

Abstract
A general method is presented for the synthesis of alkylated arenes by the chemoselective combination of two electrophilic carbons. Under the optimized conditions, a variety of aryl and vinyl bromides are reductively coupled with alkyl bromides in high yields. Under similar conditions, activated aryl chlorides can also be coupled with bromoalkanes. The protocols are highly functional-group tolerant (−OH, −NHTs, −OAc, −OTs, −OTf, −COMe, −NHBoc, −NHCbz, −CN, −SO2Me), and the reactions are assembled on the benchtop with no special precautions to exclude air or moisture. The reaction displays different chemoselectivity than conventional cross-coupling reactions, such as the Suzuki–Miyaura, Stille, and Hiyama–Denmark reactions. Substrates bearing both an electrophilic and nucleophilic carbon result in selective coupling at the electrophilic carbon (R–X) and no reaction at the nucleophilic carbon (R–[M]) for organoboron (−Bpin), organotin (−SnMe3), and organosilicon (−SiMe2OH) containing organic halides (X–R–[M]). A Hammett study showed a linear correlation of σ and σ(−) parameters with the relative rate of reaction of substituted aryl bromides with bromoalkanes. The small ρ values for these correlations (1.2–1.7) indicate that oxidative addition of the bromoarene is not the turnover-frequency determining step. The rate of reaction has a positive dependence on the concentration of alkyl bromide and catalyst, no dependence upon the amount of zinc (reducing agent), and an inverse dependence upon aryl halide concentration. These results and studies with an organic reductant (TDAE) argue against the intermediacy of organozinc reagents.
1. Introduction
The transition-metal-catalyzed union of nucleophilic organoboronic acids with electrophilic organic halides has become the dominant approach to carbon–carbon (C–C) bond formation in discovery research1 and increasingly in production as well.2These conventional cross-coupling reactions catalytically join nucleophilic carbon (Cδ– or “R–[M]”) with electrophilic carbon (Cδ+ or “R–X”), but direct coupling of two electrophilic carbons has been much less investigated (Figure 1).
Figure 1.

Conventional transition-metal-catalyzed C–C bond formation (Cδ– + Cδ+) compared to direct reductive C–C bond formation (Cδ+ + Cδ+).
The impetus for developing a reductive alternative is that the nucleophilic carbon reagents continue to present some of the largest challenges in conventional cross-coupling. For example, the most widely used nucleophilic carbon reagents, organoboron compounds, have limited commercial availability,3 and some are unstable.4 As a consequence, organoboron (as well as others: RMgX, RZnX, RSnR′3, RSiR′3) reagents are frequently synthesized when needed, and considerable efforts continue to be made in this area.5 Many organometallic reagents or the intermediates used in their synthesis require special care to exclude oxygen and moisture. Similarly, the inherent reactivity of the reagents (RMgX and RZnX) or basic reagents required to facilitate transmetalation (RB(OR′)2, RSnR′3, and RSiR′3) can place limitations on the use of functional groups that are electrophilic or that have acidic protons. Accordingly, the development of methods to alleviate some of these limitations has attracted considerable attention.6−8
By changing the locus of reduction from the substrate to the catalyst, reductive cross-coupling avoids the intermediacy of conventional carbon nucleophiles (R–[M]) and directly joins two electrophilic organic halides (Cδ+ + Cδ+, Figure 1, bottom). The only organometallic intermediates formed during the course of the reaction are comparatively more stable and short-lived catalytic intermediates. As a consequence, functional-group compatibility can be improved because there is no stoichiometric strong bases or nucleophiles. Additionally, the organic halide starting materials are comparatively stable, easy to handle, and readily available. As a result, procedures are simplified, and a large excess of one reagent becomes unnecessary because the organic halide starting materials are stable and easy to handle. Finally, given the different mechanisms and reaction conditions, the direct reductive approach offers the opportunity for synthetic orthogonality to conventional approaches.
This manuscript details a new catalyst system that enables the coupling of aryl bromides, vinyl bromides, and activated aryl chlorides with alkyl bromides in high yield and selectivity. This work builds upon results with aryl iodides that have been previously reported.9 In addition to a large improvement of substrate scope, we demonstrate compatibility with a variety of functional groups, and we demonstrate the chemoselective coupling of two electrophilic organic halides over the coupling of a nucleophilic carbon with an organic halide. Details on the background, the development of selective conditions, and the substrate scope are given below. The potential for the intermediacy of nucleophilic carbon reagents (R–[M]), additive and ligand effects, and the dependence of the rate of reaction on the concentration of reagents and catalyst are discussed.
2. Background
The direct reductive coupling of two electrophilic organic halides (Cδ+ + Cδ+) can avoid many of the ongoing challenges of conventional cross-coupling methods (Cδ– + Cδ+, vida supra), yet this approach has received less attention. While the direct coupling of haloarenes with haloalkanes by the action of stoichiometric sodium metal (Wurtz–Fittig reaction) predates conventional cross-coupling,10 the development of more mild, transition-metal-catalyzed approaches has largely11 been limited to the electrochemical coupling of activated alkyl halides such as α-halogenated carbonyls,12 allylic acetates,13 and benzyl halides.14 A majority of the studies are electrochemical, and in many cases these methods required a substantial excess of one organic halide,12a,12e,12f,13c,13e slow addition of one reactant,14b or both.11,12b,13a These measures were required to minimize the formation of dimeric products and inherent selectivity was low. Indeed, the general challenge of coupling two electrophiles is the difficulty in achieving selectivity for the cross-coupled product over dimerization products. As the two substrates become more similar (both electrophiles), different mechanisms of achieving cross-selectivity must be developed.
The major approach that has been used in previous studies is the flooding of the reaction with an excess of one reagent.15 While this can result in a high yield with respect to the limiting reagent, it requires wasting a large amount of material (Figure 2). As an illustration, a coupling that uses a 2:1 ratio of starting materials (n = 2), but has only statistical selectivity, provides a maximum yield of cross-coupled product (R–R′) of 80%. However, 1.25 mol of dimer byproducts (R–R, R′–R′) are generated for every 1 mol of product! A more difficult yet less wasteful approach is the development of catalysts able to differentiate the two electrophilic reagents.
Figure 2.

Effects of increasing the equivalents of one substrate on maximum statistical yield and waste.
There is currently a poor mechanistic understanding of the selectivity-determining step in direct reductive cross-coupling reactions of aromatic halides with unactivated alkyl halides. Given that this reaction is comparatively new and mechanistically unexplored, a review of related reductive reactions and their proposed mechanisms is presented (Scheme 1).
Scheme 1. Possible Mechanisms for the Direct Cross-Coupling of Aryl Halides with Alkyl Halides.

Off-cycle generation of a nucleophilic carbon reagent concurrent with transition-metal-catalyzed cross-coupling (Scheme 1A) is an active area of research.7 The coupling may also proceed in analogy to the dimerization of aryl halides, by the disproportionation of two [NiII](R)(X) intermediates (Scheme 1B).16 Similarly, Kochi proposed a metathesis of [NiIII]ArX2 and [NiII](Ar)(X) in a radical chain mechanism in nonpolar solvents, but in polar solvents, such as nitromethane, disproportionation of [NiII](Ar)(X) intermediates occurs.17In contrast Colon suggested that the coupling of aryl chlorides by nickel catalysis and reducing metals in polar solvents proceeds via reduction of [NiII](Ar)(X) to [NiI](Ar) followed by subsequent oxidative addition of Ar–X (Scheme 1C), giving privilege to [NiI] and [NiIII] intermediates.18 Amatore and Jutand have made a similar proposal for the electrochemical biaryl synthesis.19,20 On the other hand, stoichiometric21 and electrochemical11 studies of [NiII](Ar)(X) complexes have suggested that intermediate reduction is not required for product formation when coupling with an alkyl halide (Scheme 1D). In this case, two different mechanisms were proposed: two sequential oxidative addition steps to form a [NiIV] intermediate22 or a radical chain mechanism.23 At present, the exact nature of the nickel-catalyzed Csp2–Csp3 bond formation is under active investigation in our lab.
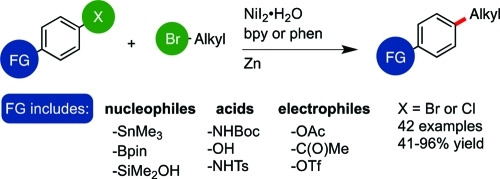
As we reported in 2010, the challenges associated with reductive cross-coupling can be overcome and iodoarenes can be coupled with unactivated iodoalkanes and bromoalkanes in high yield,9 but the use of bromoarenes resulted in lower yield and selectivity for product.24 Gosmini and Amatore published a related cobalt-catalyzed reaction that provided high yields with activated aryl bromides.25 A single example of an activated aryl chloride was provided, but the yield and selectivity were low.26 Being restricted to only iodoarenes is a serious limitation because bromo- and chloroarenes are more readily available than iodoarenes. In addition, no examples of vinyl halides in these couplings have been reported. Finally, the functional-group scope has not been widely explored, even though it is anticipated to differ from conventional cross-coupling reactions. We report here a new catalyst system that for the first time enables the coupling of activated and unactivated bromoarenes, vinyl bromides, and activated chloroarenes with bromoalkanes in good yield and selectivity (Figure 3). We also highlight several key differences in functional-group compatibility between conventional and reductive cross-coupling reactions.
Figure 3.

This work: reductive coupling of aryl bromides, vinyl bromides, and aryl chlorides with alkyl bromides.
3. Results
3.1. Optimization: Catalysts and Reaction Conditions
We began by modifying our previously reported catalyst and reaction conditions for aryl iodide couplings,9 optimizing for the cross-coupling of bromobenzene (1) with 1-bromooctane (2) (Table 1). As before, the major challenges to overcome are the development of a truly cross-selective process and minimization of dimerization, β-hydride elimination, and hydrodehalogenation. Three important changes led to generally high yields: (1) the addition of catalytic amounts of sodium iodide; (2) changing the reducing agent from Mn0 to Zn0; and (3) changing the ligands used. The addition of substoichiometric amounts of sodium iodide to the reaction reduced the amount of dimeric byproduct 8 (Table 1 entry 1 vs entry 2). Changing the reducing agent from Mn0 to Zn0 further decreased dimerization of the aryl halide (8) and suppressed dimerization of the alkyl halide (9) (Table 1, entries 2 vs 3 and 6 vs 7). The synergism we previously observed between bipyridine and bisphosphine ligands9 is not observed in bromide coupling reactions (Table 1, entries 1–5), and the best yields are obtained by using only bipyridine ligands. During early reaction development 4,4′-dimethoxy-2,2′-bipyridine (6) gave higher yields than the previously used 4,4′-di-tert-butyl-2,2′-bipyridine (4), and reactions run with ligand 6 more consistently resulted in complete conversion of starting material.27 For these reasons ligand 6 was chosen for further study. After these optimized conditions were developed, ligand 4 was found to perform about as well as ligand 6 for the coupling of bromobenzene (1) with 1-bromooctane (2) (Table 1, entry 5 vs entry 6). 1,10-Phenanthroline (7) also proved to be an effective ligand and was carried forward in further studies (Table 1, entry 8). In applications where ligand cost is a major consideration, ligands 4 and 7 merit serious consideration.28 A reaction run without ligand produced very little cross-coupled product (Table 1, entry 9). The low reactivity of unligated nickel and the variable stoichiometry of NiI2·xH2O (x ≈ 3.5 by elemental analysis) led us to use a slight excess of nickel (see Supporting Information).
Table 1. Reaction Optimizationa.

| entry | ligand | additives | reductant | yield 3a (%)b | 8 (A%)c | 9 (A%)c | 10 (A%)c | 11 (A%)c | 12 (A%)c |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 4 + 5d,e | py | Mn | 39 | 17 | 13 | 1 | 3 | 0 |
| 2 | 4 + 5 | py, NaI | Mn | 50 | 7 | 11 | 2 | 5 | 3 |
| 3 | 4 + 5 | py, NaI | Zn | 65 | 3 | 2 | 1 | 8 | 8 |
| 4 | 4f | py, NaI | Zn | 67 | 8 | 3 | 3 | 6 | 5 |
| 5 | 4 | py, NaI | Zn | 75 | 2 | 4 | 2 | 10 | 3 |
| 6 | 6 | py, NaI | Zn | 77 | 1 | 1 | 1 | 6 | 3 |
| 7 | 6 | py, NaI | Mn | 39 | 25 | 15 | 4 | 3 | 4 |
| 8 | 7 | py, NaI | Zn | 73 | 2 | 3 | 1 | 3 | 1 |
| 9 | py, NaI | Zn | 11 | 0.5 | 0 | 0 | 2 | 2 | |
| 10 | 6g | py, NaI | Zn | NR | 0 | 0 | 0 | 0 | 0 |
| 11 | 6h | py, NaI | Zn | 53 | 7 | 6 | 4 | 8 | 3 |
| 12 | 6i | py, NaI | Zn | 49 | 10 | 6 | 6 | 11 | 6 |
| 13 | 6 | py | Zn | 66 | 2 | 3 | 2 | 6 | 5 |
| 14 | 6 | NaI | Zn | 75 | 3 | 5 | 2 | 3 | 2 |
| 15 | 6j | py, NaI | Zn | 76 | 2 | 5 | 4 | 5 | 4 |
| 16 | 6 | py, NaI | NR | 0 | 0 | 0 | 0 | 0 |
Reactions were assembled on the benchtop on 0.5 mmol scale in 2 mL of 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (DMPU). The reaction mixtures were heated for 3.5–36 h, and reaction progress was monitored by GC analysis. See Supporting Information for full details.
Yield of 3a was determined by GC analysis vs an internal standard and is corrected.
The amounts of these products are area % (A%) data.
Reaction conducted with 5 mol % 4 and 5 mol % 5.
Reaction run on 1 mmol scale; yield reported is the isolated yield.
Reaction run with 5 mol % 4/NiI2·xH2O/pyridine.
Reaction run with no nickel.
Reaction run at 70 °C.
Reaction run at 80 °C.
TMSCl and 1,2-dibromoethane (4 μL each) were added sequentially as the last two reagents to the reaction vial.
A reaction run without nickel did not consume starting materials, suggesting that direct insertion of zinc into the organic bromides is not likely (Table 1, entry 10), but reactions conducted at 70 and 80 °C (Table 1, entries 11 and 12) contained larger amounts of reduced products (11 and 12), consistent with increased amounts of direct zinc insertion. These results suggest that direct insertion is, in fact, detrimental to yield (Scheme 1A).
While the beneficial effect of sodium iodide is obvious from the data presented in Table 1 (entries 1 vs 2 and 6 vs 13), the improvement due to pyridine is less clear (Table 1 entries 6 vs 14). The addition of pyridine does not dramatically affect the yield, but omission of pyridine has led to slow and/or partial conversion of starting materials. Since catalytic amounts of pyridine appear to make the reaction more robust, it was used throughout the examples in this manuscript. Lastly, the use of catalytic amounts of chlorotrimethylsilane and 1,2-dibromoethane to activate the reducing agent resulted in reaction times as short as 3.5 h (compared to 18 h) with no change in selectivity (Table 1, entries 6 vs 15). The omission of zinc results in no reaction (Table 1, entry 16).
Because these conditions are derived from those developed for the coupling of organic iodides, the specificity of these conditions for the cross-coupling of organic bromides was examined next (Table 2). When one or both organic bromides are replaced with the corresponding organic iodides (Table 2, entries 2–4), yields are diminished compared to the coupling of two bromides. These data demonstrate the complementarity of these two catalyst systems: organic iodides are coupled in the highest yield with the first-generation catalyst system,9 and the coupling of two organic bromides is best accomplished with the new conditions reported here. The large decrease in yield observed when an alkyl iodide is used is accompanied by formation of large amounts of alkyl iodide-derived byproducts, primarily the parent alkane (12). Lastly, the corresponding homocoupling reactions (Table 2, entries 5 and 6) are appreciably slower than the cross-coupling reactions (53 h vs 18 h), and the dimeric products are accompanied by large amounts of reduction byproduct (11 or 12).
Table 2. Specificity of Conditions for Cross-Coupling of Organic Bromidesa.

| entry | organic halides | time (h) | yield (%)b |
|---|---|---|---|
| 1 | Br–C8H17 + Br–Ph | 18 | 77 |
| 2 | I–C8H17 + Br–Ph | 12 | 45 |
| 3 | Br–C8H17 + I–Ph | 47 | 61 |
| 4 | I–C8H17 + I–Ph | 12 | 49 |
| 5 | Br–C8H17 only | 53 | 45c (dimer) |
| 6 | Br–Ph only | 53 | 24c (dimer) |
See Table 1 for reaction conditions.
GC yield corrected vs dodecane internal standard.
Yields are for the corresponding dimeric product.
3.2. Aryl and Vinyl Bromides
The optimized conditions were then tested with a wide range of functionalized substrates. While initial optimization was conducted with 10 mol % catalyst, the catalyst loading could generally be lowered to 5 or 7 mol % (Scheme 2). These lower catalyst loadings represent progress for reductive cross-coupling, which has previously been reported with 10–20 mol % catalyst.9,25 Even with these lower catalyst loadings, the electron-rich aryl bromides 4-bromoanisole and 4-bromo-N,N-dimethylaniline were coupled successfully for the first time, affording alkylated arene products 3c and 3e in high yield. The single ortho-substituent29 on 2-bromotoluene does not inhibit formation of 3f, but 2-bromomesitylene failed to react even under more forcing conditions (80 °C for 3 days), consistent with reports by Klein and others.30 This result could reflect the high stability of (diamine)Ni(Br)(mesityl) complexes and/or the slow oxidative addition of 2-bromomesiltylene to our nickel catalyst. Alkyl bromides with β-branching couple well (3g), but neopentyl bromide gave only trace product. Neopentylated products can be obtained in moderate yield from aryl iodides and our first-generation conditions.9 Electrophilic and electron-withdrawing functionalities, including a base-sensitive methyl ketone and a potentially coordinating nitrile group, also coupled efficiently to afford products 3d and 3h in good yield. Medicinally important fluorine substituents31 are also well tolerated (3i–j).
Scheme 2. Substrate Scope of Aryl and Alkyl Bromides for the Nickel-Catalyzed Reductive Cross-Coupling.

Reaction conditions: organic halides (0.75 mmol each), NiI2·xH2O (0.054–0.078 mmol), ligand (0.05–0.075 mmol), pyridine (0.05–0.075 mmol), sodium iodide (0.19 mmol), zinc dust (>10 μm, 1.5 mmol), and DMPU (3 mL) were assembled on the bench in a 1 dram vial and heated for 5–41 h under air. Yields are of isolated and purified product.
Average of two runs.
Used 1.25 equiv of alkyl bromide (0.94 mmol).
The 2-bromoheptane contained 11% 3-bromoheptane (NMR). Product 3n was isolated as an 83:17 ratio of 3n:heptan-3-ylbenzene (NMR).
Isolated as an inseparable mixture with benzyl butyrate; yields determined by NMR analysis of this mixture.
Isolated as an inseparable mixture of (E) and (Z) isomers.
Isomer ratio determined by NMR analysis.
Starting material (2-bromo-2-butene) was an 88:12 ratio of (Z) and (E) isomers.
Functional group tolerance on the alkyl bromide includes the common nitrogen protecting groups tert-butoxycarbonyl (−Boc) and benzyloxycarbonyl (−Cbz), which also each contain a relatively acidic secondary carbamate proton (3k, 3l). Trisubstituted alkenes were tolerated, giving 3m, but terminal monosubstituted alkenes suffer from isomerization.32At present only select secondary alkyl bromides (3n–3p) couple in moderate yield, but these yields are comparable to those obtained with nickel catalysts for similar conventional cross-coupling reactions.33
Di- and trisubstituted olefins 3q–3t could be made by the direct coupling of vinyl bromides with alkyl halides. The coupling of vinyl bromides in this way has not been previously reported. Olefin migration was not observed in any of these examples, but some loss of stereochemical purity was observed in the formation of 3s (9%) and 3t (17%). Future work will build on these promising results with the aim of further suppressing olefin scrambling.
Prior to this work, substitution at the meta-position of haloarenes had not been explored in a direct reductive cross-coupling with alkyl halides, and we observe interesting ligand effects in the two examples presented in Scheme 2 (3u, 3v, see Discussion section).
3.3. Chloroarenes
Aryl chlorides are often more readily available than aryl bromides, and usually at lower cost, but our first-generation catalyst failed with chloroarenes. A slight modification of the conditions used for the coupling of electron-rich aryl bromides (Scheme 2) proved to be general for the coupling of electron poor aryl chlorides with alkyl bromides (Scheme 3). Omission of sodium iodide, higher reaction temperature (80 °C), and a slight excess of alkyl bromide (1.25 equiv) combined to provide generally high yields of alkylated arene products. In general, there is less biaryl formation and hydrodehalogenation of the chloroarenes compared to reductive couplings with bromoarenes (see Supporting Information for details).
Scheme 3. Substrate Scope of Aryl Chlorides for the Nickel-Catalyzed Reductive Cross-Coupling,

Reaction conditions: aryl chloride (0.75 mmol), alkyl bromide (0.94 mmol), NiI2·xH2O (0.054 mmol), ligand 6 (0.05 mmol), pyridine (0.05 mmol), zinc dust (>10 μm, 1.5 mmol), and DMPU (3 mL) were assembled on the bench in a 1 dram vial and heated for 18–23 h under air.
Yields are of isolated and purified product.
Average of two runs.
Used 1.0 equiv alkyl bromide (0.75 mmol).
Technical grade 1-chloronaphthalene was used (87:13 1-chloronaphthalene/2-chloronaphthalene).
As observed with the aryl bromides, the functional-group tolerance is high. While unactivated aryl chlorides presently do not couple in good yields (Scheme 3, 3b and 3w), chloroarenes bearing electron-withdrawing substituents such as p-trifluoromethyl (Scheme 3, 3i) couple in high yield. Electrophilic functionalities such as a methyl ketone (Scheme 3, 3d) and a nitrile (Scheme 3, 3h) were all tolerated, and sometimes yields were superior to those of reactions with the analogous bromoarenes (Scheme 3, 3h vs Scheme 2, 3h). A methylsulfone substituent also endured the reaction conditions without reduction to the thioether (3x). Finally, while an o-cyano group was reported to be problematic under cobalt-catalyzed conditions (38% yield),25 a good yield of product 3y was obtained (78%).
3.4. Chemoselectivity and Functional-Group Compatibility
A variety of functional groups that are sensitive or reactive under the conditions employed for conventional cross-coupling reactions were tested under these reductive conditions (Scheme 4). In addition to substrates with acidic or electrophilic functional groups, several bifunctional substrates bearing both an electrophilic carbon (C–Br) and a nucleophilic carbon (C–B, C–Si, C–Sn) were tested in order to probe the selectivity of these conditions for the coupling of two electrophiles versus the coupling of an electrophile with a nucleophile.
Scheme 4. Substrates That Demonstrate the Complementarity of Direct Reductive Cross-Coupling to Conventional Cross-Coupling,

Reaction conditions: organic bromides (0.75 mmol each), NiI2.xH2O (0.054 mmol), ligand (0.05 mmol), pyridine (0.05 mmol), sodium iodide (0.19 mmol), zinc dust (>10 μm, 1.5 mmol), and DMPU (3 mL) were assembled on the bench in a 1 dram vial and heated for 3.5–23 h under air.
Yields are of isolated and purified product.
Average of two runs.
Run at 80 °C and with 1 equiv of sodium iodide.
Run with 1.25 equiv of alkyl bromide (0.94 mmol).
Zinc was activated in situ with TMS-Cl and 1,2-dibromoethane (6 μL each).
Several functional groups bearing acidic protons were examined because organozinc and organomagnesium reagents react rapidly with such protons, requiring workarounds such as prior or in situ deprotonation,34 protection,35 or syringe-pump addition (RZnX·LiCl only).36 Not only 4-bromophenol (pKa ≈ 18, DMSO)37 but also 4-bromo-N-p-toluenesulfonylaniline (pKa ≈ 11.9, DMSO)38 were coupled to form the expected alkylated arenes 3z and 3aa in good yield (Scheme 4). The pKa of the sulfonamide is comparable to that of acetic acid (pKa = 12.6, DMSO).37 However, all attempts to cross couple 4-bromobenzoic acid (pKa < 11.0, DMSO)39 resulted in no product formation.
The use of organometallic reagents with β-leaving groups can be plagued by competing β-elimination processes, but the reductive coupling of 4-bromophenol with (2-bromoethoxy)(tert-butyl)dimethylsilane produced product 3ab in high yield (Scheme 4). This product has been made by other methods but requires extra protection/deprotection steps.40 Additionally, the reaction time could be reduced from 15–20 h to only 3.5 h by activating the zinc with chlorotrimethylsilane and dibromoethane.6b The α-arylation of acetaldehyde is currently not possible,41 but 2-(bromoethyl)-1,3-dioxolane42 can be cross-coupled with bromoarenes under these conditions as an alternative method to access α-arylated acetaldehydes (3ac).
The chemoselectivity of these conditions for reaction with organic bromides over pseudohalides was examined next. Many nickel catalysts are excellent at activating aryl triflates,43 tosylates, and even acetates,44,45 but under these conditions products 3ad–3af are formed in high yield with exclusive reaction at the C–Br bond and minimal hydrolysis (Scheme 4). These electrophiles could be used in subsequent conventional cross-coupling reactions or directed functionalizations29 to form polysubstituted arenes.
Finally, the chemoselectivity of these conditions for electrophilic C–Br bonds in the presence of nucleophilic C–B, C–Si, and C–Sn bonds was examined. Similar to our previous report on C–I bonds,9 the reductive coupling of aryl bromides with alkyl bromides is selective for functionalization of C–Br bonds over both CAr–B and CAlkyl–B bonds to form products 3ag–3ah (Scheme 4). Additionally, dimethyl-4-bromophenylsilanol could be coupled with ethyl 4-bromobutryate to form 3ai, which could later be deprotonated and coupled by the conventional cross-coupling method developed by Denmark.46 Finally, 4-trimethylstannylbromobenzene and ethyl 4-bromobutyrate also reacted selectively at the C–Br bonds and not at the C–Sn bond (3aj). Only a small amount of destannylation was observed.
Nitrogen heterocycles are pervasive in medicinal chemistry but often represent a challenge for metal-catalyzed reactions. Here N-acyl-5-bromoindole (Scheme 4, 3ak) coupled in high yield, but unprotected indoles, imidazoles, and pyridines do not couple in acceptable yields under these conditions. To date we have achieved only 26% yield for the cross-coupling of 2-chloro-6-methylpyridine (3al). While this is the first example of an unprotected basic-nitrogen-containing heterocycle in a direct reductive cross-coupling with an alkyl halide, the nickel-catalyzed coupling of halopyridines with haloarenes has been reported.47 More robust and general conditions are currently being developed for the reductive cross-coupling of heteroaromatic halides with alkyl halides.
3.5. Preliminary Mechanistic Analysis
In order to shed light on the apparent relationship between ligand, substrate electronics, and yield revealed in Schemes 2–4, a competitive rate study was undertaken (Table 3). Excess aryl bromide48 was used to approximate pseudo-first-order conditions in alkyl bromide.49 Assuming that reactions with different ligands and aryl halides have the same rate expressions, then krel = kArBr/kPhBr. The krel values were obtained by fitting the data to ln([ArBr]0/[ArBr]) = krel ln([PhBr]0/[PhBr]).50 Table 3 presents the krel data for Ni/6 and Ni/7 for six representative substrates, as well as literature data for three different σ parameters that were used in a Hammett analysis (see Section 4.7).67
Table 3. Competitive Rate Study of Aryl Bromidesa.

| entry | substituent | (6)[Ni] krel | (7)[Ni] krel | σ | σ(−) | σ(•) |
|---|---|---|---|---|---|---|
| 1 | 4-H | 1 | 1 | 0 | 0 | 0 |
| 2 | 4-OMe | 0.81 | 0.71 | –0.268 | –0.26 | 0.24 |
| 3 | 4-F | 1.34 | 1.84 | 0.062 | –0.03 | –0.08 |
| 4 | 3-OMe | 1.15 | 1.19 | 0.12 | 0.115 | –0.02 |
| 5 | 3-CO2Et | 4.21 | 3.62 | 0.37 | 0.315 | 0.35 |
| 6 | 4-CF3 | 10.06 | 8.42 | 0.54 | 0.65 | 0.08 |
| 7 | 4-C(O)Me | 9.21 | 8.43 | 0.50 | 0.84 | 0.54 |
Reaction conditions: aryl bromides (0.375 mmol each), ethyl-4-bromobutyrate (0.25 mmol), NiI2·xH2O (0.078 mmol), ligand (0.075 mmol), pyridine (0.075 mmol), sodium iodide (0.19 mmol), chlorotrimethylsilane (0.034 mmol), 1,2-dibromoethane (0.07 mmol), zinc dust (>10 μm, 1.25 mmol), and DMPU (3 mL) were assembled on the bench in a 1 dram vial and heated until consumption of alkyl bromide. See Supporting Information for full procedures and calculations.
To further understand the roles of each reaction component, we followed formation of product (3a) for a series of reactions in which we sequentially doubled the concentration (or amount) of (i) bromobenzene (1), (ii) 1-bromooctane (2), (iii) Ni/6/pyridine, and (iv) zinc dust. This data was plotted as −ln(1 – f) versus time, where f is the fraction of product as a function of time (Figure 4a).51a These data indicate there is only a positive dependence on catalyst and no dependence the amount of zinc or alkyl bromide (2). Interestingly, there is an apparent inverse dependence on aryl bromide (1). The lack of dependence on reducing agent might be attributed to the heterogeneity of the reaction and the fact that there are far more unoccupied surface sites on the zinc than there are catalyst molecules.52 This also suggests that under the standard reaction conditions reduction is the turnover-frequency determining step.
Figure 4.

(a) Plot of −ln(1 – f), where f is the fraction of product as a function of time (see Supporting Information for full details and linear fits): standard conditions (□, −ln(1 – f) = 0.00327t, R2 = 0.9957); 2 equiv of bromobenzene (1) (+, −ln(1 – f) = 0.000905t, R2 = 0.9951); 2 equiv of 1-bromooctane (2) (○, −ln(1 – f) = 0.0031t, R2 = 0.9817); 4 equiv of Zn0 (Δ, −ln(1 – f) = 0.00336t, R2 = 0.9916); 20 mol % Ni/6/pyridine, (◇, −ln(1 – f) = 0.00479t, R2 = 0.9971). (b) As in panel a, but with activated zinc (TMSCl and 1,2-dibromoethane): standard conditions (□, −ln(1 – f) = 0.00502t, R2 = 0.9944); 2 equiv of bromobenzene (1) (+, −ln(1 – f) = 0.00160t, R2 = 0.9896); 2 equiv of 1-bromooctane (2) (○, −ln(1 – f) = 0.00954t, R2 = 0.9859), 4 equiv of Zn0 (Δ, −ln(1 – f) = 0.00482t, R2 = 0.9987); 20 mol % Ni/6/pyridine (◇, −ln(1 – f) = 0.0105t, R2 = 0.9945); with 1 equiv of benzene (●, −ln(1 – f) = 0.00376t, R2 = 0.9987).
In order to probe the reaction under conditions in which reduction might not be turnover-frequency limiting, the same series of reactions were conducted with activated zinc (Figure 4b). Under these conditions we still observe no dependence on the amount of zinc, but now there is a clear positive dependence on alkyl bromide (2), suggesting that reduction is no longer turnover-frequency limiting. Because additional bromobenzene (1) appears to slow down the rate at which product is formed (Figure 4), 1 equiv of benzene was added to a standard reaction. While this reaction was also slower, hinting at π-complexation as a mechanism for slowing the reaction, the reaction with additional bromobenzene was slower still.
4. Discussion
4.1. Functional Group Compatibility and Chemoselectivity of Reductive Coupling
Several general trends for the functional-group compatibility of reductive cross-coupling are clear from the examples in this and our previous manuscript.9 First, reductive coupling displays good chemoselectivity for carbon–halogen bonds over other electrophiles, such as acidic protons, esters, ketones, and electrophilic phenol derivatives (R–OTs, R–OTf, and R–OAc). This is in contrast to conventional cross-coupling reactions and provides an alternative when these types of functional groups are required.
Second, unlike conventional cross-coupling reactions, reductive cross-coupling strongly prefers the coupling of two electrophilic carbon-halogen bonds. Competition experiments between C–Br and C–B, C–Si, and C–Sn bonds illustrate this remarkable selectivity (Scheme 5).53 Although protonolysis of the C–B, C–Si, and C–Sn bonds was a minor side product, no products of C–C bond formation at these sites were observed. Bifunctionalized substrates such as these are well suited to the synthesis of polysubstituted arenes, and in some cases this strategy could be an alternative to protection/deprotection approaches.8
Scheme 5. Comparison of Reductive Cross-Coupling and Iterative Cross-Coupling for Synthesis of Functionalized Carbon Nucleophiles.

Finally, reductive cross-coupling also has complementary limitations to conventional cross-coupling reactions. The use of zinc reductant complicates the use of easily reduced organic molecules, such as bromopyrene and 4-nitrobromobenzene. We observe no cross-coupled products in reactions with these substrates.55
4.2. Selectivity for Cross-Coupling
The present conditions are highly selective for the formation of the cross-coupled product. For the coupling of bromoarenes with bromoalkanes, 32 of the 34 examples displayed <10 A% alkyl dimer based on GC analysis of crude reaction mixtures. Formation of biaryl is a larger problem, but it is generally a minor side product as well, with 25 of 34 examples displaying <10 A% aryl dimer. These results are much better than statistical selectivity for a 1:1 ratio of starting materials, which would give 1:1 cross product to combined dimers (See Figure 2). Reduction and β-hydride elimination of the alkyl bromide was a problem for couplings with secondary alkyl bromides, vinyl bromides, and 2-chloropyridine, but not for the other substrates. Arene reduction was usually a minor side product except with aryl bromides that were less reactive (electron-rich bromoarenes, electron-neutral chloroarenes, and hindered bromoarenes) or those that contained acidic functional groups. In general, reactions with electron-poor chloroarenes suffered from fewer side products. More alkyl dimer is observed, but this may be due to the presence of a slight excess of alkyl bromide.
4.3. Iodide Additives
A key finding during optimization studies was that the addition of catalytic amounts of sodium iodide (25 mol %) provided higher yields of cross-coupled product (Tables 1 and 4). Reactions conducted with different sources of iodide (tetrabutylammonium iodide, sodium iodide, potassium iodide) all showed the same beneficial effect. The role of the iodide may be to (1) help facilitate reduction of the nickel catalyst by acting as a bridging ligand with zinc,56,18 (2) promote formation of more reactive nickelate complexes,57,18 (3) generate a small amount of the more reactive alkyl iodide in situ,58 and/or (4) facilitate ligand exchange reactions.59 The addition of more sodium iodide (>25 mol %) was not helpful except in the synthesis of 3ac, which was slow to react and for which SN2 reactions are a challenge.60 The effect of sodium iodide on reactions to form 3b–3d are shown below (Table 4). These substrates were chosen to represent electron-poor, electron-neutral, and electron-rich bromoarenes. In all cases the addition of sodium iodide suppressed aryl halide and alkyl halide dimerization. Dimer products are particularly deleterious to yield because they consume 2 equiv of starting material.
Table 4. Effect of Catalytic Sodium Iodide on Yieldab.

| entry | FG | ligand | NaI (mol %) | yield (%)b | product (A%)c | Ar-H (A%)c | alkene/alkanes (A%)c | aryl dimer (A%)c | alkyl dimer (A%)c |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 4-H | 6 | 25 | 86 (3b) | 86 | 3 | 2 | 8 | 1 |
| 2 | 4-H | 6 | 74 (3b) | 79 | 2 | 3 | 13 | 3 | |
| 3 | 4-OMe | 6 | 25 | 73 (3c) | 81 | 4 | 2.5 | 9.5 | 3 |
| 4 | 4-OMe | 6 | 68 (3c) | 75 | 2 | 2 | 16 | 5 | |
| 5 | 4-C(O)Me | 7 | 25 | 84 (3d) | 88 | 4 | 2 | 4 | 2 |
| 6 | 4-C(O)Me | 7 | 63 (3d) | 70.5 | 8 | 3 | 11 | 7.5 |
Reaction conditions: organic halides (0.75 mmol each), NiI2·xH2O (0.078 mmol), ligand (0.075 mmol), pyridine (0.075 mmol), sodium iodide (0.19 mmol), zinc dust (>10 μm, 1.5 mmol), and DMPU (3 mL) were assembled on the bench in a 1 dram vial and heated for 15–41 h under air until complete consumption of starting materials.
Yield of isolated and purified product.
The amounts of these products are area % (A%) data.
In contrast to the results with bromoarenes, the addition of sodium iodide was not beneficial for reactions conducted with chloroarenes (Scheme 3). In these reactions, we propose that formation of a more reactive alkyl iodide by halogen exchange may result in too large a difference in reactivity between the two electrophiles. This, in turn, results in rapid consumption of alkyl bromide and no consumption of aryl chloride. Reactions of iodoalkanes with chloroarenes under our first-generation catalyst system resulted in no cross-coupled product and no consumption of chloroarene, consistent with this hypothesis.
Although in related dimerizations of alkyl halides we have found the use of iodide salts to allow the coupling of alkyl chlorides and sulfinate esters,61 these substrates are poorly reactive in the present coupling with aryl bromides.
4.4. Potential Intermediacy of Organozinc Reagents
Among the many mechanisms proposed for the direct coupling of two organic halides, perhaps the most obvious is the concurrent reduction of one of the two organic halides to a nucleophilic carbon reagent (RZnBr). This mechanism (Scheme 1A) appears to be operative in several recent reports7 and has not been ruled out in most non-electrochemical coupling reactions of organic halides. The tolerance observed in this study for acidic protons, other electrophiles, and a β-silyloxy substituent (3ab) argue against the intermediacy of an organozinc reagent under these reaction conditions. However, in some cases hydrodehalogenation products were observed in this study, and some organic zinc reagents are remarkably tolerant of water.7c Finally, there is strong precedent for the in situ formation of organozinc reagents either by direct insertion of zinc62 or by a nickel-catalyzed route.63
Direct insertion of activated zinc in to the C–Br bond of alkyl bromides typically requires higher temperatures,62 and our own direct insertion study with both zinc and activated zinc6b indicates that direct insertion of zinc into the C–Br bonds of 1 and 2 is slow compared to the reductive cross-coupling (Scheme 6).64
Scheme 6. Direct Insertion of Zinc and Activated Zinc.

GC yield at 24 h based on unreacted 1 or 2, corrected vs dodecane internal standard.
Further evidence against the intermediacy of nucleophilic RZnX intermediates that might arise from nickel-catalyzed organozinc reagent synthesis is the fact that zinc powder could be replaced with tetrakis(dimethylamino)ethylene (TDAE), a nonmetallic reducing agent.65 A reaction with TDAE as the reductant provided an appreciable yield of the cross-coupled product (Scheme 7), which indicates that an organozinc reagent is not necessary for the coupling reaction to proceed.66
Scheme 7. Nickel-Catalyzed Reductive Cross-Coupling with a Nonmetallic Reducing Agent.

GC yield corrected vs dodecane internal standard. TDAE = tetrakis(dimethylamino)ethylene.
4.5. Reductant
One of the major differences between reductive cross-coupling of electrophiles and cross-coupling of nucleophiles with electrophiles is the existence of a reduction step. A variety of reductants have been utilized for reductive reactions in the literature, including electrochemical reduction, organic reductants, and metal powders, but zinc is particularly attractive. On a cost/electron (∼1$/mol of e–) basis or mass/electron basis (∼33 g/mol of e–), zinc dust is very economical. Because zinc dust is a readily available and easy-to-handle reagent, the reactions require no specialized techniques or equipment to run.
Because a reductant is present in the reaction flask, a NiII precatalyst can be used in place of more sensitive Ni0precatalysts. This same process, reduction of NiII to Ni0, likely serves to rescue any nickel intermediates that are oxidized by oxygen, resulting in the observed tolerance to air. If this is the case, then reactions conducted under air could be expected to proceed more slowly than those under inert atmosphere. Indeed, reactions run under argon (8–10 h) are faster than reactions that are run with air in the vessel headspace (15–18 h). The longer reaction time is due to an induction period during which no product is formed. It is important to note that only the total reaction time is affected. Under air or argon, the same selectivity and yield are obtained. We observed that if air was continually introduced into a reaction vessel by repeated piercing of a rubber septum, then the induction period extended for long periods, up to 18 h, and cross-coupling would begin only after the introduction of air ceased. Again, these reactions then proceeded normally and afforded good yields of cross-coupled product.
If high-quality zinc powder or dust (Alfa, 6–9 μm and Aldrich <10 μm) is used, reactions are complete within the standard times reported. We did find, however, one batch of zinc dust from Aldrich that resulted in longer induction periods, presumably due to a thicker oxide coating, but afforded normal yields of product. Activating this material with chlorotrimethylsilane and 1,2-dibromoethane6b in situ or washing the zinc with HClaq restored activity (see Supporting Information for details).67 If zinc dust was overactivated, however, yields were diminished and large amounts of hydrodehalogenated alkyl products were observed. In these cases, competing direct insertion of zinc into the bromoalkane is likely. Zinc powder with a larger particle size (−325 mesh, 44 μm diameter) also worked well, but a slightly lower yield was obtained (Table 5).
Table 5. Effect of Different Zn0 Sources on Yield of 3aa.

| entry | Zn0 type/source | yield (%)b |
|---|---|---|
| 1 | >10 μm/Aldrich | 82 |
| 2 | 6–9 μm/Alfa | 86 |
| 3 | –325 mesh (44 μm)/Alfa | 76 |
See Table 1 for conditions.
GC yield corrected vs dodecane internal standard.
Finally, Amatore and Jutand had suggested that the turnover-frequency limiting step of reductive coupling reactions would be the heterogeneous reduction step when metal powders were used.19 Consistent with this hypothesis, we observed the rate of reactions run with zinc dust could be substantially increased by activating the zinc with chlorotrimethylsilane and 1,2-dibromoethane (from 15 h down to 2.5 h) without altering yield or selectivity (Table 1, entry 15). However, reactions run with double the amount of activated zinc did not produce product at a faster rate (Figure 4b). Under these conditions, reduction may no longer be turnover-frequency limiting.
4.6. Ligand Effects
Reactions conducted with 4,4′-dimethoxy-2,2′-bipyridine (6) as the ligand provide the highest yields of product for electron-neutral and electron-rich aryl bromides, and 1,10-phenanthroline (7) provides better results for aryl bromides as electron-poor as 3-methoxybromobenzene. Figure 5 shows the yield of product for a series of substituted bromoarenes ranked by their relative ability to donate electron density (extrapolated from σ(−) Hammett parameters68). All aryl chlorides worked better with ligand 6, even electron-poor ones, consistent with the idea that oxidative addition underlies this trend. Even without a complete understanding of the origin of these effects, the optimal ligand can be chosen on the basis of the known or inferred donicity of functional groups on the bromoarene.
Figure 5.

Yield of product versus bromoarene substituent for 4,4′-dimethoxy-2,2′-bipyridine (6, solid bars) and 1,10-phenanthroline (7, white speckled bars). In general, ligand 6 is superior for electron-rich arenes, such as 4-methoxybromobenzene and all chloroarenes (data not on chart). Ligand 7 works best for electron-poor bromoarenes.
4.7. Hammett Study
Because 4,4′-dimethoxy-2,2′-bipyridine (6) worked better for reactions with electron-rich bromoarenes and chloroarenes but 1,10-phenanthroline (7) was superior for reactions with electron-poor bromoarenes (Figure 5), we investigated whether these differences were evident in the relative reaction rates as well (Figure 6). Plots of log(krel) versus σ(−) and σ were linear, but a plot versus σ(•) was not linear (see page S20, Supporting Information). The linear fits versus σ(−) and σ were of only moderate quality (R2 ∼ 0.9), and the slope (ρ) was between 1.2 and 1.7.
Figure 6.

Hammett plots of (a) log(krel) versus σ(−) for ligand 6 (■), krel = 1.235σ(−), R2 = 0.9259; (b) log(krel) vs σ(−) for ligand 6 (■), krel = 1.635σ(−), R2 = 0.9531; (c) log(krel) vs σ(−) for ligand 7 (●), krel = 1.264σ, R2 = 0.9387; (d) log(krel) vs σ for ligand 7 (●), krel = 1.657σ, R2 = 0.9468.
The ρ values in Figure 6, 1.2–1.7, are smaller than those reported for stoichiometric studies of the oxidative addition of aryl halides to Pd69 (2.3–5.2) or Ni70 (4.4–8.8). The observation of smaller ρ values for catalytic reactions71 has been interpreted as a sign that oxidative addition is not turnover-frequency limiting.49a Interpretation of Hammett plots for catalytic systems is challenging, especially when the fits might have some curvature.72 These data are not consistent with turnover-frequency determining oxidative addition of bromoarene and appear to hint at another role for the bromoarene in the reaction.
4.8. Substrate, Catalyst, and Reducing Agent Effects on Rate
Although the order of each reactant was not determined, the effect of doubling the amount of each reaction component on the rate of product formation shows that υ ∝ [bromoalkane]x[catalyst]y/[bromoarene]z (x, y, and z are positive numbers and could be non-integers). The apparent positive dependence of the rate upon the concentration of bromoalkane and catalyst suggests that the turnover-frequency limiting step involves the interaction of a nickel species with the bromoalkane.
Although reactions run with activated zinc form product faster than reactions run with unactivated zinc (see product 3ab in Scheme 4, for example), no increase in rate was observed for reactions containing double the usual amount of zinc (Figure 4). Additionally, the reaction displays a positive dependence on alkyl bromide when activated zinc is used, but no dependence on alkyl bromide is observed when unactivated zinc is used. The rate of reactions that contain unactivated zinc is likely limited by a reduction step, as predicted by Amatore and Jutand,19 but reactions with activated zinc make it possible to kinetically observe the next slowest step in the catalytic cycle. This appears to be a change in rate-determining step, not a change in mechanism, because the yield of product and selectivity for product remain essentially unchanged (Table 1, entry 6 vs entry 15).
Finally, the apparent inverse dependence of the rate on bromoarene concentration and the dependence upon bromoalkane concentration provides an explanation for the low ρ value obtained in the Hammett study: oxidative addition of bromoarene is not turnover-frequency determining. Additionally, the bromoarene could have a secondary interaction with the catalyst that has a completely different correlation to electron donicity.
5. Conclusions
By enabling the direct coupling of alkyl bromides with aryl bromides, vinyl bromides, and electron-poor aryl chlorides for the first time, these new catalysts increase the pool of commercially available substrates by more than an order of magnitude and provide increased flexibility in synthetic planning. The reducing nature of the conditions, along with the absence of highly reactive or basic intermediates, imparts resiliency to adventitious water and oxygen and allows benchtop reaction assembly. The chemoselectivity observed for the coupling of carbon–halogen bonds in the presence of a variety of electrophilic functional groups, acidic protons, and nucleophilic C–B, C–Si, and C–Sn bonds presents new opportunities in the synthesis of complex molecules. Challenges that remain are the development of conditions that will allow the coupling of electron-rich aryl chlorides, more general conditions for secondary alkyl bromides, conditions that can further minimize the olefin isomerization, and catalysts that can couple basic-nitrogen-containing substrates in higher yield.
With regards to the mechanism by which these results are achieved, several findings reported here narrow down the list of potential mechanisms. The intermediacy of organozinc reagents appears unlikely at this point, which contrasts with the majority of cross-coupling methods that require a nucleophilic carbon reagent. Hammett analysis found that oxidative addition of the aryl bromide is probably not turnover-frequency determining, and this was confirmed by observing the effect of bromoarene concentration on the rate of product formation. The rate of product formation is in fact proportional to [bromoalkane]x[catalyst]y/[bromoarene]z (x, y, and z are positive numbers and could be non-integers). While the origin of the inverse dependence on bromoarene is not yet known, it appears to be more than simple complexation. Studies that aim to elucidate the complete mechanism are ongoing.
6. Experimental Section
Representative Procedure for Reductive Coupling Reactions. Ethyl 4-(4-(4-Methylphenylsulfonamido)phenyl)butanoate (3aa)
On the benchtop with no precautions to exclude air or moisture, NiI2·xH2O (15.1 mg, 0.040 mmol, 0.053 equiv), 4,4′-dimethoxy-2,2′-bipyridine (8.1 mg, 0.038 mmol, 0.050 equiv), sodium iodide (28.5 mg, 0.190 mmol, 0.250 equiv), and N-(4-bromophenyl)-4-methylbenzenesulfonamide (245 mg, 0.75 mmol, 1.00 equiv) were weighed on weigh paper and transferred to a 1 dram vial equipped with a magnetic stir bar. DMPU (1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone, 3.0 mL), pyridine (3 μL, 0.039 mmol, 0.052 equiv), ethyl 4-bromobutanoate (107 μL, 0.75 mmol, 1.00 equiv), and zinc dust (98 mg, 1.50 mmol, 2.00 equiv) were added. The reaction vial was capped with a PTFE-faced silicone septum, and the green solution was stirred at room temperature for approximately 5 min before heating to 60 °C in a reaction block on the benchtop. Upon completion (judged by GC analysis and color change to black), the reaction mixture was directly applied to the top of a chromatography column. The product was eluted with a 75:25 hexanes/ethyl acetate mixture, and any mixed fractions were further purified by preparative TLC (75:25 hexanes/ethyl acetate, 1500 μm). Sulfonamide 3aa was isolated as viscous colorless oil: first run 221 mg (93%, 5 mol % catalyst, 23 h); second run 233 mg (98%, 5 mol % catalyst, 16 h). 1H NMR (400 MHz; CDCl3): δ 7.62 (d, J = 8.1 Hz, 2H), 7.22 (d, J = 7.9 Hz, 2H), 7.04 (d, J = 8.1 Hz, 2H), 6.96 (d, J = 8.1 Hz, 2H), 6.34 (s, 1H), 4.11 (q, J = 7.2 Hz, 2H), 2.57 (t, J = 7.6 Hz, 2H), 2.38 (s, 3H), 2.27 (t, J = 7.4 Hz, 2H), 1.89 (quintet, J = 7.5 Hz, 2H), 1.25 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz; CDCl3): δ 173.4, 148.0, 145.4, 140.6, 132.6, 129.81, 129.62, 128.6, 122.3, 60.4, 34.6, 33.6, 26.4, 21.8, 14.3. GC–MS m/z (% relative intensity, ion): 362.15 (18.4, M + H), 317.15 (12.6, M + H – C2H5O), 274.10 (33.32, M+ – C4H7O2), 207.1 (10.3, M+ – C7H7O2), 155.05 (43.9, M+ – C12H16NO2), 91.05 (100.0, M+ – C12H16NO4S).
Acknowledgments
This work was supported by the University of Rochester, the National Institutes of Health (R01 GM097243), and the NSF (Graduate Research Fellowship to D.A.E. and Research Experience for Undergraduates Fellowship to B.A.J.). Acknowledgment is also made to the donors of the American Chemical Society Petroleum Research Fund for partial support of this work. Analytical data were obtained from the CENTC Elemental Analysis Facility at the University of Rochester, funded by NSF CHE-0650456. The authors thank Michael R. Prinsell (University of Rochester) for assistance with Figure 2.
Supporting Information Available
Experimental procedures, supporting tables, and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Author Present Address
† Materia, Inc., 60 N. San Gabriel Blvd., Pasadena, CA 91107.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- For a recent reviews of the methods used in drug discovery, see: ; a Roughley S. D.; Jordan A. M. J. Med. Chem. 2011, 54, 3451–3479. [DOI] [PubMed] [Google Scholar]; b Cooper T. W. J.; Campbell I. B.; Macdonald S. J. F. Angew. Chem., Int. Ed. 2010, 49, 8082–8091. [DOI] [PubMed] [Google Scholar]
- For reviews of the reactions used in process chemistry, see: ; a Dugger R.; Ragan J.; Ripin D. Org. Process Res. Dev. 2005, 9, 253–258. [Google Scholar]; b Carey J.; Laffan D.; Thomson C.; Williams M. Org. Biomol. Chem. 2006, 4, 2337–2347. [DOI] [PubMed] [Google Scholar]; c Laird T. Org. Process Res. Dev. 2006, 10, 851–852. [Google Scholar]
- The most commercially available nucleophilic carbon source used in cross-coupling today is the boronic acid. A search of Scifinder Scholar on 9/7/2011 found 5,484 commercially available substances of the general structure C*B(OH)2, where “C*” was any substituents off of carbon. Similar exhaustive searches for R–I and R–Br produced over 80,000 and 700,000 substances in 2010.
- For a discussion of unstable organobrononic acids and a proposed solution, see: Knapp D. M.; Gillis E. P.; Burke M. D. J. Am. Chem. Soc. 2009, 131, 6961–6963 and references therein.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Molander G. A.; Ellis N. Acc. Chem. Res. 2007, 40, 275–286. [DOI] [PubMed] [Google Scholar]; Carbon–boron bonds can be prepared directly from C–H bonds by transition metal catalysis:; b Mkhalid I. A. I.; Barnard J. H.; Marder T. B.; Murphy J. M.; Hartwig J. F. Chem. Rev. 2009, 110, 890–931. [DOI] [PubMed] [Google Scholar]; Preparation of aryl boronic acids from aryl halides can be accomplished by transition metal catalysis. For selected examples, see:; c Haddenham D.; Bailey C. L.; Vu C.; Nepomuceno G.; Eagon S.; Pasumansky L.; Singaram B. Tetrahedron 2011, 67, 576–583. [Google Scholar]; d Wilson D. A.; Wilson C. J.; Moldoveanu C.; Resmerita A.-M.; Corcoran P.; Hoang L. M.; Rosen B. M.; Percec V. J. Am. Chem. Soc. 2010, 132, 1800–1801. [DOI] [PubMed] [Google Scholar]; e Molander G. A.; Trice S. L. J.; Dreher S. D. J. Am. Chem. Soc. 2010, 132, 17701–17703. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Moldoveanu C.; Wilson D. A.; Wilson C. J.; Corcoran P.; Rosen B. M.; Percec V. Org. Lett. 2009, 11, 4974–4977. [DOI] [PubMed] [Google Scholar]; g Murata M.; Oyama T.; Watanabe S.; Masuda Y. J. Org. Chem. 1999, 65, 164–168. [DOI] [PubMed] [Google Scholar]; h Ishiyama T.; Murata M.; Miyaura N. J. Org. Chem. 1995, 60, 7508–7510. [Google Scholar]; For preparation of boronic acids from organolithiums or Grignards and trialkylborates, see:; i Brown H. C.; Rangaishenvi M. V. Tetrahedron Lett. 1990, 31, 7113–7114. [Google Scholar]; j Brown H. C.; Bhat N. G.; Srebnik M. Tetrahedron Lett. 1988, 29, 2631–2634. [Google Scholar]; k Brown H. C.; Cole T. E. Organometallics 1983, 2, 1316–1319. [Google Scholar]; l Matteson D. S.; Liedtke J. D. J. Am. Chem. Soc. 1965, 87, 1526–1531. [Google Scholar]
- For selected examples of the one-pot generation and cross-coupling of organometallic reagents, see: ; a Amatore M.; Gosmini C. Chem. Commun. 2008, 5019–5021. [DOI] [PubMed] [Google Scholar]; b Sase S.; Jaric M.; Metzger A.; Malakhov V.; Knochel P. J. Org. Chem. 2008, 73, 7380–7382. [DOI] [PubMed] [Google Scholar]; c Alessi M.; Larkin A. L.; Ogilvie K. A.; Green L. A.; Lai S.; Lopez S.; Snieckus V. J. Org. Chem. 2007, 72, 1588–1594. [DOI] [PubMed] [Google Scholar]
- For in situ organozinc synthesis concurrent with cross-coupling, see: ; a Krasovskaya V.; Krasovskiy A.; Bhattacharjya A.; Lipshutz B. H. Chem. Commun. 2011, 47, 5717–5719. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Duplais C.; Krasovskiy A.; Wattenberg A.; Lipshutz B. H. Chem. Commun. 2010, 46, 562–564. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Krasovskiy A.; Duplais C.; Lipshutz B. H. J. Am. Chem. Soc. 2009, 131, 15592–15593. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Bégouin J. M.; Gosmini C. J. Org. Chem. 2009, 74, 3221–3224. [DOI] [PubMed] [Google Scholar]; e Amatore M.; Gosmini C. Chem. Commun. 2008, 5019–5021. [DOI] [PubMed] [Google Scholar]; For in situ Grignard synthesis concurrent with cross-coupling, see:; f Czaplik W. M.; Mayer M.; Jacobi von Wangelin A. ChemCatChem 2011, 3, 135–138. [Google Scholar]; g Czaplik W.; Mayer M.; Jacobi Von Wangelin A. Angew. Chem., Int. Ed. 2009, 48, 607–610. [DOI] [PubMed] [Google Scholar]
- Several groups have focused on using protected carbon nucleophiles to impart stability. For examples using MIDA boronates, see: ; a Gillis E. P.; Burke M. D. J. Am. Chem. Soc. 2008, 130, 14084–14085. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gillis E. P.; Burke M. D. J. Am. Chem. Soc. 2007, 129, 6716–6717. [DOI] [PubMed] [Google Scholar]; For examples using 1,8-dimainonaphthyl (dan) derivatives, see:; c Noguchi H.; Shioda T.; Chou C.-M.; Suginome M. Org. Lett. 2008, 10, 377–380. [DOI] [PubMed] [Google Scholar]; d Noguchi H.; Hojo K.; Suginome M. J. Am. Chem. Soc. 2007, 129, 758–759. [DOI] [PubMed] [Google Scholar]; For a silicon-based approach, see:; e Nakao Y.; Chen J.; Tanaka M.; Hiyama T. J. Am. Chem. Soc. 2007, 129, 11694–11695. [DOI] [PubMed] [Google Scholar]
- Everson D. A.; Shrestha R.; Weix D. J. J. Am. Chem. Soc. 2010, 132, 920–921. [DOI] [PubMed] [Google Scholar]
- a Fittig R.; König J. Liebigs Ann. Chem. 1867, 144, 277–294. [Google Scholar]; b Kwa T. L.; Boelhouwer C. Tetrahedron 1969, 25, 5771–5776. [Google Scholar]
- For an example of a nickel-catalyzed electrochemical coupling of methyl 3-bromopropionate with bromobenzene that produces the product in 38% yield, see: Durandetti M.; Nédélec J.-Y.; Périchon J. J. Org. Chem. 1996, 61, 1748–1755. [DOI] [PubMed] [Google Scholar]
- For selected electrochemical approaches: ; a Durandetti M.; Gosmini C.; Périchon J. Tetrahedron 2007, 63, 1146–1153. [Google Scholar]; b Durandetti M.; Périchon J.; Nédélec J. Y. J. Org. Chem. 1997, 62, 7914–7915. [DOI] [PubMed] [Google Scholar]; c Durandetti M.; Devaud M.; Périchon J. New J. Chem. 1996, 20, 659–667. [Google Scholar]; d Durandetti M.; Sibille S.; Nédélec J. Y.; Périchon J. Synth. Commun. 1994, 24, 145–151. [Google Scholar]; e Conan A.; Sibille S.; Dincan E.; Périchon J. Chem. Commun. 1990, 48–49. [Google Scholar]; f Folest J. C.; Périchon J.; Fauvarque J. F.; Jutand A. J. Organomet. Chem. 1988, 342, 259–261. [Google Scholar]
- For selected electrochemical approaches, see: ; a Durandetti M.; Périchon J. Synthesis 2004, 3079–3083. [Google Scholar]; b Gomes P.; Buriez O.; Labbe E.; Gosmini C.; Périchon J. J. Electroanal. Chem. 2004, 562, 255–260. [Google Scholar]; c Gomes P.; Gosmini C.; Périchon J. J. Org. Chem. 2003, 68, 1142–1145. [DOI] [PubMed] [Google Scholar]; d Buriez O.; Labbe E.; Périchon J. J. Electroanal. Chem. 2003, 543, 143–151. [Google Scholar]; e Gomes P.; Gosmini C.; Périchon J. Org. Lett. 2003, 5, 1043–1045. [DOI] [PubMed] [Google Scholar]
- For selected electrochemical approachs, see ref (11) and ; a Durandetti M.; Hardou L.; Clement M.; Maddaluno J. Chem. Commun. 2009, 4753–4755. [DOI] [PubMed] [Google Scholar]; b Begouin J. M.; Claudel S.; Gosmini C. Synlett 2009, 3192–3194. [Google Scholar]
- For example, the cross-McMurry reaction: ; a McMurry J. E. Chem. Rev. 1989, 89, 1513–1524. [Google Scholar]; b Ladipo F. T. Curr. Org. Chem. 2006, 10, 965–980. [Google Scholar]
- a Parshall G. W. J. Am. Chem. Soc. 1974, 96, 2360–2366. [Google Scholar]; b Nakamura A.; Otsuka S. Tetrahedron Lett. 1974, 15, 463–466. [Google Scholar]; c Matsumoto H.; Inaba S.; Rieke R. D. J. Org. Chem. 1983, 48, 840–843. [Google Scholar]; d Yamamoto T.; Wakabayashi S.; Osakada K. J. Organomet. Chem. 1992, 428, 223–237. [Google Scholar]; e Osakada K.; Yamamoto T. Coord. Chem. Rev. 2000, 198, 379–399. [Google Scholar]
- For electron transfer mechanisms, see: ; a Kochi J. Acc. Chem. Res. 1974, 7, 351–360. [Google Scholar]; b Morrell D. G.; Kochi J. K. J. Am. Chem. Soc. 1975, 97, 7262–7270. [Google Scholar]; c Tsou T. T.; Kochi J. K. J. Am. Chem. Soc. 1979, 101, 7547–7560. [Google Scholar]
- Colon I.; Kelsey D. R. J. Org. Chem. 1986, 51, 2627–2637. [Google Scholar]
- Amatore C.; Jutand A. Organometallics 1988, 7, 2203–2214. [Google Scholar]
- For a review on the Ullmann coupling, see: ; Hassan J.; Sevignon M.; Gozzi C.; Schulz E.; Lemaire M. Chem. Rev. 2002, 102, 1359–1470. [DOI] [PubMed] [Google Scholar]
- For examples of L2ArNi(II)X complexes reacting with R–X to give Ar–R and L2Ni(0), see: ; a Uchino M.; Yamamoto A.; Ikeda S. J. Organomet. Chem. 1970, 24, C63–C64. [Google Scholar]; b Folest J. C.; Périchon J.; Fauvarque J. F.; Jutand A. J. Organomet. Chem. 1988, 342, 259–261. [Google Scholar]; c Kim Y.-J.; Sato R.; Maruyama T.; Osakada K.; Yamamoto T. Dalton Trans. 1994, 943–948. [Google Scholar]
- a Semmelhack M. F.; Helquist P. M.; Jones L. D. J. Am. Chem. Soc. 1971, 93, 5908–5910. [Google Scholar]; b Hegedus L. S.; Miller L. L. J. Am. Chem. Soc. 1975, 97, 459–460. [Google Scholar]; c Bradley J. S.; Connor D. E.; Dolphin D.; Labinger J. A.; Osborn J. A. J. Am. Chem. Soc. 1972, 94, 4043–4044. [Google Scholar]
- For a similar proposal involving the coupling of alkyl halides with π-allyl nickel bromide, see: Hegedus L. S.; Miller L. L. J. Am. Chem. Soc. 1975, 97, 459–460. [Google Scholar]
- The coupling of iodobenzene with iodooctane proceeded in 88% GC yield, iodobenzene with bromooctane in 85% GC yield, bromobenzene with iodooctane in 77% GC yield, and bromobenzene with bromooctane in 65% GC yield.
- Amatore M.; Gosmini C. Chem.–Euro. J. 2010, 16, 5848–5852. [DOI] [PubMed] [Google Scholar]
- The coupling of 4-chlorobenzonitrile with 1-bromo-3-phenylpropane required 2 equiv of the bromoalkane and 20 mol % catalyst and formed the product with a 57% yield.
- For example, the coupling of bromobenzene with 1-bromooctane in in DMPU with 4,4′-di-tert-butyl-2,2′-dipyridyl (4) at 80 °C failed to consume all starting materials after 42 h.
- Ligand prices (Aldrich Chemical Co.): 4,4′-di-tert-butyl-2,2′-dipyridyl (4) $7.86/g, 4,4′-dimethoxy-bipyridine (6) $23.40/g, 1,10-phenanthroline (7) $2.66/g.
- Coordinating substituents ortho to the halogen, such as −C(O)R and −CO2R, resulted in quantative hydrodehalogenation of the bromoarene. These products are best synthesized by a directed-ortho-metalation/cross-coupling strategy. For metalation using ester and ketone groups as directors, see: ; a Wunderlich S.; Knochel P. Org. Lett. 2008, 10, 4705–4707. [DOI] [PubMed] [Google Scholar]; For metalation using carboxylates as directors see:; b Nguyen T.-H.; Castanet A.-S.; Mortier J. Org. Lett. 2006, 8, 765–768. [DOI] [PubMed] [Google Scholar]; For recent examples of directed-ortho-metalation/cross-coupling strategy using amides or esters as directors, see:; c Hurst T. E.; Macklin T. K.; Becker M.; Hartmann E.; Kügel W.; Parisienne-La Salle J.-C.; Batsanov A. S.; Marder T. B.; Snieckus V. Chem. −Eur. J. 2010, 16, 8155–8161. [DOI] [PubMed] [Google Scholar]; d Quasdorf K. W.; Riener M.; Petrova K. V.; Garg N. K. J. Am. Chem. Soc. 2009, 131, 17748–17749. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Alessi M.; Larkin A. L.; Ogilvie K. A.; Green L. A.; Lai S.; Lopez S.; Snieckus V. J. Org. Chem. 2007, 72, 1588–1594. [DOI] [PubMed] [Google Scholar]
- a Klein A.; Budnikova Y. H.; Sinyashin O. G. J. Organomet. Chem. 2007, 692, 3156–3166. [Google Scholar]; b Yakhvarov D. G.; budnikova Y. H.; Sinyashin O. G. Russ. Chem. Bull. Int. Ed. 2003, 52, 567–569. [Google Scholar]; c Yakhvarov D. G.; Budnikova Y. G.; Sinyashin O. G. Russ. J. Electrochem. 2003, 39, 1261–1269. [Google Scholar]; d Yakhvarov D. G.; Samieva E. G.; Tazeev D. I.; Budnikova Y. G. Russ. Chem. Bull. Int. Ed. 2002, 51, 796–804. [Google Scholar]
- a Purser S.; Moore P. R.; Swallow S.; Gouverneur V. Chem. Soc. Rev. 2008, 37, 320–330. [DOI] [PubMed] [Google Scholar]; b Kirk K. L. Org. Process Res. Dev. 2008, 12, 305–321. [Google Scholar]
- Under the reaction conditions listed in Scheme 2, the coupling of 8-bromo-1-octene with ethyl 3-bromobenzoate resulted in a mixture (46:37:13 ratio based on GC A%) of three olefin isomers. Olefin isomerization could be suppressed by the addition of 3 equiv of N, N-diisopropylethylamine. Under these reaction conditions the rate of product formation was slower, but at partial conversion a single product was observed by GC analysis of the crude reaction mixture. At longer reaction times (41 h) a mixture of isomers was again observed (19:36:16 ratio based on GC A%, accompanied by 24 A% hydrodehalogenated aryl bromide).
- a Ren P.; Vechorkin O.; Allmen K. v.; Scopelliti R.; Hu X. J. Am. Chem. Soc. 2011, 133, 7084–7095. [DOI] [PubMed] [Google Scholar]; b Molander G. A.; Argintaru O. A.; Aron I.; Dreher S. D. Org. Lett. 2010, 12, 5783–5785. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Vechorkin O.; Proust V. r.; Hu X. J. Am. Chem. Soc. 2009, 131, 9756–9766. [DOI] [PubMed] [Google Scholar]; d Vechorkin O.; Hu X. Angew. Chem., Int. Ed. 2009, 48, 2937–2940. [DOI] [PubMed] [Google Scholar]; e Strotman N. A.; Sommer S.; Fu G. C. Angew. Chem., Int. Ed. 2007, 46, 3556–3558. [DOI] [PubMed] [Google Scholar]; f Gonzalez-Bobes F.; Fu G. C. J. Am. Chem. Soc. 2006, 128, 5360–5361. [DOI] [PubMed] [Google Scholar]; g Powell D. A.; Maki T.; Fu G. C. J. Am. Chem. Soc. 2005, 127, 510–511. [DOI] [PubMed] [Google Scholar]; h Powell D. A.; Fu G. C. J. Am. Chem. Soc. 2004, 126, 7788–7789. [DOI] [PubMed] [Google Scholar]; i Netherton M.; Fu G. Ad. Synth. Catal. 2004, 346, 1525–1532. [Google Scholar]
- For selected examples of Suzuki–Miyaura coupling reactions of substrates bearing −OH groups that employ excess base, see: ; a Petronzi C.; Filosa R.; Peduto A.; Monti M. C.; Margarucci L.; Massa A.; Ercolino S. F.; Bizzarro V.; Parente L.; Riccio R.; de Caprariis P. Eur. J. Med. Chem. 2011, 46, 488–496. [DOI] [PubMed] [Google Scholar]; b Baxendale I. R.; Griffiths-Jones C. M.; Ley S. V.; Tranmer G. K. Chem.– Eur. J. 2006, 12, 4407–4416. [DOI] [PubMed] [Google Scholar]; c Handy S. T.; Zhang Y.; Bregman H. J. Org. Chem. 2004, 69, 2362–2366. [DOI] [PubMed] [Google Scholar]; For a Ni-catalyzed Negishi cross-coupling with 2.5 equiv sec-butyl-zinc bromide or isopropyl-zinc bromide and 4-bromophenol, see:; d Joshi-Pangu A.; Ganesh M.; Biscoe M. R. Org. Lett. 2011, 13, 1218–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a palladium-catalyzed Negishi coupling with 4-bromophenol protected as the tert-butyldimethylsilyl derivative, see: ; a Johnson A. T.; Wang L.; Gillett S. J.; Chandraratna R. A. S. Bioorg. Med. Chem. Lett. 1999, 9, 573–576. [DOI] [PubMed] [Google Scholar]; The organozinc reagent of 4-bromophenol has been previously made by cobalt catalysis in 6% corrected GC yield along with 94% phenol:; b Fillon H.; Gosmini C.; Périchon J. J. Am. Chem. Soc. 2003, 125, 3867–3870. [DOI] [PubMed] [Google Scholar]
- a Manolikakes G.; Schade M. A.; Hernandez C. M. o.; Mayr H.; Knochel P. Org. Lett. 2008, 10, 2765–2768. [DOI] [PubMed] [Google Scholar]; b Sase S.; Jaric M.; Metzger A.; Malakhov V.; Knochel P. J. Org. Chem. 2008, 73, 7380–7382. [DOI] [PubMed] [Google Scholar]
- Bordwell F. G. Acc. Chem. Res. 1988, 21, 456–463. [Google Scholar]
- The pKa of PhSO2NHPh as been reported to be 11.9 in DMSO: Cheng J.-P.; Zhao Y. Tetrahedron 1993, 49, 5267–5276. [Google Scholar]
- The pKa of benzoic acid is reported to be 11.0. Inductive effects should make 4-bromobenzoic acid more acidic. Olmstead W. N.; Bordwell F. G. J. Org. Chem. 1980, 45, 3299–3305. [Google Scholar]
- a Prakash C.; Saleh S.; Blair I. A. Tetrahedron Lett. 1994, 35, 7565–7568. [Google Scholar]; b Shah S. T. A.; Singh S.; Guiry P. I. J. Org. Chem. 2009, 74, 2179–2182. [DOI] [PubMed] [Google Scholar]
- The α-arylation of linear aldehydes is still a challenge, with no reports on the arylation of simple acetaldehyde: ; a Vo G. D.; Hartwig J. F. Angew. Chem., Int. Ed. 2008, 47, 2127–2130. [DOI] [PubMed] [Google Scholar]; b Martín R.; Buchwald S. L. Org. Lett. 2008, 10, 4561–4564. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Martín R.; Buchwald S. L. Angew. Chem., Int. Ed. 2007, 46, 7236–7239. [DOI] [PubMed] [Google Scholar]; d Terao Y.; Fukuoka Y.; Satoh T.; Miura M.; Nomura M. Tetrahedron Lett. 2002, 43, 101–104. [Google Scholar]
- For a nickel-catalyzed Suzuki–Miyaura cross-coupling of the corresponding alkyl chloride with an aryl potassium trifluoroborate, see ref (33)b.
- a Meijere A. d.; Diederich F.. Metal-Catalyzed Cross Coupling Reactions, 2nd ed.; Wiley-VCH: Weinhein, 2004. [Google Scholar]; b Negishi E.-i.; Meijere A. d.. Handbook of Organopalladium Chemistry for Organic Synthesis, 1st ed.; Wiley-VCH: Weinhein, 2002. [Google Scholar]
- a Guan B.-T.; Wang Y.; Li B.-J.; Yu D.-G.; Shi Z.-J. J. Am. Chem. Soc. 2008, 130, 14468–14470. [DOI] [PubMed] [Google Scholar]; b Quasdorf K. W.; Tian X.; Garg N. K. J. Am. Chem. Soc. 2008, 130, 14422–14423. [DOI] [PubMed] [Google Scholar]
- Rosen B. M.; Quasdorf K. W.; Wilson D. A.; Zhang N.; Resmerita A.-M.; Garg N. K.; Percec V. Chem. Rev. 2010, 111, 1346–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denmark S. E.; Smith R. C.; Chang W.-T. T.; Muhuhi J. M. J. Am. Chem. Soc. 2009, 131, 3104–3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosmini C.; Bassene-Ernst C.; Durandetti M. Tetrahedron 2009, 65, 6141–6146. [Google Scholar]
- Reactions conducted with more than 3 equiv of total aryl bromide provided very low conversion.
- a Weissman H.; Milstein D. Chem. Commun. 1999, 1901–1902. [Google Scholar]; b Böhm V. P. W; Herrmann W. A. Chem.–Eur. J. 2001, 7, 4191–4197. [DOI] [PubMed] [Google Scholar]
- Kleimark J.; Hedström A.; Larsson P.-F.; Johansson C.; Norrby P.-O. ChemCatChem 2009, 1, 152–161. [Google Scholar]
- a Grisso B. A.; Johnson J. R.; Mackenzie P. B. J. Am. Chem. Soc. 1992, 114, 5160–5165. [Google Scholar]; b Blackmond D. G. Angew. Chem., Int. Ed. 2005, 44, 4302–4320. [DOI] [PubMed] [Google Scholar]
- Masel R. I.Principles of Adsorption and Reaction on Solid Surfaces; Wiley-VCH: New York, 1996. [Google Scholar]
- For an example of selective reaction with Ar–Br over Ar–Bpin in a coupling with a secondary alkylzinc reagent, see ref (34d).
- For a similar observation see ref (22a). Nitrobenzene is easily reduced by alkyl radicals: ; a Jagannadham V.; Steenken S. J. Am. Chem. Soc. 1984, 106, 6542–6551. [Google Scholar]; In aprotic polar media nitrobenzene has a reversible one electron reduction potential of −1.2V versus Ag:; b Silvester D. S.; Wain A. J.; Aldous L.; Hardacre C.; Compton R. G. J. Electroanal. Chem. 2006, 596, 131–140. [Google Scholar]; c Smith W. H.; Bard A. J. J. Am. Chem. Soc. 1975, 97, 5203–5210. [Google Scholar]
- Zembayashi M.; Tamao K.; Yoshida J.-i.; Kumada M. Tetrahedron Lett. 1977, 18, 4089–4091. [Google Scholar]
- a Terao J.; Watanabe H.; Ikumi A.; Kuniyasu H.; Kambe N. J. Am. Chem. Soc. 2002, 124, 4222–4223. [DOI] [PubMed] [Google Scholar]; b Cassar L.; Foà M. J. Organomet. Chem. 1973, 51, 381–393. [Google Scholar]
- a Finkelstein H. Chem. Ber. 1910, 43, 1528–1532. [Google Scholar]; b Takahashi H.; Inagaki S.; Nishihara Y.; Shibata T.; Takagi K. Org. Lett. 2006, 8, 3037–3040. [DOI] [PubMed] [Google Scholar]
- Klein A.; Kaiser A.; Wielandt W.; Belaj F.; Wendel E.; Bertagnolli H.; Záliš S. Inorg. Chem. 2008, 47, 11324–11333. [DOI] [PubMed] [Google Scholar]
- 2-(Bromoethyl)-1,3-dioxolane can be converted to 2-(iodoethyl)-1,3-dioxolane by reaction with sodium iodide in refluxing diethylketone (bp = 102 °C) for 24 h: Pudleiner H.; Laatsch H. Liebigs Ann. Chem. 1990, 423–432. [Google Scholar]
- Prinsell M. R.; Everson D. A.; Weix D. J. Chem. Commun. 2010, 46, 5743–5745. [DOI] [PubMed] [Google Scholar]
- Huo S. Org. Lett. 2003, 5, 423–425. [DOI] [PubMed] [Google Scholar]
- For selected examples of electrochemical nickel-catalyzed synthesis of RZnX, see: ; a Mellah M.; Labbe E.; Nédélec J. Y.; Périchon J. New J. Chem. 2002, 26, 207–212. [Google Scholar]; b Mellah M.; Labbe E.; Nédélec J. Y.; Périchon J. New J. Chem. 2001, 25, 318–321. [Google Scholar]; c Gosmini C.; Nédélec J. Y.; Périchon J. Tetrahedron 1998, 54, 1289–1298. [Google Scholar]; d Gosmini C.; Nédélec J. Y.; Périchon J. Tetrahedron Lett. 1997, 38, 1941–1942. [Google Scholar]; e Sibille S.; Ratovelomanana V.; Nédélec J. Y.; Périchon J. Synlett 1993, 425–426. [Google Scholar]; f Sibille S.; Ratovelomanana V.; Périchon J. Chem. Commun. 1992, 283–284. [Google Scholar]; g Conan A.; Siblle S.; Périchon J. J. Org. Chem. 1991, 56, 2018–2024. [Google Scholar]; h McHarek S.; Siblle S.; Nédélec J. Y.; Périchon J. J. Organomet. Chem. 1991, 401, 211–215. [Google Scholar]
- Most of the reductive coupling reactions were complete within 24 h.
- a Kuroboshi M.; Waki Y.; Tanaka H. J. Org. Chem. 2003, 68, 3938–3942. [DOI] [PubMed] [Google Scholar]; b Wiberg N. Angew. Chem., Int. Ed. 1968, 7, 766–779. [Google Scholar]
- We introduced this test in our earlier reductive cross-coupling reaction; see ref (9). See also: Shrestha R.; Weix D. J. Org. Lett. 2011, 13, 2766–2769. [DOI] [PubMed] [Google Scholar]
- Ocampo R.; Dolbier W. R. Jr Tetrahedron 2004, 60, 9325–9374. [Google Scholar]
- The Hammett parameters used in Table 3 and Figure 5 are from anilinium pKa data: ; a Tseng K. C. Acta Chim. Sin. 1966, 32, 107–121. [Google Scholar]; These data have been used previously for the study of oxidative addition of aryl halides to rhodium:; b Puri M.; Gatard S.; Smith D. A.; Ozerov O. V. Organometallics 2011, 30, 2472–2482. [Google Scholar]; For a review on Hammett, parameters see:; c Hansch C.; Leo A.; Taft R. W. Chem. Rev. 1991, 91, 165–195. [Google Scholar]; d Hansch C.; Gao H. Chem. Rev. 1997, 97, 2995–3060. [DOI] [PubMed] [Google Scholar]
- ρ = 2.3 for Ar–Cl: ; a Biscoe M.; Fors B.; Buchwald S. J. Am. Chem. Soc. 2008, 130, 6686–6687. [DOI] [PMC free article] [PubMed] [Google Scholar]; ρ = 5.2 for Ar–Cl:; b Portnoy M.; Milstein D. Organometallics 1993, 12, 1665–1673. [Google Scholar]; ρ = 2.3 for Ar–I:; c Amatore C.; Pfluger F. Organometallics 1990, 9, 2276–2282. [Google Scholar]; ρ = 2 for Ar–I:; d Fauvarque J.-F.; Pflüger F.; Troupel M. J. Organomet. Chem. 1981, 208, 419–427. [Google Scholar]
- ρ = 8.8 for Ar–Cl with EWG: ; a Foa M.; Cassar L. Dalton Trans. 1975, 2572–2576. [Google Scholar]; ρ = 4.4 for Ar–Br:; b Tsou T. T.; Kochi J. K. J. Am. Chem. Soc. 1979, 101, 6319–6332. [Google Scholar]
- ρ = 2.7 for Ar–Br:; a Cassar L.; Foà M. J. Organomet. Chem. 1973, 51, 381–393. [Google Scholar]; b Saito S.; Oh-tani S.; Miyaura N. J. Org. Chem. 1997, 62, 8024–8030. [DOI] [PubMed] [Google Scholar]
- Wells P. R. Chem. Rev. 1963, 63, 171–219. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.