multi-reads). Below, we discuss how current short-read alignment tools handle these reads and what problems remain unresolved.
Problems when mapping multi-reads
For computational tools that align NGS reads to a genome, the most commonly encountered problem arises when reads align to multiple locations. For convenience, these reads that map to multiple locations are often called multi-reads. Although the specific type of repeat does not directly influence the read-mapping program, it can influence downstream analyses (such as SNP calling) that rely on unique regions that flank the repeats. The percentage of short reads (25 bp or longer) that map to a unique location on the human genome is typically reported to be 70–80%, although this number varies depending on the read length, the availability of paired-end reads and the sensitivity of the software used for alignment. The repeat content in the human genome, by contrast, is around 50%. The main reason for the discrepancy is that most repeats are inexact, which means that many reads will have a unique ‘best match’, even though the same sequence might occur with slight variations in other locations (FIG. 1a). Assigning reads to the location of their best alignment is the simplest way to resolve repeats, although it is not always correct.
Figure 1. Ambiguities in read mapping.
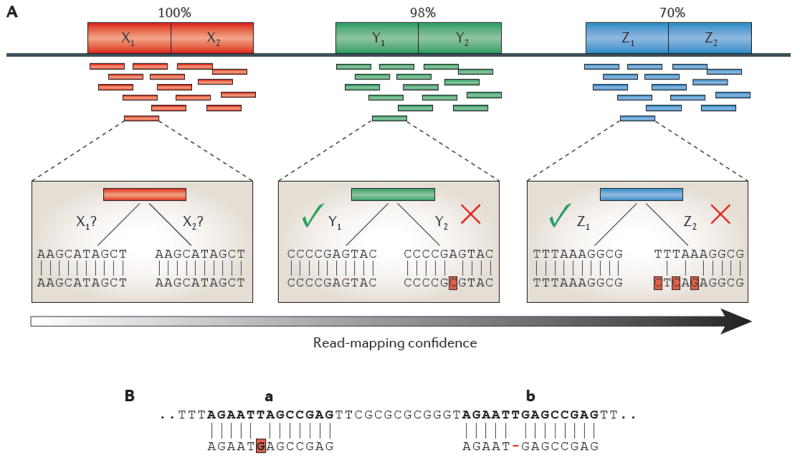
A Read-mapping confidence versus repeat-copy similarity. As the similarity between two copies of a repeat increases, the confidence in any read placement within the repeat decreases. At the top of the figure, we show three different tandem repeats with two copies each. Directly beneath these tandem repeats are reads that are sequenced from these regions. For each tandem repeat, we have highlighted and zoomed in on a single read. Starting with the leftmost read (red) from tandem repeat X, we have low confidence when mapping this read within the tandem repeat, because it aligns equally well to both X1 and X2. In the middle example (tandem repeat Y, green), we have a higher confidence in the mapping owing to a single nucleotide difference, making the alignment to Y1 slightly better than Y2. In the rightmost example, the blue read that is sequenced from tandem repeat Z aligns perfectly to Z1, whereas its alignment to Z2 contains three mismatches, giving us a high confidence when mapping the read to Z1. B | Ambiguity in read mapping. The 13 bp read shown along the bottom maps to two locations, a and b, where there is a mismatch at location a and a deletion at b. If mismatches are considered to be less costly, then the alignment program will put the read in location a. However, the source DNA might have a true deletion in location b, meaning that the true position of the read is b.
For example, suppose that a read maps to two locations, A and B, where the read aligns with one mismatch at location A and with one deletion at B (FIG. 1b). If the alignment program considers a mismatch to be less ‘costly’ than a gap (that is, if it assumes that substitutions are more likely than deletions), then the aligner will put the read in location A. However, if the source DNA has a true deletion in location B, then the read would perfectly match position B. This illustrates a problem that is inherent in the process of aligning reads to a reference genome: the source DNA is virtually never identical to the reference (and, in fact, the differences are the whole reason why the source is being sequenced).
Another example to consider is the following. Suppose that a human genome sample is sequenced, but only analysis of the variants that are present in part of the genome is required: for example, analysis of chromosome 14. The most straightforward approach would be to use a short-read aligner to map reads directly to that chromosome. Unfortunately, this strategy would lead to a large pile up of reads from repetitive regions, because all reads from those repeats would have to go to the same chromosome. To avoid this bias, we must map the reads against the entire genome and use a strategy of random placement of multi-reads to scatter them uniformly across all repeat copies. TABLE 1 lists some of the most useful parameters for dealing with repeats within the most popular alignment programs.
Multi-read mapping strategies
Systematic alignment of reads to incorrect positions in the genome can lead to false inferences of SNPs and CNVs. For example, FIG. 1b illustrates how a SNP would be erroneously identified after a mistake by the alignment program. Essentially, an algorithm has three choices for dealing with multi-reads38 (FIG. 2). The first is to ignore them, meaning that all multi-reads are discarded. The second option is the best match approach, in which the alignment with the fewest mismatches is reported. If there are multiple, equally good best match alignments, then an aligner will either choose one at random or report all of them. The third choice is to report all alignments up to a maximum number, d, regardless of the total number of alignments found. A variant on this strategy is to ignore multi-reads that align to >d locations.
Figure 2. Three strategies for mapping multi-reads.
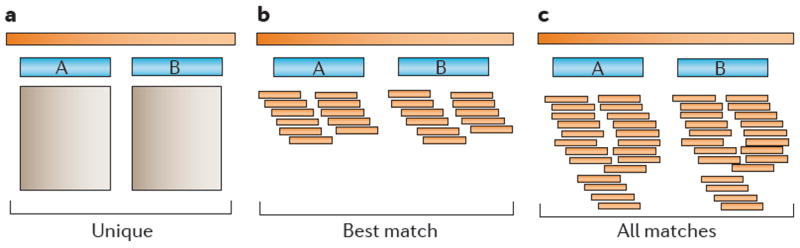
The shaded rectangles at the top represent intervals along a chromosome. The two blue rectangles below each region represent an identical two-copy repeat containing the paralogous genes A and B. The small orange bars represent reads aligned to specific positions. a | The ‘unique’ strategy reports only those reads that are uniquely mappable. Because A and B are identical, no alignments are reported. b | The ‘best match’ alignment strategy reports the best possible alignment for each read, which is determined by the scoring function of the alignment algorithm. In the case of ties, this strategy randomly distributes reads across equally good loci, as shown here. c | The ‘all matches’ strategy simply reports all alignments for each multi-read, including lower-scoring alignments.
To simplify the analysis, some alignment protocols prefer the ‘ignore’ strategy for multi-reads. However, this strategy limits analysis to unique regions in the genome, discarding many multi-gene families as well as all repeats, which might result in biologically important variants being missed. An example in which this occurred is a recent study of retinitis pigmentosa, wherein Tucker et al.39 performed exome sequencing of induced pluripotent stem cells that were derived from a patient with autosomal recessive retinitis pigmentosa. They discovered that the cause of the disease in this patient was a novel, homozygous insertion of a 353 bp Alu repeat in the middle of exon 9 of male germ-cell-associated kinase (MAK). The software used for aligning the reads to the genome trimmed off Alu sequences from the ends of reads, which created a MAK gene that appeared to be normal and initially prevented the discovery of the mutation. Only through a fortunate accident did the investigators discover the presence of the Alu insertion39. The two alternative strategies listed above will ‘fill in’ repetitive regions, although only the best match approach will provide a reasonable estimate of coverage (FIG. 2b). Allowing multi-reads to map to all possible positions (FIG. 2c) avoids making a possibly erroneous choice about read placement. Multi-reads can sometimes be manually resolved with tools such as IGV40 and SAMtools41, which allow users to choose which read placements to keep and which to discard. However, this is not usually a feasible strategy for very large NGS data sets.
Genotyping and SNP detection
After mapping the reads, the next step in the computational pipeline is to call SNPs using a program such as GATK42, MAQ43, SAMtools41, SOAPsnp44 or VarScan45. If multi-reads are handled using the ‘best match’ alignment method, SNPs should be found in at least some repetitive regions. Some methods attempt to handle multi-reads more explicitly. For example, Sniper38 assumes that some multi-reads will align unambiguously owing to slight sequence variations, and it also assumes that SNPs will occur in different locations in different paralogous genes. It uses these assumptions to compute an alignment probability for each multi-read. The probability is computed using a Bayesian genotyping model that decomposes the likehood of a read mapping to a given locus into its component likelihoods. This strategy offers some help for repeats that have few copies, but computation of these probabilities comes at a cost: Sniper would require ~3 central processing unit months to analyse data for a 70-fold coverage of the human genome.
Structural and copy number variant detection
Computational tools can discover multiple types of variants in NGS data, including deletions, insertions, inversions, translocations and duplications (reviewed in REF. 23). Although the software methods that are available can find variants in unique regions reliably, the short NGS read lengths prevent them from detecting variation in repetitive regions with comparable sensitivity. When repeats are longer than the length of a read, methods must rely on depth of coverage or paired-end data to determine whether a repeat region is a variant — neither of these options provides a perfect indictation of structural and CNVs. For example, suppose that a genome of interest is sequenced to an average depth of 30-fold coverage but that a particular tandem repeat that has two copies in the reference genome has 60-fold coverage. These data suggest that the tandem repeat has four copies in the genome of interest — twice the number seen in the reference. However, depth of coverage varies across a genome, which makes it difficult to distinguish N versus N + 1 copies of a repeat with high confidence.
With this caveat, one of the first algorithms to incorporate both read-depth and read-pair data for accurate CNV discovery was VariationHunter13, which has been updated to allow it to find transposons46. Recently, He et al.47 described a new method that was designed to find CNVs even in repeat-rich regions; this method also used information from read pairs and depth of coverage. These authors attempt to account for all mappings of each multi-read, and their method uses this information to improve the estimation of the true copy number of each repeat.
In general, the mapping strategies used for resequencing projects apply to any NGS application in which reads need to be mapped to a reference genome, although some customizations are needed to address the demands of particular applications. For example, in a methyl-seq experiment, analysis is customized to account for C-to-T changes.
De novo genome assembly
Genome assembly algorithms begin with a set of reads and attempt to reconstruct a genome as completely as possible without introducing errors. NGS read lengths (50–150 bp) are considerably shorter than the 800–900 bp lengths that capillary-based (Sanger) sequencing methods were achieving more than 5 years ago, and these short read lengths make assembly more difficult. NGS technology generates higher depth of coverage at far lower cost than Sanger sequencing and, as a result, current strategies for assembly attempt to use deeper coverage to compensate for shorter reads. However, repetitive sequences create substantial difficulties that coverage depth cannot always overcome.
Problems caused by repeats
For de novo assembly, repeats that are longer than the read length create gaps in the assembly. This fact, coupled with the short length of NGS sequences, means that most recent genome assemblies are much more fragmented than assemblies from a few years ago, as evidenced by recent surveys48,49. In addition to creating gaps, repeats can be erroneously collapsed on top of one another and can cause complex, misassembled rearrangements50,51. The degree of difficulty (in terms of correctness and contiguity) that repeats cause during genome assembly largely depends on the read length: if a species has a common repeat of length N, then assembly of the genome of that species will be far better if read lengths are longer than N. As illustrated in BOX 1, the human genome has millions of copies of repeats in the range of 200–500 bp, which is longer than the reads that are produced by today’s most efficient NGS technologies. Until read lengths are greater than 500 bp, assemblies of large plant and animal genomes will need to use other strategies to assemble these types of repeats correctly. Even Sanger read lengths (800–900 bp) cannot resolve longer repeats such as LINEs (BOX 1), and these will continue to require long-range linking information (or exceptionally long-range reads, perhaps generated by future technologies) if they are to be resolved.
Despite these challenges, many new de novo assemblers have emerged to tackle this problem, a selection of which are shown in TABLE 1. All of these assemblers fall into one of two classes: overlap-based assemblers and de Bruijn graph assemblers, both of which create graphs (of different types) from the read data. The algorithms then traverse these graphs in order to reconstruct the genome. From a technical perspective, repeats cause branches in these graphs, and assemblers must then make a guess as to which branch to follow (FIG. 3). Incorrect guesses create false joins (chimeric contigs) and erroneous copy numbers. If the assembler is more conservative, it will break the assembly at these branch points, leading to an accurate but fragmented assembly with fairly small contigs.
Figure 3. Assembly errors caused by repeats.
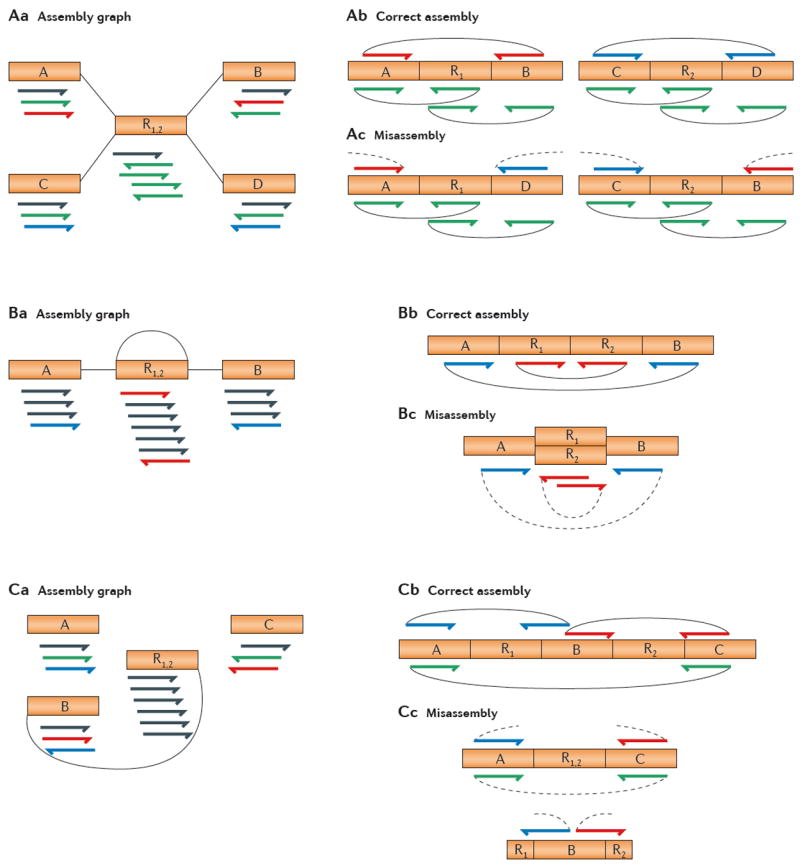
A | Rearrangement assembly error caused by repeats. Aa | An example assembly graph involving six contigs, two of which are identical (R1 and R2). The arrows shown below each contig represent the reads that are aligned to it. Ab | The true assembly of two contigs, showing mate-pair constraints for the red, blue and green paired reads. Ac | Two incorrectly assembled chimeric contigs caused by the repetitive regions R1 and R2. Note that all reads align perfectly to the misassembled contigs, but the mate-pair constraints are violated. B | A collapsed tandem repeat. Ba | The assembly graph contains four contigs, where R1 and R2 are identical repeats. Bb | The true assembly, showing mate-pair constraints for the red and blue paired reads, which are oriented correctly and spaced the correct distance apart. Bc | A misassembly that is caused by collapsing repeats R1 and R2 on top of each other. Read alignments remain consistent, but mate-pair distances are compressed. A different misassembly of this region might reverse the order of R1 and R2. C | A collapsed interspersed repeat. Ca | The assembly graph contains five contigs, where R1 and R2 are identical repeats. Cb | In the correct assembly, R1 and R2 are separated by a unique sequence. Cc | The two copies of the repeat are collapsed onto one another. The unique sequence is then left out of the assembly and appears as an isolated contig with partial repeats on its flank.
The essential problem with repeats is that an assembler cannot distinguish them, which means that the regions flanking them can easily be misassembled. The most common error is that an assembler will create a chimaera by joining two chromosomal regions that do not belong near one another, as illustrated in FIG. 3. As shown in the figure, all of the reads may align well to the misassembled genome; the only hint of a problem is found in the paired-end links. Paired-end reads are generated from a single DNA fragment of a fixed size, from which both ends are sequenced. An assembler uses both the expected distance and the orientation of the reads when reconstructing a genome. If the sequence data do not contain paired ends that span a particular repeat, then it might be impossible to assemble the data unambiguously.
Two recent studies illustrate the difficulty of assembling large genomes from very short reads. Alkan et al.52 looked at recent human genome assemblies and found that they were 16% shorter than the reference genome, primarily owing to missing repetitive sequences. In particular, the NGS assemblies were lacking 420 Mbp of common repeats, including LINE 1 elements, Alu elements and a large majority of segmental duplications. Ye et al.48 compared two NGS assemblies of the chicken genome to its reference genome, which was generated by Sanger sequencing. The chicken genome has a much lower repeat content than the human genome (10% versus 50%), making it considerably easier to assemble. Although their analysis did not look at recent segmental duplications at the level of detail of Alkan et al., they found only 37 long (>10 kb) contigs that were misassembled in total from the two assemblies. Visual inspection indicated that most of these errors were caused by the collapse of interspersed repeats flanking unique sequences (FIG. 3c).
Tandem repeats present another common assembly problem. Near-identical tandem repeats are often collapsed into fewer copies, and it is difficult for an assembler to determine the true copy number. Notably, the investigation into the 2001 Bacillus anthracis attacks in the United States identified isolates of the attack strain that only differed in the presence of two- and three-copy tandem repeats, which the genome assembler had initially collapsed incorrectly53,54. After the assembly errors were detected, the CNVs were correctly reconstructed. These CNVs were present in only minor ‘morphotypes’ from the anthrax-containing letters, which contained a mixture of slight variants on the Ames strain of B. anthracis. The tandem repeat copies were 822, 2,023 and 2,607 nucleotides in length, and these unique markers provided crucial forensic evidence that led investigators back to a single source for the attacks53. FIGURE 3b illustrates a collapsed repeat in which two identical copies are assembled into one. Note that all of the reads may align perfectly, but the coverage depth and the mate-pair information will be inconsistent.
Strategies for handing repeats
In either an overlap graph or a de Bruijn graph, all copies of a repeat will initially be represented by a single node. Repeat boundaries and sequencing errors show up as branch points in the graph, and complex repeats appear as densely connected ‘tangles’ (REF. 55). Assemblers use two main strategies to resolve these tangles. First and most importantly, they use mate-pair information from reads that were sequenced in pairs. A variety of protocols are available for producing two reads from opposite ends of a longer fragment of DNA; these fragments range in length from 200 bp up to 20,000 bp. Even longer stretches can be produced using fosmid clones (30 to 40 kbp) and bacterial artificial chromosome (BAC) clones (up to 150 kbp), although efficient ways of sequencing the ends of these clones are still under development. If a read pair spans a repeat, then the assembler can use that information to decide how to move from a unique region in the graph through a repeat node and into the correct unique region on the other side. Longer fragments allow assemblers to span longer repeats. Because paired-end information is imperfect, most assemblers require two or more pairs of reads to confirm each decision about how to assemble a repeat region.
A good illustration of this strategy is the recently assembled potato genome56. Potato is highly repetitive and has repeats covering an estimated 62% of its genome. The first assembly of this 844 Mbp genome, which was generated with a combination of Illumina and 454 reads, produced tiny contigs that had an N50 size of just 697 bp and also produced scaffolds with an N50 size of 8 kb. As the genome was reassembled using Illumina mate-pair libraries with increasingly large fragment sizes (2 to 10 kb), the scaffolds grew linearly with the insert size, as shown in FIG. 4. The final scaffold N50 size, after using Sanger sequencing to generate paired ends from 40 kb fosmids and 100 kb BACs, was 1.3 Mbp — a 100-fold improvement over the initial statistics. This is a good example of how long fragment libraries can be used to ‘jump’ across repetitive DNA and link together many more contigs.
Figure 4. Longer paired-end libraries improved assembly contiguity in the repetitive potato genome.
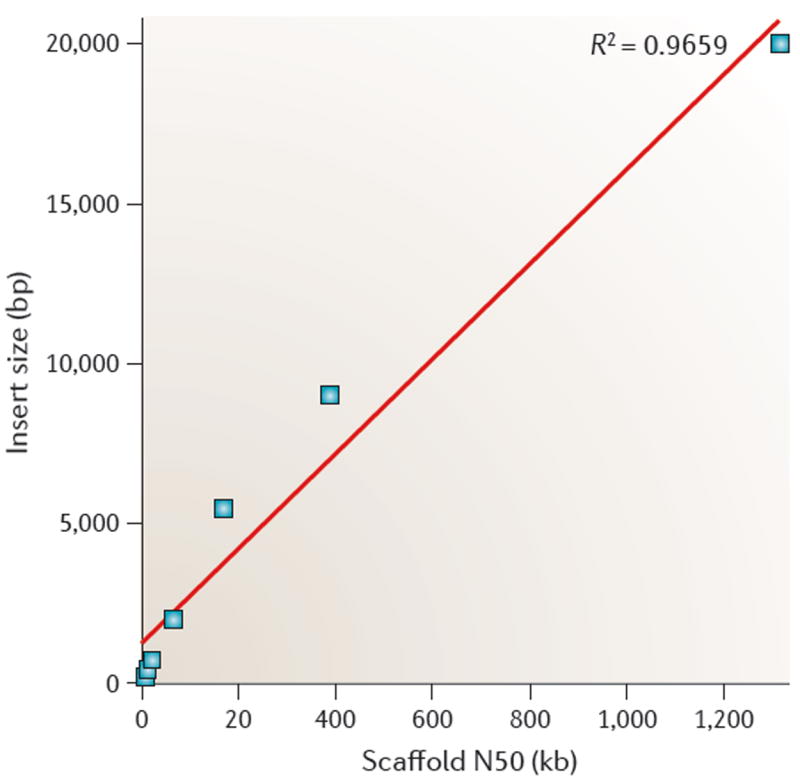
Each point represents the scaffold N50 size of an assembly of the potato genome that was built using paired-end reads from inserts of a specific size and smaller. Successive points moving from left to right used all previous data plus one additional, longer paired-end library size, which is plotted on the y axis. With the addition of the final, 20 kb library, the scaffold N50 size reached 1.3 Mb. The data in this figure are taken from REF. 56.
The second main strategy for handling repeats is to compute statistics on the depth of coverage for each contig. These statistics do not tell assemblers exactly how to assemble each repeat, but they do identify the repeats themselves. In order to make use of this information, assembly programs must assume that the genome is uniformly covered; this means that if a genome is sequenced to 50-fold (50×) coverage, then the assembler assumes that most contigs should also be covered at 50×. A repetitive region, by contrast, will have substantially deeper coverage, which allows the algorithm to identify it as a repeat and to process it differently. In particular, repeats are usually assembled after unique regions, and assemblers may require multiple paired ends to link a repetitive contig to a unique one. One recent study57 suggested that paired-end libraries can be ‘tuned’ to the specific genome being assembled; in it, a strategy is described that uses a preliminary sequence assembly from unpaired reads to estimate repeat structure, which, in turn, can be used to design appropriate paired-end libraries.
A combination of strategies exists for resolving problems that are caused by repetitive DNA, including sequencing strategies that use fragment libraries of varying sizes57, post-processing software that is designed for detecting misassemblies51, analysing coverage statistics and detecting and resolving tangles in a de Bruijn graph. One of the leading NGS assemblers, Allpaths-LG, has specific requirements for the types of paired-end reads that it needs for optimal performance20. None of these requirements completely solves the problems, however, and the ultimate solution may require much longer read lengths.
Alignment and assembly of RNA sequences
High-throughput sequencing of the transcriptome provides a detailed picture of the genes that are expressed in a cell. RNA-seq experiments capture a huge dynamic range of expression levels, and they also detect novel transcripts and alternative splicing events. In response to the rapid growth of these experiments, many new computational tools have emerged, some of which are shown in TABLE 1. RNA-seq analysis centres around three main computational tasks: mapping the reads to a reference genome, assembling the reads into full-length or partial transcripts and quantifying the amount of each transcript. Above, we discussed the first two tasks in the context of genome resequencing projects and de novo assembly, and the problems caused by repeats are largely the same in transcriptome assembly and alignment.
Splicing
A distinct challenge posed by RNA-seq data is the need for spliced alignment of NGS reads. Simply put, this is the problem of aligning a read to two physically separate locations on the genome, which is made necessary by the presence of introns. RNA-seq aligners, such as TopHat58, MapSplice59, rnaSeqMap60, RUM61 and SpliceMap62 are capable of aligning a short read to two distinct locations. Other aligners, including TopHat-Fusion63, FusionSeq64, ShortFuse65 and SplitSeek61 have been designed to scan RNA-seq data and to detect fusion genes that are caused by chromosome breakage and rejoining: a common event in cancer cells. Because a read must be split into pieces before alignment, spliced alignments are shorter, which, in turn, means that repeats present a greater problem than in full-length alignments. For example, if an intron interrupts a read so that only 5 bp of that read span the splice site, then there may be many equally good locations to align the short 5 bp fragment.
Spliced alignment algorithms address this problem by requiring additional, confirming alignments in which longer sequences align on both sides of each splice site. This strategy works well for alignments that span normal genes but, for fusion genes, repeats are particularly problematic. Fusion gene discovery algorithms must allow a pair of reads to align anywhere in the genome; this means that the normal constraints on the distance and orientation of a mate pair cannot be used. When one of the reads falls in a repeat sequence, the algorithm may be faced with thousands of false positives. Collectively, this becomes millions of false positives when extended to all of the data from an RNA-seq experiment. Most fusion gene aligners address this problem by excluding any read with more than one alignment, although some allow a small, fixed number of alignments. Without this restriction, algorithms for fusion gene detection might become computationally unfeasible.
Gene expression
Another challenge that is unique to RNA-seq data is the measurement of gene expression levels, which can be estimated from the number of reads mapping to each gene. The standard approach for estimating expression levels is to count the number of reads or read pairs (also known as fragments) that are aligned to a given gene and to normalize the count based on gene length and sequencing depth. (The measurement is usually expressed as reads or fragments per kilobase of transcript per million reads or fragments sequenced, abbreviated as RPKM or FPKM.)
For gene families and genes containing repeat elements (BOX 1), multi-reads can introduce errors in estimates of gene expression. For example, suppose that a gene exists in two slightly different copies, A and B, and suppose that A is expressed at a much higher level than B is expressed. If the genes are very close paralogues, then most of the reads will map equally well to either copy. In regions where A and B diverge, reads will preferentially map to the correct version of the gene, but this might only be a small portion of the total transcript. Thus, the overall estimate of expression of A will be biased downwards, and the estimate of expression of B will be biased upwards. This error will increase as the sequence similarity between A and B increases.
One way to avoid this bias in the placement of multi-reads is the strategy implemented in ERANGE5 and related methods: these approaches distribute multi-reads in proportion to the number of reads that map to unique regions of each transcript. A similar idea was developed into a more sophisticated statistical model by Jiang and Wong66, who used it to allocate reads among different splice variants. A method that was developed by Chung et al.67 also places multi-reads proportionally, after first estimating expression levels using an expectation maximization algorithm. They demonstrated that, in contrast to methods that only considered uniquely mapped reads, their method can markedly increase coverage in ChIP–seq data, which, in turn, allows for detection of signals that would otherwise be missed67. Li et al.68 developed a software tool called RNA-seq by Expectation Maximization (RSEM) to address the uncertainty that is inherent in multi-read mapping by modelling both isoform levels and non-uniform read distributions; this method produced improved expression estimates in the highly repetitive maize genome. Although it is not clear whether any of these methods is substantially superior to the others, what is clear is that ignoring multi-reads can seriously interfere with accurate scientific analysis.
Conclusions
Advances in DNA-sequencing technology, coupled with novel, efficient computational analysis tools, have made it possible to analyse sequencing-based experimental data on an unprecedented scale. In many of these studies, if not most of them, repetitive DNA sequences present major obstacles to accurate analysis. Repetitive sequences, which permeate the genomes of species from across the tree of life, create ambiguities in the processes of aligning and assembling NGS data. Prompted by this challenge, algorithm developers have designed a variety of strategies for handling the problems that are caused by repeats. For alignment of reads to existing genomes, focusing on uniquely mapped reads addresses some problems, such as SNP discovery, but more sophisticated approaches are necessary to avoid ignoring possibly important sections of a genome: for example, regions containing copy number variation. For de novo genome assembly, shorter read lengths mean that repeats create much greater problems than they did in the era of Sanger sequencing.
Current algorithms rely heavily on paired-end information to resolve the placement of repeats in the correct genome context. This dependency may entail a substantial increase in cost, particularly for large insert sizes in fosmids or BACs (such as those used in the potato genome project), which can be difficult to obtain. Highly repetitive genomes continue to present a serious hurdle to assembly, and these genomes might remain difficult to assemble until read lengths increase substantially. The maize and potato genome projects, both of which were dealing with highly repetitive genomes, were able to avoid generating highly fragmented assemblies by using multiple sequencing technologies, creating multiple large insert libraries and using Sanger sequencing to create the longest insert libraries. Recent human genome assemblies that relied solely on Illumina technology and small insert libraries were less successful, leaving out hundreds of megabases of genomic sequence52. Finally, efforts for estimating gene expression in the presence of repeats have made important strides owing to sophisticated modelling techniques, which use the unique regions of each gene to estimate expression levels and then allocate multi-reads based on statistical estimates. All of these strategies will probably rapidly evolve in response to changing sequencing technologies, which are producing ever-greater volumes of data while slowly increasing read lengths. As it becomes easier to analyse repeats, we will probably learn much more about their role in disease and their contributions to gene function, genome structure and evolution.
Acknowledgments
We thank K. Hansen for useful comments on an earlier draft.
Glossary
- Next-generation sequencing (NGS)
Any of several technologies that sequence very large numbers of DNA fragments in parallel, producing millions or billions of short reads in a single run of an automated sequencer. By contrast, traditional Sanger sequencing only produces a few hundred reads per run
- Interspersed repeats
Identical or nearly identical DNA sequences that are separated by hundreds, thousands or even millions of nucleotides in the source genome. Repeats can be spread out through the genome by mechanisms such as transposition
- Tandem repeats
DNA repeats, (≥2bp in length) that are adjacent to each other and can involve as few as two copies or many thousands of copies. Centromeres and telomeres are largely comprised of tandem repeats
- Short interspersed nuclear elements
(SINEs). Repetitive DNA elements that are typically 100–300 bp in length and spread throughout the genome (such as Alu repeats)
- Long interspersed nuclear elements
(LINEs). Repetitive DNA elements that are typically >300 bp in length and spread throughout the genome (such as L1 repeats)
- Multi-read
A DNA sequence fragment (a ‘read’) that aligns to multiple positions in the reference genome and, consequently, creates ambiguity as to which location was the true source of the read
- Paired-end reads
Reads that are sequenced from both ends of the same DNA fragment. These can be produced by a variety of sequencing protocols, and paired-end preparation is specific to a given sequencing technology. Some recent sequencing vendors use the terms ‘paired end’ and ‘mate pair’ to refer to different protocols, but these terms are generally synonymous
- De Bruijn graph
A directed graph data structure representing overlaps between sequences. In the context of genome assembly, DNA sequence reads are broken up into fixed-length subsequences of length k, which are represented as nodes in the graph. Directed edges are created between nodes i and j if the last k–1 nucleotides of i match the first k–1 nucleotides of j. Reads become paths in the graph, and contigs are assembled by following longer paths
- Contigs
Contiguous stretches of DNA that are constructed by an assembler from the raw reads produced by a sequencing machine
- DNA fragment
In the sequencing process, millions of small fragments are randomly generated from a DNA sample. In paired-end sequencing, both ends of each fragment are sequenced, and the fragment length becomes the ‘library’ size
- N50
A widely used statistic for assessing the contiguity of a genome assembly. The N50 value is computed by sorting all contigs in an assembly from largest to smallest, then cumulatively adding contig sizes starting with the largest and reporting the size of the contig that makes the total greater than or equal to 50% of the genome size. The N50 value is also used for scaffolds
- Scaffold
A scaffold is a collection of contigs that are linked together by paired end information with gaps separating the contigs
Footnotes
Competing interests statement
The authors declare no competing financial interests.
References
- 1.Weigel D, Mott R. The 1001 genomes project for Arabidopsis thaliana. Genome Biol. 2009;10:107. doi: 10.1186/gb-2009-10-5-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.The 1000 Genomes Project Consurtium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Genome 10K Community of Scientists. Genome 10K: a proposal to obtain whole-genome sequence for 10,000 vertebrate species. J Hered. 2009;100:659–674. doi: 10.1093/jhered/esp086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nagalakshmi U, et al. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008;320:1344–1349. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-seq. Nature Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 6.Lister R, et al. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell. 2008;133:523–536. doi: 10.1016/j.cell.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cloonan N, et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nature Methods. 2008;5:613–619. doi: 10.1038/nmeth.1223. [DOI] [PubMed] [Google Scholar]
- 8.Park PJ. ChIP–seq: advantages and challenges of a maturing technology. Nature Rev Genet. 2009;10:669–680. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schmidt D, et al. Five-vertebrate ChIP–seq reveals the evolutionary dynamics of transcription factor binding. Science. 2010;328:1036–1040. doi: 10.1126/science.1186176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein–DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- 11.Garber M, Grabherr MG, Guttman M, Trapnell C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nature Methods. 2011;8:469–477. doi: 10.1038/nmeth.1613. [DOI] [PubMed] [Google Scholar]
- 12.Brunner AL, et al. Distinct DNA methylation patterns characterize differentiated human embryonic stem cells and developing human fetal liver. Genome Res. 2009;19:1044–1056. doi: 10.1101/gr.088773.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hormozdiari F, Alkan C, Eichler EE, Sahinalp SC. Combinatorial algorithms for structural variation detection in high-throughput sequenced genomes. Genome Res. 2009;19:1270–1278. doi: 10.1101/gr.088633.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meyerson M, Gabriel S, Getz G. Advances in understanding cancer genomes through second-generation sequencing. Nature Rev Genet. 2010;11:685–696. doi: 10.1038/nrg2841. [DOI] [PubMed] [Google Scholar]
- 15.Medvedev P, Stanciu M, Brudno M. Computational methods for discovering structural variation with next-generation sequencing. Nature Methods. 2009;6:S13–S20. doi: 10.1038/nmeth.1374. [DOI] [PubMed] [Google Scholar]
- 16.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, Hu Y, Bolund L, Wang J. State of the art de novo assembly of human genomes from massively parallel sequencing data. Hum Genomics. 2010;4:271–277. doi: 10.1186/1479-7364-4-4-271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roberts A, Pimentel H, Trapnell C, Pachter L. Identification of novel transcripts in annotated genomes using RNA-seq. Bioinformatics. 2011;27:2325–2329. doi: 10.1093/bioinformatics/btr355. [DOI] [PubMed] [Google Scholar]
- 19.Trapnell C, et al. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotech. 2010;28:511–515. doi: 10.1038/nbt.1621. This paper describes transcript assembly and abundance estimation from RNA-seq data, including statistical corrections for multi-reads. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gnerre S, et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci USA. 2011;108:1513–1518. doi: 10.1073/pnas.1017351108. This paper presents a highly effective NGS genome assembler that integrates several effective strategies for handling repeats. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grabherr MG, et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nature Biotech. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lunter G, Goodson M. Stampy: a statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res. 2011;21:936–939. doi: 10.1101/gr.111120.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alkan C, Coe BP, Eichler EE. Genome structural variation discovery and genotyping. Nature Rev Genet. 2011;12:363–376. doi: 10.1038/nrg2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmid CW, Deininger PL. Sequence organization of the human genome. Cell. 1975;6:345–358. doi: 10.1016/0092-8674(75)90184-1. [DOI] [PubMed] [Google Scholar]
- 25.Batzer MA, Deininger PL. Alu repeats and human genomic diversity. Nature Rev Genet. 2002;3:370–379. doi: 10.1038/nrg798. [DOI] [PubMed] [Google Scholar]
- 26.Jurka J, Kapitonov VV, Kohany O, Jurka MV. Repetitive sequences in complex genomes: structure and evolution. Annu Rev Genomics Hum Genet. 2007;8:241–259. doi: 10.1146/annurev.genom.8.080706.092416. [DOI] [PubMed] [Google Scholar]
- 27.Britten RJ. Transposable element insertions have strongly affected human evolution. Proc Natl Acad Sci USA. 2010;107:19945–19948. doi: 10.1073/pnas.1014330107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hua-Van A, Le Rouzic A, Boutin TS, Filee J, Capy P. The struggle for life of the genome’s selfish architects. Biol Direct. 2011;6:19. doi: 10.1186/1745-6150-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim PM, et al. Analysis of copy number variants and segmental duplications in the human genome: evidence for a change in the process of formation in recent evolutionary history. Genome Res. 2008;18:1865–1874. doi: 10.1101/gr.081422.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang L, Lu HH, Chung WY, Yang J, Li WH. Patterns of segmental duplication in the human genome. Mol Biol Evol. 2005;22:135–141. doi: 10.1093/molbev/msh262. [DOI] [PubMed] [Google Scholar]
- 31.Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- 32.Schnable PS, et al. The B73 maize genome: complexity, diversity, and dynamics. Science. 2009;326:1112–1115. doi: 10.1126/science.1178534. [DOI] [PubMed] [Google Scholar]
- 33.Reichwald K, et al. High tandem repeat content in the genome of the short-lived annual fish Nothobranchius furzeri: a new vertebrate model for aging research. Genome Biology. 2009;10:R16. doi: 10.1186/gb-2009-10-2-r16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cho NH, et al. The Orientia tsutsugamushi genome reveals massive proliferation of conjugative type IV secretion system and host-cell interaction genes. Proc Natl Acad Sci USA. 2007;104:7981–7986. doi: 10.1073/pnas.0611553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shen Y, et al. A SNP discovery method to assess variant allele probability from next-generation resequencing data. Genome Res. 2010;20:273–280. doi: 10.1101/gr.096388.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mu XJ, Lu ZJ, Kong Y, Lam HY, Gerstein MB. Analysis of genomic variation in non-coding elements using population-scale sequencing data from the 1000 Genomes Project. Nucleic Acids Res. 2011;39:7058–7076. doi: 10.1093/nar/gkr342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gravel S, et al. Demographic history and rare allele sharing among human populations. Proc Natl Acad Sci USA. 2011;108:11983–11988. doi: 10.1073/pnas.1019276108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Simola DF, Kim J. Sniper: improved SNP discovery by multiply mapping deep sequenced reads. Genome Biol. 2011;12:R55. doi: 10.1186/gb-2011-12-6-r55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tucker BA, et al. Exome sequencing and analysis of induced pluripotent stem cells identify the cilia-related gene male germ cell-associated kinase (MAK) as a cause of retinitis pigmentosa. Proc Natl Acad Sci USA. 2011;108:E569–E576. doi: 10.1073/pnas.1108918108. This study shows a striking example of why multi-reads should not be discarded. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robinson JT, et al. Integrative genomics viewer. Nature Biotech. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.DePristo MA, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li R, et al. SNP detection for massively parallel whole-genome resequencing. Genome Res. 2009;19:1124–1132. doi: 10.1101/gr.088013.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koboldt DC, et al. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics. 2009;25:2283–2285. doi: 10.1093/bioinformatics/btp373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hormozdiari F, et al. Next-generation VariationHunter: combinatorial algorithms for transposon insertion discovery. Bioinformatics. 2010;26:i350–i357. doi: 10.1093/bioinformatics/btq216. The authors of this paper present variation detection software that explicitly searches for repetitive transposon sequences. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.He D, Hormozdiari F, Furlotte N, Eskin E. Efficient algorithms for tandem copy number variation reconstruction in repeat-rich regions. Bioinformatics. 2011;27:1513–1520. doi: 10.1093/bioinformatics/btr169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ye L, et al. A vertebrate case study of the quality of assemblies derived from next-generation sequences. Genome Biol. 2011;12:R31. doi: 10.1186/gb-2011-12-3-r31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schatz MC, Delcher AL, Salzberg SL. Assembly of large genomes using second-generation sequencing. Genome Res. 2010;20:1165–1173. doi: 10.1101/gr.101360.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pop M, Salzberg SL. Bioinformatics challenges of new sequencing technology. Trends Genet. 2008;24:142–149. doi: 10.1016/j.tig.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Phillippy AM, Schatz MC, Pop M. Genome assembly forensics: finding the elusive mis-assembly. Genome Biol. 2008;9:R55. doi: 10.1186/gb-2008-9-3-r55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alkan C, Sajjadian S, Eichler EE. Limitations of next-generation genome sequence assembly. Nature Methods. 2011;8:61–65. doi: 10.1038/nmeth.1527. This is an excellent review that highlights the difficulties repeats pose for NGS assemblers. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Read TD, et al. Comparative genome sequencing for discovery of novel polymorphisms in Bacillus anthracis. Science. 2002;296:2028–2033. doi: 10.1126/science.1071837. [DOI] [PubMed] [Google Scholar]
- 54.Rasko DA, et al. Bacillus anthracis comparative genome analysis in support of the Amerithrax investigation. Proc Natl Acad Sci USA. 2011;108:5027–5032. doi: 10.1073/pnas.1016657108. This paper provides a description of how scientists used DNA sequencing to discover a few rare variants in the anthrax-causing bacterium, which led US Federal Bureau of Investigation FBI investigators to the original source of the mailed anthrax from the 2001 attacks. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pevzner PA, Tang H, Waterman MS. An Eulerian path approach to DNA fragment assembly. Proc Natl Acad Sci USA. 2001;98:9748–9753. doi: 10.1073/pnas.171285098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu X, et al. Genome sequence and analysis of the tuber crop potato. Nature. 2011;475:189–195. doi: 10.1038/nature10158. [DOI] [PubMed] [Google Scholar]
- 57.Wetzel J, Kingsford C, Pop M. Assessing the benefits of using mate-pairs to resolve repeats in de novo short-read prokaryotic assemblies. BMC Bioinformatics. 2011;12:95. doi: 10.1186/1471-2105-12-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang K, et al. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 2010;38:e178. doi: 10.1093/nar/gkq622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lesniewska A, Okoniewski MJ. rnaSeqMap: a Bioconductor package for RNA sequencing data exploration. BMC Bioinformatics. 2011;12:200. doi: 10.1186/1471-2105-12-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grant GR, et al. Comparative analysis of RNA-seq alignment algorithms and the RNA-seq unified mapper (RUM) Bioinformatics. 2011;27:2518–2528. doi: 10.1093/bioinformatics/btr427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Au KF, Jiang H, Lin L, Xing Y, Wong WH. Detection of splice junctions from paired-end RNA-seq data by SpliceMap. Nucleic Acids Res. 2010;38:4570–4578. doi: 10.1093/nar/gkq211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim D, Salzberg SL. TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol. 2011;12:R72. doi: 10.1186/gb-2011-12-8-r72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sboner A, et al. FusionSeq: a modular framework for finding gene fusions by analysing paired-end RNA-sequencing data. Genome Biol. 2010;11:R104. doi: 10.1186/gb-2010-11-10-r104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kinsella M, Harismendy O, Nakano M, Frazer KA, Bafna V. Sensitive gene fusion detection using ambiguously mapping RNA-seq read pairs. Bioinformatics. 2011;27:1068–1075. doi: 10.1093/bioinformatics/btr085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jiang H, Wong WH. Statistical inferences for isoform expression in RNA-seq. Bioinformatics. 2009;25:1026–1032. doi: 10.1093/bioinformatics/btp113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chung D, et al. Discovering transcription factor binding sites in highly repetitive regions of genomes with multi-read analysis of ChIP–seq data. PLoS Comput Biol. 2011;7:e1002111. doi: 10.1371/journal.pcbi.1002111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li B, Ruotti V, Stewart RM, Thomson JA, Dewey CN. RNA-seq gene expression estimation with read mapping uncertainty. Bioinformatics. 2010;26:493–500. doi: 10.1093/bioinformatics/btp692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Homer N, Merriman B, Nelson SF. BFAST: an alignment tool for large scale genome resequencing. PLoS ONE. 2009;4:e7767. doi: 10.1371/journal.pone.0007767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li H, Durbin R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Alkan C, et al. Personalized copy number and segmental duplication maps using next-generation sequencing. Nature Genet. 2009;41:1061–1067. doi: 10.1038/ng.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li R, et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25:1966–1967. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
- 73.Miller JR, et al. Aggressive assembly of pyrosequencing reads with mates. Bioinformatics. 2008;24:2818–2824. doi: 10.1093/bioinformatics/btn548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zerbino DR, McEwen GK, Margulies EH, Birney E. Pebble and rock band: heuristic resolution of repeats and scaffolding in the velvet short-read de novo assembler. PLoS ONE. 2009;4:e8407. doi: 10.1371/journal.pone.0008407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Robertson G, et al. De novo assembly and analysis of RNA-seq data. Nature Methods. 2010;7:909–912. doi: 10.1038/nmeth.1517. [DOI] [PubMed] [Google Scholar]
- 77.Garg R, Patel RK, Tyagi AK, Jain M. De novo assembly of chickpea transcriptome using short reads for gene discovery and marker identification. DNA Res. 2011;18:53–63. doi: 10.1093/dnares/dsq028. [DOI] [PMC free article] [PubMed] [Google Scholar]