

Abstract
Disturbances in gene expression as a result of perturbed transcription or posttranscriptional regulation is one of the main causes of cellular dysfunction that underlies different disease states. About a decade ago, the discovery of microRNAs (miRNAs) in mammalian cells has renewed our focus on posttranscriptional regulatory mechanisms during pathogenesis. These tiny posttranscriptional regulators are differentially expressed in almost every disease that has been studied-to-date and can modulate a gene’s expression via specifically binding to its messenger RNA. Due to their capacity to simultaneously target multiple, functionally-related, genes, they are proving to be potentially powerful therapeutic agents/targets. In this review we will focus on the miRNAs that are differentially regulated in the more common cardiovascular pathologies, their targets, and potential function.
Keywords: microRNA, hypertrophy, failure, ischemia, angiogenesis
Introduction
Lin-4, the first miRNA to be discovered, is a 22 ribonucleotide long molecule that negatively regulates the translation of lin-14 and, thereby, the timing of the larva-to-adult switch of Caenorhabditis elegans (C. elegans) 1. It functions by binding to complementary sequences in the 3’ untranslated region (3’UTR) of lin-14 mRNA, via which it inhibits its translation without affecting its stability 2. A similar mechanism was later ascribed to let-7 and its regulation of a set of genes that included lin-14, lin-28, lin-41, lin-42, and daf-12 in C. elegans, where if over- or under-expressed, results in premature development or reiteration of cell lineages, respectively 3. These findings define 2 aspects of miRNA’s unique functionality; 1) precise regulation of the timing of a cellular event, 2) via synchronous posttranscriptional inhibition of a cadre of genes that are functionally interdependent, thus, operating as an efficient molecular switch.
MiRNAs are predominantly known to negatively regulate mRNA translation through assembling into a complex known as miRNA RNA-inducing silencing complex (RISC or miRISC). This complex includes the RNA-specific endonuclease Dicer, which is involved in the processing of pre-miRNA into the mature form 4; the argonaute (Ago) protein, onsisting of 4 isoforms, of which Ago2 or slicer, can cleave the target mRNA 5, 6; the P-body protein PW182 7; the human immunodeficiency virus transactivating response RNA-binding protein (TRBP), known to recruit Ago2 to RISC 8; and fragile X mental retardation protein (FMRP1) 9, which associates with polyribosomes 10; among others whose RISC-related functions have not been fully elucidated yet 11. This complex binds a target gene via partial sequence complementarity between the miRNA and, preferentially, a conserved site within the 3’UTR of the gene. Through this interaction they can inhibit translation by any of 3 different mechanisms: 1) inhibiting translation at a post-initiation step without reducing mRNA abundance, polyadenylation, or polyribsomal content 2; 2) inhibiting translation initiation via a cap- 12–14 and poly(A)-dependent dependent mechanism 12; 3) inducing deadenylation of mRNAs independently of inhibition of translation 15–17.
Perturbation in gene expression is one of the main underlying mechanisms that contribute to disease development. While transcriptional regulation of some genes has been recorded others are regulated by posttranscriptional means. The discovery of miRNAs has recently advanced our understanding regarding the latter mode of gene regulation in development and disease 18, as will be detailed below for cardiovascular diseases, in particular.
MiRNAs in cardiac hypertrophy and failure
Cardiac-enriched miR-1 and miR-133 in cardiac hypertrophy and failure
MiRNAs are differentially regulated during cardiac hypertrophy and failure in both rodents and humans 19–27 (Fig. 1). Reprogramming of gene expression in the heart to recapitulate the pattern of the fetal/neonatal heart is an established mechanism that contributes to the development of cardiac hypertrophy 28–30. Therefore, an analogous change in the expression pattern of miRNAs supports their role in the underlying pathogenesis 31. MiR-1 is one of the earliest miRNAs that are downregulated in the heart after the induction of pressure overload, which precedes the increase in cardiac mass, suggesting that it may potentially be involved in initiating the process 23. In accordance, miR-1 levels are also lower in genetic models of hypertrophy that include the calcineurin- 32 and AKT-overexpressing 33 transgenic mice hearts. Furthermore, miR-1 is reduced in the hypertrophied left ventricle of acromegalic patients 23 and in those who suffer from aortic stenosis with normal ejection fraction 20. Experiments in cultured myocytes 23 and those involving inhibition of miR-1 in vivo 33, suggest that a decrease in miR-1 is necessary for an increase in cell size and mass. Some of its targets that were validated in these studies include insulin-like growth factor-1 34, calmodulin, and myocyte enhancer factor 2A 32, all of which are known to contribute to the development of cardiac hypertrophy. In contrast, the fluctuation in miR-1 levels were less consistent in the failing heart. For example, as Matkovich et al 21 and Thum et al 26 recorded an increase in miR-1, Ikeda et al 20, Sucharov et al 24, and Naga Prasad et al 22 detected a reduction in miR-1, in ischemic and non-ischemic dilated cardiomyopathies that were associated with a significant reduction in percent ejection fraction. Therefore, it would appear that an increase in cardiac mass is accompanied by downregulation of miR-1, however, as hypertrophy progresses into dilatation and contractile dysfunction, miR-1 concentrations return to normal or above normal levels.
Figure 1. A diagram showing miRNAs and their targets in cardiac hypertrophy.
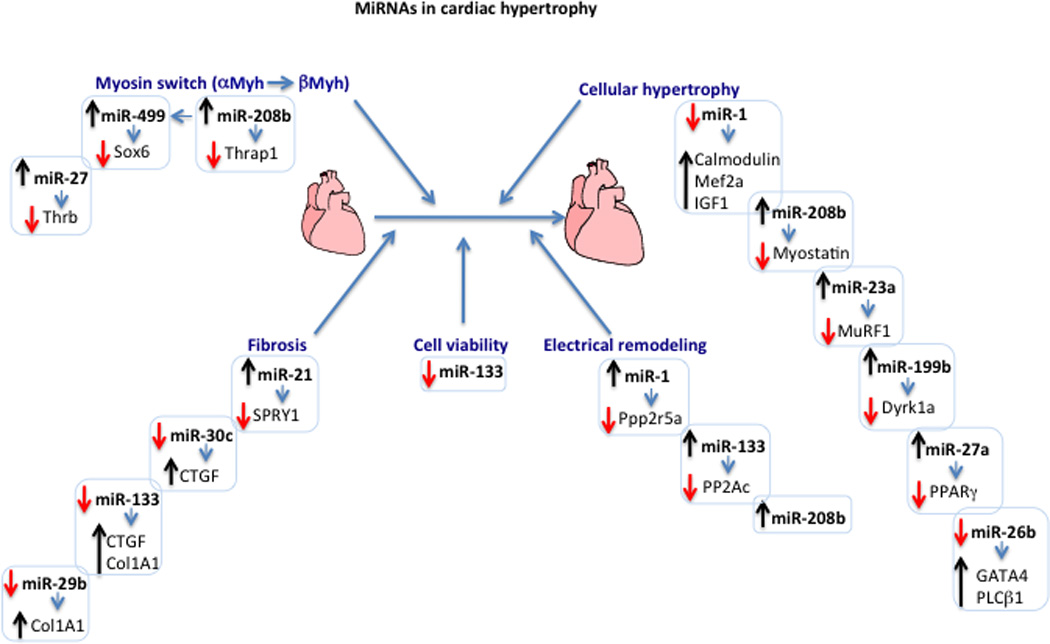
The diagram displays the different miRNAs and their targets that are involved in gene switching, cellular hypertrophy, fibrosis, and electrical remodeling during cardiac hypertrophy. Upregulation or downregulation of a specific miRNA is represented by an upward (black) or a downward (red) arrow, respectively. The change in the expression levels of target genes inversely correlates with that of the targeting miRNA and is similarly represented by an up or down arrow. All listed targets have been validated. The listed targets include: thyroid hormone receptor-associated protein 1 (Thrap1), SRY-box containing gene 6 (Sox6), thyroid hormone receptor beta (Thrb), calmodulin, myocyte enhancer factor 2A (Mef2a), insulin-like growth factor 1 (IGF1), myostatin, muscle-specific RING finger protein 1 (MuRF1), dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 1A (Dyrk1a), sprouty 2/4 (SPRY2/4), connective tissue growth factor (CTGF), collagen IA1 (Col1A1), GATA binding protein 4 (GATA4), phospholipase C beta 1 (PLCβ1), protein phosphatase 2, regulatory subunit B (B56) alpha isoform (Ppp2r5a), protein phosphatase 2 catalytic subunit (PP2Ac) and, the indirect targets alpha and beta myosin heavy chain (αand βMyh),
MiR-133, which is also a muscle-enriched miRNA that shares the same primary transcript as miR-1, is likewise downregulated during cardiac hypertrophy 19, 27, 33. Moreover, like miR-1, its knockdown via antagomirs was sufficient for inducing cardiac hypertrophy and the characteristic increase in some of the fetal genes 33. In contrast to these results, genomic knockout of miR-133a-1 or miR-133a-2, each of which contribute to ~50% reduction in miR-133 levels, which is equivalent to the decrease observed in cardiac hypertrophy, exhibited normal cardiac growth and function under quiescent or pressure overload conditions 35. This discrepancy in outcomes is not readily explainable. On the other hand, a transgenic mouse model overexpressing miR-133 that compensates for its downregulation during hypertrophy failed to inhibit cardiac enlargement, suggesting that downregulation of miR-133 in the heart is not required for the induction of cardiac hypertrophy 36.
The regulation of contractile fibers by myomiRs during cardiac hypertrophy and failure
While miR-1 and miR-133 are enriched in both skeletal and cardiac muscle, miR-208 is cardiac restricted 37. It has 2 isoforms, miR-208a and miR-208b, that are contained within the introns of Myh6 (α myosin heavy chain) and Myh7 (β myosin heavy chain) genes, respectively. Homozygous miR-208a mice (miR-208a−/−) are developmentally normal with the expected normal levels of α and β myosinheavychain (Myh) at birth, although the adult mice fail to upregulate βMyh in response to thyroid hormone inhibition 38. Likewise, pressure overload on the heart did not induce the expected increase in βMyh. In addition, there was absence of hypertrophic growth and fibrosis following an increase in workload, accompanied by reduced levels of miR-499 38. Interestingly, transgenic overexpression of miR-499 in the miR-208a−/− background restored the normal response to thyroid hormone inhibition, suggesting that it is a downstream effector of miR-208 and is sufficient for mediating its effect 39. MiR-499 was shown to target Sox6 39, a negative regulator of βMyh 40, and accordingly the transgene reduced its levels in the miR-208−/− mice. Paradoxically, though, ablation of miR-499 did not recapitulate the miR-208a−/− phenotype.
Similar to miR-208a−/−, miR-208b−/− mice has no overt developmental defects 39. Mirroring the expression pattern of their host genes, miR-208a is predicted to be the main source of mature miR-208 in the adult mouse heart, whereas miR-208b is mainly expressed during fetal and early postnatal cardiac development. Accordingly, the lack of a phenotype after targeted deletion of the latter, offers a more conclusive result regarding the role of miR-208 in cardiac development. Thus, we can conclude that miR-208 is not required for the expression of βMyh during cardiac development, but is necessary for the increase in its expression that occurs during cardiac hypertrophy and hypothyroidism. In contrast, in vivo delivery of antimiR-208 or antimiR-499 results in almost complete elimination of endogenous βMyh in the adult mouse, where it appeared uncharacteristically abundant in this study 41. In accordance, antimiR-208a increased the survival rate of hypertensive rats and reduced the isovolumic relaxation time, which could be attributable to the reduction in βMyh. In addition, myocyte hypertrophy and perivascular fibrosis were also reduced.
In contrast to the absence of any spontaneous defects associated with the miR-208a/b knockout mice, cardiac specific transgenic expression of miR-208a induced cardiac hypertrophy within 4 months of age 42. The phenotype, though, did not exhibit downregulation of αMyh, upregulation of atrial natriuretic factor, or changes in any of the hypertrophy-related miRNAs. It was, however, associated with a decrease in myostatin, a negative regulator of hypertrophy 43 and a validated target of miR-208a, which could potentially explain its anti-hypertrophic effect 42. Additionally, these mice exhibited defects in conduction in the form of first- or second-degree atrioventricular bundle block, compared to the miR-208a−/− mice, which suffered from atrial fibrillation. The latter could be attributed to a reduction in connexin 40 and its transcriptional regulator Hop, however, these proteins were unchanged in the miR-208a transgenic mice. Meanwhile, GATA4, a validated target of miR-208a and a positive regulator of connexin 40 (Cx40) 44, is more abundant in the miR-208a−/− mice, although this obviously does not reconcile with the observed downregulation of Cx40 in this model. Similar to the miR-208 transgenic, a cardiac-specific transgenic mouse model of miR-208’s downstream effector, miR-499, is sufficient for inducing cardiac hypertrophy 45. However, under these conditions, its validated target, Sox6, was not altered in the transgenic hearts.
MiRNAs that regulate cardiac fibrosis
One of the most highly and consistently increased miRNAs during cardiac hypertrophy is miR-21 19, 23, 25, 27. Its function in this context has been elusive, as cardiac-specific transgenic mice appeared structurally and functionally normal, and did not differ from wild type mice in its response to pressure overload 46, 47. While miR-21 increases in the whole heart during pathological hypertrophy, it was suggested that its increase in myofibroblasts is more pronounced than it is in the myocytes 47. In the former cell type, it was shown to directly target and suppress sprouty1, resulting in enhanced extracellular signal-regulated kinase 1/2 phosphorylation and myofibroblast survival, which, in turn, indirectly contributes to the increase in fibrosis during cardiac hypertrophy 47. This was confirmed by treating mice with a miR-21 antagomir, which resulted in the reduction of both fibrosis and cardiac hypertrophy. A recent report, though, disputes these results, as it shows that global genomic ablation of miR-21, or knockdown with LNA-modified antisense oligonucleotides, has no impact on the development of fulminant cardiac hypertrophy in response to pressure overload, angiotensin II, calcineurin, or infarction 48. It is plausible that an unidentified gene/mechanism compensated for the genomic ablation of miR-21. Thus, the jury is still out regarding the role of miR-21 during hypertrophy.
Although cardiac hypertrophy is not inhibited as a result of normalizing miR-133 levels; apoptosis and fibrosis were restored to normal levels 36. Connective tissue growth factor (CTGF) is one of the main factors that mediate fibrosis in the heart 49, 50. While one study did show that it is a direct target of miR-133, counter to prediction, the miR-133 transgenic mouse exhibits an increase in CTGF mRNA in the heart 51. Alternatively, miR-133 directly targets collagen 1A1 and is downregulated during angiotensin II-induced myocardial fibrosis, along with miR-29b 52. MiR-29b was also shown to target multiple collagen isoforms and its inhibition in vivo was sufficient for inducing fibrosis in the heart 53. On the other hand, CTFG was also identified as a target of the miR-17~92 cluster 54, as miR-18 and -19 inversely correlate with the expression of CTGF, thrombospondin, and fibrosis in the, aging, failing heart in mice and human subjects 55. However, there is no data that show that normalizing the levels of miR-29, miR-18, or miR-19 in the heart during hypertrophy or failure would inhibit the development of fibrosis.
MiRNAs that influence cardiac size
MiR-23a is clustered with miR-27a and miR-24, and increases during pressure overload- 19, 23, 25, 27 and isoproterenol-induced cardiac hypertrophy 56. It was found to be directly regulated via nuclear factor of activated T-cells, cytoplasmic, calcineurin-dependent 1 NFATc3 and is both required and sufficient for mediating the effects of the calcineurin-3 (NFATc3) pathway during cardiac hypertrophy 56. Its effects are partly mediated via the direct targeting and suppression of the negative regulator, muscle ring finger 1 (MuRF1). In accordance, inhibiting miR-23a with antagomirs results in the upregulation of MuRF1 in the heart, and virtually complete inhibition of isoproterenol-induced cardiac hypertrophy and the accompanied changes in gene expression and cardiac functions. This reconciles with a MuRF1-dependent function that was revealed in the MuRF1 knockout mice, as they developed an exaggerated form of pressure-induced hypertrophy 57. The other member of the cluster, miR-27a, is also upregulated during cardiac hypertrophy, and, likewise, was found both necessary and sufficient for the induction of cardiac hypertrophy and contractile dysfunction in a mouse model 58. In this context it targets peroxisome proliferator-activated receptor gamma (PPARγ), a well-known regulator of fatty acid and glucose metabolism 59. The finding is consistent with the role of PPARγ in inhibiting cardiac hypertrophy 60. It also has been shown to positively regulate βMyh through targeting the thyroid hormone receptor β1 in cardiac myocytes 61. However, there is no evidence yet as to whether this contributes to the upregulation of βMyh and contractile dysfunction during cardiac hypertrophy in vivo.
More recently, da Costa Martins et al demonstrated that mir-199b, which is upregulated during cardiac hypertrophy, is also a direct transcriptional target of NFATc 62. In this case, miR-199b targets and suppresses the NFATc kinase dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 1a (Dyrk1a), which is a negative regulator of NFAT. Accordingly, mutant mice overexpressing miR-199b in the heart, or those hemizygous for Dyrk1a, exhibited exaggerated hypertrophy in response to pressure overload. Conversely, antisense ‘antagomir’ treatment normalized miR-199a levels, and not only prevented hypertrophy and failure, but also almost completely reversed them.
The transcription factor GATA4 plays an essential role in the development of cardiac hypertrophy 63, 64 as it regulates the expression of some of the fetal genes that are re-expressed in this condition 65. In accordance, its DNA-binding activity and abundance increase during hypertrophy 66, 67. Recent data utilizing state-of-the-art RNA polymerase II immunoprecipitation followed by deep sequencing established that the increase in GATA4 expression in cardiac myocytes is not due to an increase in the transcription of its gene 68. This prompted the search for miRNAs, which were downregulated during cardiac hypertrophy that targeted GATA4 3’UTR. MiR-26a/b, which is highly expressed in the heart and other organs, inversely correlated with the expression of GATA4 levels in the developing and hypertrophied hearts. It was proven to directly target GATA4 and, subsequently, reduce cardiac growth. However, although knockdown of miR-26 was sufficient for inducing GATA4 in the heart in vivo, it was insufficient for inducing cardiac hypertrophy. Another validated target of miR-26b is phospholipase C beta1, which proved to be a major negative regulator of miR-26b, creating a double negative feedback loop characteristic of many miRNAs and their targets 68. This is the first proven example of a gene that is primarily regulated by a posttranscriptional miRNA-dependent mechanism during disease, underscoring the functional relevance of miRNAs.
MiRNAs in ischemic heart disease
MiRNAs that regulate apoptosis during ischemia
Myocardial ischemia results from insufficient blood flow to the myocardium, which leads to apoptotic, autophagic, and necrotic cell death, with subsequent inflammatory cell infiltration, fibrosis and contractile dysfunction 69. MiRNAs’ expression is rapidly perturbed during myocardial ischemia or cellular hypoxia (Fig. 2). MiR-1 and miR-133, which are reduced during cardiac hypertrophy, are upregulated when myocytes are subjected to hypoxia. Under these conditions, miR-1 was found to target heat shock proteins (HSP) HSP60 and HSP70, while miR-133 targeted caspase-9 70. In accordance, miR-1 has a proapoptotic effect, whereas miR-133 is antiapoptotic. In contrast, miR-320 is downregulated during ischemia/reperfusion (I/R) and is responsible for upregulation of its target HSP20 71. Further suppression of this miRNA with an antagomir successfully reduces infarct size after I/R.
Figure 2. A diagram showing miRNAs and their targets in cardiac ischemia.
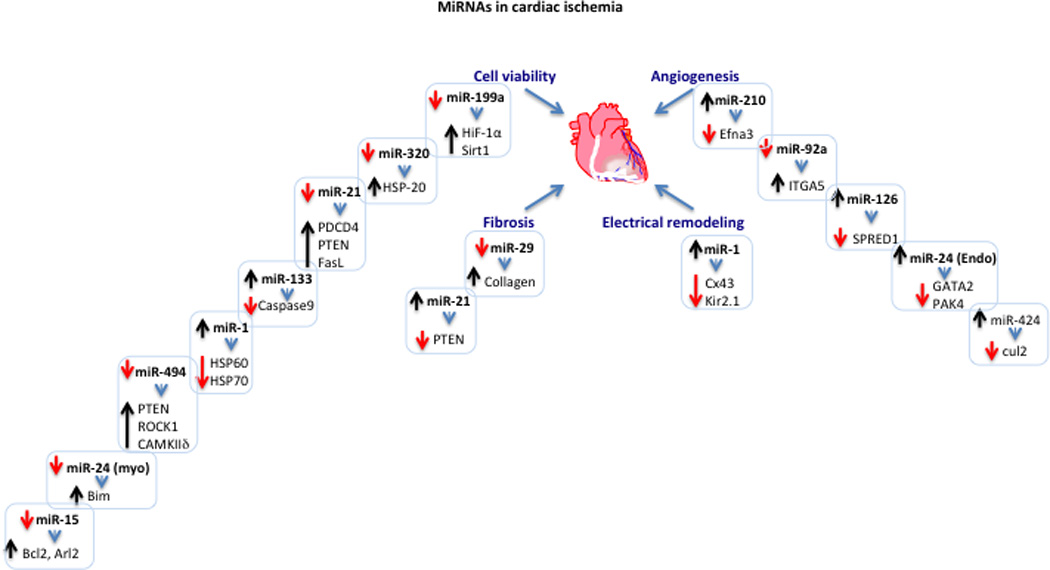
The diagram displays the different miRNAs and their targets that are involved in cell viability, angiogenesis, fibrosis, and electrical remodeling during cardiac ischemia. Upregulation or downregulation of a specific miRNA is represented by an upward (black) or a downward (red) arrow, respectively. The change in the expression levels of target genes inversely correlates with that of the targeting miRNA and is similarly represented by an up or down arrow. All listed targets have been validated. The listed targets include: hypoxia-inducible factor 1 alpha (Hif-1α), sirtuin 1 (Sirt1), heat shock protein 20, 60, and 70 (HSP20, HSP60, HSP70), programmed cell death 4 (PDCD4), phosphatase and tensin homolog (PTEN), fas ligand (FasL), caspase 9, Rho-associated coiled-coil containing protein kinase 1 (Rock1), calmodulin kinase II delta (CAMKIIδ), BCL2-like 11 (Bim), ADP-ribosylation factor-like 2 (Arl2), B-cell CLL/lymphoma 2 (Bcl2), ephrin A3 (Ephna3), integrin, alpha 5 (ITGA5), sprouty-related, EVH1 domain containing 1 (SPRED1), GATA binding protein 2 (GATA2), p21 protein (Cdc42/Rac)-activated kinase 4 (PAK4), cullin 2 (cul2), connexin 43 (Cx43), inward rectifier potassium channel 2 (Kir2.1).
Notably, miR-21, which is ubiquitously expressed, has emerged as a major inhibitor of cell apoptosis in myocytes as well as a plethora of other cell types. It is acutely downregulated during myocardial ischemia, specifically within the ischemia zone, where replenishing it reduces the infarct size and retards the development of heart failure 46, 72. These results were corroborated by experiments conducted in isolated cardiac myocytes, in which miR-21 was proven to inhibit hypoxia-induced apoptosis by targeting Fas ligand (FasL) 46 and programmed cell death 4 (PDCD4) 73. Furthermore, miR-21 is induced by the AKT pathway and partly mediates its antiapoptotic effect via suppression of FasL 46. Conversely, miR-21 activates AKT through suppression of phosphatase and tensin homolog (PTEN) via a double negative feedback loop 46. In addition to myocytes, miR-21 is enriched in myofibroblasts and infiltrating cells within the infarct zone. In these cells it was shown to inhibit PTEN and induce upregulation of metalloprotease-2 74, thereby, promoting fibrosis. As we discussed earlier, miR-21also has a profibrotic effect during cardiac hypertrophy, via promoting the survival of myofibroblast 47, however, this particular mechanism was not examined in the ischemia heart.
Similar to miR-21, miR-494 is also reduced in the infarct zone, where normalizing its levels by transgenic overexpression reduces infarct size and improves contractility 75. Intriguingly, miR-494 targets both proapoptotic genes that include PTEN, Rho-associated coiled-coil containing protein kinase 1, and calcium/calmodulin-dependent protein kinase II delta, as well as, antiapoptotic genes, such as fibroblast growth factor receptor 2 and leukemia inhibitory factor. However, as mentioned, the net result is a cardioprotective effect that is associated with enhanced AKT phosphorylation, possibly due to inhibition of PTEN. Likewise, miR-24 is downregulated in the peri-infarct zone, where replenishing it reduces myocyte apoptosis, infarct size, and cardiac dysfunction after myocardial ischemia by targeting Bim 76. Additionally, miR-24, along with miR-21 and miR-221, inhibit Bim in cardiac progenitor cells and improve their survival and contribution to functional recovery after coronary artery ligation 77.
Other miRNAs are upregulated within the infarct zone. One example includes miR-15b, which is upregulated in the infarct zone of a porcine model after ischemia (60 min) / reperfusion (24 h) 78. When neutralized in cardiac myocytes it enhances their viability during hypoxia in association with an increase of 2 of its validated targets, ADP-ribosylation factor-like 2 79 and Bcl2 80. In vivo, suppressing miR-15b with locked nucleic acid-modified “tiny” antimiRs resulted in reducing the infarct size after ischemia (75 min) / reperfusion (24 h) and improved % ejection fraction and left ventricular end diastolic volume measured 2 weeks after I/R 78. The mechanism underlying this protection is presumed to be improved myocyte survival possibly due to an increase in the expression of suppressors of cell death.
The response of a cell to hypoxia or ischemia is biphasic; initially ischemia induces an adaptive response, which then transitions into a damaging effect upon the persistence of ischemia. Accordingly, exposing cells to brief periods of hypoxia or ischemia elicits a protective response, which involves activation of the AKT pathway 81. This process is known as ischemia preconditioning (IPC) and has been proven to protect organs against subsequent damaging periods of ischemia 82. MiRNAs are involved in early preconditioning, which involves posttranscriptional vs. transcriptional events 83, including the upregulation of hypoxia-inducible factor 1 alpha (Hif-1α)84, 85. In specific, miR-199a-5p is downregulated within minutes of applying IPC to the porcine or mouse hearts or hypoxia preconditioning (HPC) to cardiac myocytes. At least in myocytes, this reduction proved to be both required and sufficient for upregulation of its target Hif-1α, to the same extent as that observed after HPC86. Sirt1 was also validated as a direct conserved target of miR-199a-5p, which is required for downregulation of prolyl hydroxylase 2 and, thus, stabilization of Hif-1α86. These effects are mediated via an a AKT-dependent pathway, which is sufficient for inducing downregulation of miR-199a-5p and upregulation of Hif-1α and Sirt1 87. This is a unique example of a miRNA, which functions as an immediate early switch for the translation of a gene required for immediate adaptation to an insult such as hypoxia. In addition, AKT induces upregulation of miR-21 46, which is upregulated during IPC of the heart 88, 89. It targets and inhibits proapoptotic genes that include PTEN, FasL, and PDCD4, thus, assuming a powerful antiapoptotic function during ischemia. The results implicate miRNAs in mediating the prosurvival effects of the AKT pathway through both increasing prosurvival proteins like Hif-1a and Sirt1 and suppressing proapoptotic genes such as PTEN, FasL, and PDCD4.
MiRNAs that regulate angiogenesis during ischemia
In addition to directly regulating cell viability during myocardial ischemia, miRNAs could also indirectly influence cell survival and the size of infarct zone or scar formation via regulating angiogenesis. MiR-126 is one that is highly expressed in murine lung and heart and to a lesser extent in the brain, liver, and kidney, where it is localized to endothelial 90 and epithelial cells 91. Deletion of both its alleles results in ~50% embryonic lethality due to systemic edema and widespread hemorrhage from ruptured vessels 92, 93. Those embryos that escaped the lethal phenotype developed normally with no apparent abnormalities until they were challenged with ischemia, which elicits reparative angiogenesis. In particular, when the left coronary artery was occluded, angiogenesis was compromised in the ischemic hearts, which resulted in precipitous cardiac failure 93. Similar results were observed when miR-126 antagomir was injected in mice 94. One of miR-126’s relevant and validated targets include Spred-1, for which gain- and loss-of-function experiments in an aortic ring sprouting assay proved that it is sufficient for inhibiting vascular outgrowths 93. In an opposing function, however, miR-126 was found to target stromal cell-derived factor-1 95. This study shows that silencing miR-126 during ischemia results in an increase in this protein, which mobilizes Sca-1(+)/Lin(−) progenitor cells and enhances angiogenesis in a hind limb ischemia model. It is possible that the extent of silencing of miR-126 in different studies determines which of its functions predominates.
In contrast to miR-126, forced expression of miR-92a inhibits tube formation, cell migration, and adhesion of endothelial cells 96. Conversely, inhibition of miR-92a with antagomirs enhances angiogenesis during hind limb or myocardial ischemia in mice, and improves blood flow and tissue viability in the region. MiR-92a targets alpha5 integrin, which plausibly plays a role in mediating its function, although, most likely other targets are also involved. MiR-92 is a member of the miR-17~92 cluster of miRNA. Recently, it was shown that other members of this cluster, including miR-17, miR-18a, and miR-20, also exhibited an antiangiogenic effect both in vitro and in vivo, where Janus kinase 1 was identified as a miR-17 target 97. In contrast, another study reported that myc induces upregulation of miR-17~92 and that overexpression of the cluster with Ras enhanced tumor growth by increasing neovascularization 54. In this case, it was suggested that the targeting of thrombospondin by miR-19 mediates the cluster’s effects. This does agree with the fact that c-myc induces angiogenesis by posttranscriptionally inhibiting the expression of thrombospondin-1 98–100, and provides an explanatory mechanism for it. The discrepancy between may reflect differences in the models that were investigated.
Whereas miR-24 is downregulated in the peri-infarct zone and contributes to myocyte apoptosis and cardiac dysfunction during myocardial ischemia through an increase in its target Bim 76, another study reports that miR-24 is upregulated in endothelial cells, in specific, after cardiac ischemia, where it induces apoptosis of these cells 101. Accordingly, blocking its upregulation by an antagomir, rescues endothelial cells from apoptosis, increases angiogenesis, and limits both infarct size and cardiac dysfunction. In this context, it was shown to target GATA2 and p21-activated kinase 4. Thus, it would appear that miR-24 has different targets and opposite effects in cardiac myocytes vs. endothelial cells. It should be noted here that the dose of antagomir used in vivo was relatively low and appeared to be preferentially taken up by the endothelial cells, thus, purportedly sparing the myocytes from any apoptotic effect. What should also be taken into account, especially if used for therapeutic targeting, is that miR-24 also targets histone 2Ax in differentiating hematopoietic cells and renders them more susceptible to DNA damaging agents 102. Thus, silencing miR-24 will have an beneficial effect on these cells as well.
Mir-424 is also upregulated during hypoxia in endothelial cells, in particular, and induces upregulation of Hif-1α via targetng cullin 2 103. The latter is one of the components of the von Hipple-Lindau-elongin B/C-Nedd8 complex that is responsible for the degradation of Hif-1α104. Thus, miR-424 overexpression induces migration, proliferation, and tubulogenesis of human umbilical vein endothelial cells (HUVEC). In vivo, miR-424-overexpressing HUVEC were preferentially integrated into the neovasculature of an ovarian carcinoma, however, it did not increase vascular formation. In the mouse heart, miR-322, the mouse homolog of miR-424, is upregulated after 1 wk of coronary artery ligation in the periinfarct and remote areas, although its role in promoting angiogenesis under these conditions has not been investigated yet.
Hif-1α is an established angiogenic factor that functions through directly enhancing the transcription of vascular endothelial growth factor. In was also found to induce the expression of miR-210 during hypoxia 105, 106. This miRNA was found to be sufficient for inducing tubulogenesis of endothelial cells 106. Moreover, recent findings show that supplementing the myocardium with miR-210 increases angiogenesis in the periinfarct zone, reduces apoptosis and infarct size, and results in improved cardiac function following left anterior descending coronary artery ligation 107. Two of its targets include ephrin-A3 and protein tyrosine phosphatase, non-receptor type 1, which are involved in inhibition of angiogenesis and apoptosis, respectively.
In addition to the intracellular function of miRNAs, it was recently noted that some have a paracrine quality. In specific, the saphenous vein-derived pericyte progenitor cells mediate their angiogenic effect via secretion of miR-132 108. This was confirmed when silencing of miR-132 by an antimiR reduced the ability of these progenitor cells to induce angiogenesis and reduce infarct size after coronary artery occlusion. This function is potentially mediated by repression of RasGAP and subsequently activation of Ras. Furthermore, by targeting methyl CpG binding protein 2, miR-132 may also inhibits fibrosis.
MiRNAs that regulate arrhythomogenesis
Abnormalities in electrical conduction and/or repolarization is an underlying cause of arrhythmias manifest during coronary/ischemic heart disease 109. MiRNAs not only regulate cell viability during ischemia, but have also been found to regulate the expression of molecules involved in mounting an action potential and in conductivity. MiR-1, for example, is upregulated during coronary artery disease in humans and ischemic rat hearts, which are accompanied by an increase in the incidence of arrhythmias 110. As proof of miR-1’s role in arrhythmogenesis, overexpressing or inhibiting miR-1 in the ischemic rat heart resulted in an equivalent effect on the frequency of arrhythmias. MiR-1’s validated targets include the gap junction protein Cx43 and Kir2.1 subunit of the K+ channel that mediates IK1, both of which could potentially be mediating its effects. Indeed, deletion or missense mutation of Cx43 111 or IK1 112, respectively, were shown to increase the incidence of arrhythmias. Moreover, their ablation resulted in the reversal of miR-1’s knockdown anti-arrhythmogenic effect 110. On the other hand, the downregulation of miR-133 during cardiac hypertrophy is responsible for a reduction in the Ito,f accessory subunit, Kcnip2, through an indirect effect, which could be a cause of prolonged QT-interval 36.
An increase in miR-1 in adult myocytes has been shown to regulate inward calcium current (ICa) and calcium release from the sarcoplasmic reticulum (SR) 113. Overexpression of miR-1 increases ICa, however, this does not elicit an increase in maximum SR Ca2+ release, as it possibly counteracted by a 25% lower SR Ca2+ content. Increased spark frequency and SR Ca2+ leak in the miR-1-overexpressing myocytes might be a contributing factor to the lower SR Ca2+. These effects of miR-1 could be attributed to its targeting and suppression of the protein phosphatase 2A (PP2A) regulatory subunit B56α and consequently an increase in the phosphorylation of the ryanodine (RyR) and dihydropyridine receptors. Indeed, phosphorylation of RyR2 by CAMKII has been linked to SR diastolic Ca2+ leakiness and lower SR Ca2+ content 114 and to ventricular tachycardiac 115. In concordance, overexpression of miR-1 dramatically increased the frequency of extrasystolic Ca2+ transients and afterdepolarizations in isoproterenol-stimulated, paced, rat adult myocytes 113, which was reversed by a CAMKII inhibitor. MiR-133, which is co-expressed with miR-1, collaborates in this function by targeting both the alpha and beta subunits of the catalytic subunit of PP2A, and has a similar capacity to increase arrhythmogenesis in isolated rat myocytes 116. This mechanism has not been validated in vivo, however, supportive evidence for that includes an increase in miR-1 and miR-133 and a decrease in PP2A catalytic and regulatory subunits in a canine heart failure model 116. These results would suggest that higher levels of both miR-1 and miR-133 in the failing or ischemic heart contribute to arrhythmogenesis via perturbation of the expression of genes involved in conduction, depolarization, and calcium handling.
On the other hand, miR-328, but not miR-1 or miR-133, was reported to be upregulated in a canine model of atrial fibrillation (AF) and in human AF patients with rheumatic heart disease 117. Intramyocardial injection of miR-328 into the right atrium free wall increased susceptibility to AF. Similarly, transgenic overexpression of the miRNA in the mouse heart induced spontaneous AF 28 d after birth, and vice versa, knockdown of miR-328 reduced the incidence of pacing-induced AF. In this case miR-328 targeted and suppressed both the α1c and β1 subunits of the L-type calcium channel, which accordingly explained the dramatic reduction in the L-type Ca2+ current observed in the transgenic model. Thus, it would appear that the miRNA-regulated mechanisms underlying arrhythmogenesis in the failing or ischemic heart differ from those involved with AF.
MiRNAs in vascular disease and remodeling
MiRNAs in atherosclerogenesis
Perturbation of cholesterol homeostasis is a main underlying cause of atherosclerosis. One of the regulatory factors in cholesterol metabolism include sterol regulatory element-binding protein (SREBP) transcription factors, which regulate the expression of cholesterogenic genes such as low-density lipoprotein receptor and 3-hydroxy-3-methylglutaryl coenzyme A reductase 118. The human SREBP-1 and -2 genes harbor miR-33b and miR-33a, respectively, which appear to be co-expressed with their host genes 119. Interestingly, miR-33 was found to target adenosine triphosphate–binding cassette A1 (ABCA1) cholesterol transporter 119, which mediates intracellular cholesterol efflux from cells to form plasma high-density lipoprotein that is protective against atherosclerosis 120. Thus, as expected, when antisense miR-33 is injected in western diet-fed mice, serum high-density lipoprotein (HDL) is significantly elevated. The clinical relevance of this was tested in low-density lipoprotein receptor-deficient mice with atherosclerosis. Treatment of these mice with antimiR-33 for 4 weeks resulted in upregulation of ABCA1 in the liver and macrophages, an increase in HDL levels, and ultimately reduced plaques size and inflammatory gene expression 121. Similar results were observed in African green monkeys, in which antimiR-33 resulted in elevated HDL, in addition to reduced levels of very-low-density lipoprotein-associated triglycerides 122.
Other miRNAs such as miR-145/143 cluster, miR-133, and miR-221, have a more direct role in atherogenesis. MiR-145/miR-143 cluster is enriched in visceral and vascular smooth muscle cells (VSMC) as early as E11.5 123 and throughout adulthood 123–125. Genomic deletion of this cluster resulted in viable mice with a mild vascular phenotype but no cardiac abnormalities 123–125. In particular, the smooth muscle cells were smaller and exhibited an increase in rough endoplasmic reticulum and a decrease in actin stress fibers, with a resultant thinner tunica media. In addition, there were small neointimal lesions and a megacolon phenotype, which suggested that knockout of miR-145/143 may inhibit differentiation and, subsequently, enhance proliferation and migration of smooth muscle cells 125. However, the failure to detect a consistent increase in VSMC proliferation or apoptosis, argued that the miRNA may not be directly required for differentiation but rather for the ability of VSMC to differentiate in response to the physiological contractile demand 124.
MiR-145 is downregulated in response to vascular injury 126. While the knockout models dispute the fact that downregulation of miR-145 is sufficient for reducing α-actin and myosin heavy chain 123, 124, they do show that abrogation of miR-145 induces limited neointimal lesion formation 124, 125. In particular, one study has shown that these lesions are only observed in the femoral artery of older knockout mice (18 mo), which suggested that although downregulation of miR-145/143 does not result in fulminant neointimal formation, it does promote it 124. In consensus, restoring miR-145 levels during vascular injury inhibits neointimal formation 123, 125, with promising therapeutic prospects. MiR-145’s effect in this context could be, at least partially, explained by the suppression of its target, Krupple-lik factor 5 (Klf5). Indeed, KLF5, first identified as a transcription factor that induces embryonic smooth muscle myosin heavy chain 127, is involved in enhancing smooth muscle proliferation by upregulating cyclin D1 128.
MiR-133 is mainly known to be enriched in the heart and skeletal muscle, but a recent report shows that it is also expressed in smooth muscle cells, albeit in relatively low levels 129. After balloon injury, miR-133 is downregulated in the tunica media and becomes undetectable in the neointimal layer. Overexpression of miR-133 in the carotid artery halts VSMC proliferation and prevents neointimial hyperplasia. The levels of miR-133 in this tissue inversely correlate with its targets Sp1 and moesin. The prediction is that by inhibiting Sp1, Klf4 is deactivated, which in-turn, prevents downregulation of Myh11, and VSMC phenotype switching and proliferation.
In contrast to miR-145 and miR-133, miR-221 130, 131, but not the co-clustered miR-222 130, positively regulates smooth muscle proliferation. It is induced by platelet-derived growth factor (PDGF), which is known to stimulate VSMC switching and proliferation during angiogenesis and neointimal formation 130, 131. Gain- and loss-of-function experiments proved that it is indeed required for mediating the effects of PDGF via targeting and suppressing the cell cycle inhibitors p27Kip1 130, 131 and p57Kip2 131, as well as, c-kit 130. In concordance, knockdown of miR-221 during vascular injury reduced neointimal thickness by 40% 131. The effects of miR-221 are likely to be reinforced by the concurrent upregulation of miR-21 in neointimal lesions 132. MiR-21, which is a ubiquitously expressed prosurvival miRNA, was shown to inhibit PTEN expression under these conditions 132–134. Accordingly, its abrogation increased apoptotic VSMC death and reduced neoitimal thickness. Thus, neointimal formation is the result of a combinatorial effect of changes in the expression of several miRNAs and their targets that regulate VSMC differentiation, proliferation, and survival.
MiRNAs in aneurysm formation
Patients with thoracic aortic aneurysm have reduced levels of miR-1, miR-133, miR-21, and miR-29a, all of which significantly inversely correlated with the aortic diameter 135. It was confirmed that miR-29a targeted metalloproteinase 2 (MMP2) but not MMP9, which showed a positive correlation with aneurysm size in these patients. In contrast, Boon et al found no change in miR-29a, but an increase in miR-29b in thoracic aortic aneurysm specimens obtained from a similar patient demographic (30 patients undergoing tricuspid valve replacement vs. control samples from patients undergoing coronary artery bypass) 136. The one difference, though, was that the former study’s control group included some samples from the aorta of heart donors, which may account for the difference, considering that coronary artery disease in the control group might have been associated with changes in miRNA expression in the aorta. In rodents, miR-29b was also upregulated in the dilated aorta of angiotensin II-treated aged mice and in Fibulin-4 knockdown mice 136. Treatment of the former mice with antimiR-29 induced an increase in miR-29’s targets elastic collagen 1A1 and 3A1 and reversed the phenotype. In this model MMP9 was decreased but the levels of MMP2 were undetermined. Marfan’s syndrome, which is characterized by the development of aortic aneurysms due to a defect in connective tissue, is also associated with an increase in miR-29b and a decrease in elastin in the aorta of the Fbn1C1039G/+ mouse model 137. In contrast, though, MMP2, which is also a miR-29 target, was increased, suggesting that miR-29b may not be the main regulator of its expression under these conditions. Similar to the angiotensin II-treated aged model, prenatal treatment of these mice with antimiR-29 completely inhibited aortic dilation at 4 wks of age. Overall, miR-29 emerges as a major regulator of extracellular matrix protein expression with potential therapeutic utility.
Conclusion
The discovery of miRNAs has introduced a new dimension to our understanding of gene regulation in cardiac disease. They are characterized by targeting multiple, functionally related, versus single, genes, which renders them a potentially powerful therapeutic target/tool. However, to fully exploit these molecules in diagnostics and therapeutics, more studies are necessary. Specifically, we need to discover the full spectrum of genes that a single miRNA may target in a given context, how these targets functionally interact, and the extent to which each contributes to the miRNA’s observed functionality. In addition, since a single mRNA could be simultaneously regulated by multiple miRNAs, the collaborative effects of these miRNAs, in a given function, need to also be explored. These details will further explain the functional significance of miRNAs and aid in utilizing them for therapeutic purposes.
Acknowledgements
Thanks to all past and present lab members for their hard work and contribution to microRNA research.
Funding Source
National Institute of Health Grants 2R01 HL057970 and R01 HL081381.
Non-standard Abbreviations and Acronyms
- 3’UTR
3-prime untranslated region
- ABCA1
adenosine triphosphate-binding cassette A1
- AF
atrial fibrillation
- CTGF
connective tissue growth factor
- Cx
connexin
- Dyrk1a
dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 1a
- FasL
Fas ligand
- HDL
high-density lipoprotein
- Hif-1α
hypoxia-inducible factor 1 alpha
- HPC
hypoxia preconditioning
- HUVEC
human umbilical vein endothelial cells
- I/R
ischemia/reperfusion
- IPC
ischemia preconditioning
- Klf
Krupple-like factor
- miRNA
microrRNA
- MuRF1
muscle ring finger 1
- Myh
myosin heavy chain
- NFATc
nuclear factor of activated T-cells, cytoplasmic, calcineurin-dependent
- PDCD4
programmed cell death 4
- PDGF
platelet-derived growth factor
- PP2A
protein phosphatase 2A
- PPARγ
peroxisome proliferator-activated receptor gamma
- PTEN
phosphatase and tensin homolog
- RyR
ryanodine receptor
- SR
sarcoplasmic reticulum
- SREBP
sterol regulatory element-binding protein
- VSMC
vascular smooth muscle cells
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
None
References
- 1.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- 2.Olsen PH, Ambros V. The lin-4 regulatory RNA controls developmental timing in Caenorhabditis elegans by blocking Lin-14 protein synthesis after the initiation of translation. Dev Biol. 1999;216:671–680. doi: 10.1006/dbio.1999.9523. [DOI] [PubMed] [Google Scholar]
- 3.Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR, Ruvkun G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403:901–906. doi: 10.1038/35002607. [DOI] [PubMed] [Google Scholar]
- 4.Hutvagner G, Zamore PD. A microRNA in a multiple-turnover RNAi enzyme complex. Science. 2002;297:2056–2060. doi: 10.1126/science.1073827. [DOI] [PubMed] [Google Scholar]
- 5.O'Carroll D, Mecklenbrauker I, Das PP, Santana A, Koenig U, Enright AJ, Miska EA, Tarakhovsky A. A Slicer-independent role for Argonaute 2 in hematopoiesis and the microRNA pathway. Genes Dev. 2007;21:1999–2004. doi: 10.1101/gad.1565607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meister G, Landthaler M, Patkaniowska A, Dorsett Y, Teng G, Tuschl T. Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol Cell. 2004;15:185–197. doi: 10.1016/j.molcel.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 7.Liu J, Rivas FV, Wohlschlegel J, Yates JR, 3rd, Parker R, Hannon GJ. A role for the P-body component GW182 in microRNA function. Nat Cell Biol. 2005;7:1261–1266. doi: 10.1038/ncb1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chendrimada TP, Gregory RI, Kumaraswamy E, Norman J, Cooch N, Nishikura K, Shiekhattar R. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature. 2005;436:740–744. doi: 10.1038/nature03868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jin P, Zarnescu DC, Ceman S, Nakamoto M, Mowrey J, Jongens TA, Nelson DL, Moses K, Warren ST. Biochemical and genetic interaction between the fragile X mental retardation protein and the microRNA pathway. Nat Neurosci. 2004;7:113–117. doi: 10.1038/nn1174. [DOI] [PubMed] [Google Scholar]
- 10.Feng Y, Gutekunst CA, Eberhart DE, Yi H, Warren ST, Hersch SM. Fragile X mental retardation protein: nucleocytoplasmic shuttling and association with somatodendritic ribosomes. J Neurosci. 1997;17:1539–1547. doi: 10.1523/JNEUROSCI.17-05-01539.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hock J, Weinmann L, Ender C, Rudel S, Kremmer E, Raabe M, Urlaub H, Meister G. Proteomic and functional analysis of Argonaute-containing mRNA-protein complexes in human cells. EMBO Rep. 2007;8:1052–1060. doi: 10.1038/sj.embor.7401088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Humphreys DT, Westman BJ, Martin DI, Preiss T. MicroRNAs control translation initiation by inhibiting eukaryotic initiation factor 4E/cap and poly(A) tail function. Proc Natl Acad Sci U S A. 2005;102:16961–16966. doi: 10.1073/pnas.0506482102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pillai RS, Bhattacharyya SN, Artus CG, Zoller T, Cougot N, Basyuk E, Bertrand E, Filipowicz W. Inhibition of translational initiation by Let-7 MicroRNA in human cells. Science. 2005;309:1573–1576. doi: 10.1126/science.1115079. [DOI] [PubMed] [Google Scholar]
- 14.Thermann R, Hentze MW. Drosophila miR2 induces pseudo-polysomes and inhibits translation initiation. Nature. 2007;447:875–878. doi: 10.1038/nature05878. [DOI] [PubMed] [Google Scholar]
- 15.Wu L, Fan J, Belasco JG. MicroRNAs direct rapid deadenylation of mRNA. Proc Natl Acad Sci U S A. 2006;103:4034–4039. doi: 10.1073/pnas.0510928103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Behm-Ansmant I, Rehwinkel J, Doerks T, Stark A, Bork P, Izaurralde E. mRNA degradation by miRNAs and GW182 requires both CCR4:NOT deadenylase and DCP1:DCP2 decapping complexes. Genes Dev. 2006;20:1885–1898. doi: 10.1101/gad.1424106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wakiyama M, Takimoto K, Ohara O, Yokoyama S. Let-7 microRNA-mediated mRNA deadenylation and translational repression in a mammalian cell-free system. Genes Dev. 2007;21:1857–1862. doi: 10.1101/gad.1566707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sayed D, Abdellatif M. MicroRNAs in Development and Disease. Physiol Rev. 2011;91:827–887. doi: 10.1152/physrev.00006.2010. [DOI] [PubMed] [Google Scholar]
- 19.Cheng Y, Ji R, Yue J, Yang J, Liu X, Chen H, Dean DB, Zhang C. MicroRNAs are aberrantly expressed in hypertrophic heart: do they play a role in cardiac hypertrophy? Am J Pathol. 2007;170:1831–1840. doi: 10.2353/ajpath.2007.061170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ikeda S, Kong SW, Lu J, Bisping E, Zhang H, Allen PD, Golub TR, Pieske B, Pu WT. Altered microRNA expression in human heart disease. Physiol Genomics. 2007;31:367–373. doi: 10.1152/physiolgenomics.00144.2007. [DOI] [PubMed] [Google Scholar]
- 21.Matkovich SJ, Van Booven DJ, Youker KA, Torre-Amione G, Diwan A, Eschenbacher WH, Dorn LE, Watson MA, Margulies KB, Dorn GW., 2nd Reciprocal regulation of myocardial microRNAs and messenger RNA in human cardiomyopathy and reversal of the microRNA signature by biomechanical support. Circulation. 2009;119:1263–1271. doi: 10.1161/CIRCULATIONAHA.108.813576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Naga Prasad SV, Duan ZH, Gupta MK, Surampudi VS, Volinia S, Calin GA, Liu CG, Kotwal A, Moravec CS, Starling RC, Perez DM, Sen S, Wu Q, Plow EF, Croce CM, Karnik S. Unique microRNA profile in end-stage heart failure indicates alterations in specific cardiovascular signaling networks. J Biol Chem. 2009;284:27487–27499. doi: 10.1074/jbc.M109.036541. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23.Sayed D, Hong C, Chen IY, Lypowy J, Abdellatif M. MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ Res. 2007;100:416–424. doi: 10.1161/01.RES.0000257913.42552.23. [DOI] [PubMed] [Google Scholar]
- 24.Sucharov C, Bristow MR, Port JD. miRNA expression in the failing human heart: functional correlates. J Mol Cell Cardiol. 2008;45:185–192. doi: 10.1016/j.yjmcc.2008.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tatsuguchi M, Seok HY, Callis TE, Thomson JM, Chen JF, Newman M, Rojas M, Hammond SM, Wang DZ. Expression of microRNAs is dynamically regulated during cardiomyocyte hypertrophy. J Mol Cell Cardiol. 2007;42:1137–1141. doi: 10.1016/j.yjmcc.2007.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thum T, Galuppo P, Wolf C, Fiedler J, Kneitz S, van Laake LW, Doevendans PA, Mummery CL, Borlak J, Haverich A, Gross C, Engelhardt S, Ertl G, Bauersachs J. MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation. 2007;116:258–267. doi: 10.1161/CIRCULATIONAHA.107.687947. [DOI] [PubMed] [Google Scholar]
- 27.van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, Richardson JA, Olson EN. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci U S A. 2006;103:18255–18260. doi: 10.1073/pnas.0608791103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Izumo S, Nadal-Ginard B, Mahdavi V. Protooncogene induction and reprogramming of cardiac gene expression produced in pressure overload. PNAS. 1988;85:339–343. doi: 10.1073/pnas.85.2.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mahdavi V, Lompre AM, Chambers AP, Nadal-Ginard B. Cardiac myosin heavy chain isozymic transitions during development and under pathological conditions are regulated at the level of mRNA availability. Eur Heart J. 1984;5 Suppl F:181–191. doi: 10.1093/eurheartj/5.suppl_f.181. [DOI] [PubMed] [Google Scholar]
- 30.Johnatty SE, Dyck JR, Michael LH, Olson EN, Abdellatif M. Identification of Genes Regulated During Mechanical Load-induced Cardiac Hypertrophy. J Mol Cell Cardiol. 2000;32:805–815. doi: 10.1006/jmcc.2000.1122. [DOI] [PubMed] [Google Scholar]
- 31.Rane S, Sayed D, Abdellatif M. MicroRNA with a MacroFunction. Cell Cycle. 2007;6:1850–1855. doi: 10.4161/cc.6.15.4551. [DOI] [PubMed] [Google Scholar]
- 32.Ikeda S, He A, Kong SW, Lu J, Bejar R, Bodyak N, Lee KH, Ma Q, Kang PM, Golub TR, Pu WT. MicroRNA-1 negatively regulates expression of the hypertrophy-associated calmodulin and Mef2a genes. Mol Cell Biol. 2009;29:2193–2204. doi: 10.1128/MCB.01222-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Care A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, Bang ML, Segnalini P, Gu Y, Dalton ND, Elia L, Latronico MV, Hoydal M, Autore C, Russo MA, Dorn GW, 2nd, Ellingsen O, Ruiz-Lozano P, Peterson KL, Croce CM, Peschle C, Condorelli G. MicroRNA-133 controls cardiac hypertrophy. Nat Med. 2007;13:613–618. doi: 10.1038/nm1582. [DOI] [PubMed] [Google Scholar]
- 34.Elia L, Contu R, Quintavalle M, Varrone F, Chimenti C, Russo MA, Cimino V, De Marinis L, Frustaci A, Catalucci D, Condorelli G. Reciprocal Regulation of MicroRNA-1 and Insulin-Like Growth Factor-1 Signal Transduction Cascade in Cardiac and Skeletal Muscle in Physiological and Pathological Conditions. Circulation. 2009;120:2377–2385. doi: 10.1161/CIRCULATIONAHA.109.879429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu N, Bezprozvannaya S, Williams AH, Qi X, Richardson JA, Bassel-Duby R, Olson EN. MicroRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 2008;22:3242–3254. doi: 10.1101/gad.1738708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matkovich SJ, Wang W, Tu Y, Eschenbacher WH, Dorn LE, Condorelli G, Diwan A, Nerbonne JM, Dorn GW., 2nd MicroRNA-133a Protects Against Myocardial Fibrosis and Modulates Electrical Repolarization Without Affecting Hypertrophy in Pressure-Overloaded Adult Hearts. Circ Res. 2009;106:166–175. doi: 10.1161/CIRCRESAHA.109.202176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beuvink I, Kolb FA, Budach W, Garnier A, Lange J, Natt F, Dengler U, Hall J, Filipowicz W, Weiler J. A novel microarray approach reveals new tissue-specific signatures of known and predicted mammalian microRNAs. Nucleic Acids Res. 2007;35:e52-. doi: 10.1093/nar/gkl1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress-dependent cardiac growth and gene expression by a MicroRNA. Science. 2007;316:575–579. doi: 10.1126/science.1139089. [DOI] [PubMed] [Google Scholar]
- 39.van Rooij E, Quiat D, Johnson BA, Sutherland LB, Qi X, Richardson JA, Kelm RJ, Jr, Olson EN. A family of microRNAs encoded by myosin genes governs myosin expression and muscle performance. Dev Cell. 2009;17:662–673. doi: 10.1016/j.devcel.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hagiwara N, Yeh M, Liu A. Sox6 is required for normal fiber type differentiation of fetal skeletal muscle in mice. Dev Dyn. 2007;236:2062–2076. doi: 10.1002/dvdy.21223. [DOI] [PubMed] [Google Scholar]
- 41.Montgomery RL, Hullinger TG, Semus HM, Dickinson BA, Seto AG, Lynch JM, Stack C, Latimer PA, Olson EN, van Rooij E. Therapeutic Inhibition of miR-208a Improves Cardiac Function and Survival During Heart Failure. Circulation. 2011;124:1537–1547. doi: 10.1161/CIRCULATIONAHA.111.030932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Callis TE, Pandya K, Seok HY, Tang RH, Tatsuguchi M, Huang ZP, Chen JF, Deng Z, Gunn B, Shumate J, Willis MS, Selzman CH, Wang DZ. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J Clin Invest. 2009;119:2772–2786. doi: 10.1172/JCI36154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morissette MR, Cook SA, Foo S, McKoy G, Ashida N, Novikov M, Scherrer-Crosbie M, Li L, Matsui T, Brooks G, Rosenzweig A. Myostatin regulates cardiomyocyte growth through modulation of Akt signaling. Circ Res. 2006;99:15–24. doi: 10.1161/01.RES.0000231290.45676.d4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Linhares VL, Almeida NA, Menezes DC, Elliott DA, Lai D, Beyer EC, Campos de Carvalho AC, Costa MW. Transcriptional regulation of the murine Connexin40 promoter by cardiac factors Nkx2-5, GATA4 and Tbx5. Cardiovasc Res. 2004;64:402–411. doi: 10.1016/j.cardiores.2004.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shieh JT, Huang Y, Gilmore J, Srivastava D. Elevated miR-499 levels blunt the cardiac stress response. PLoS One. 2011;6 doi: 10.1371/journal.pone.0019481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sayed D, He M, Hong C, Gao S, Rane S, Yang Z, Abdellatif M. MicroRNA-21 is a downstream effector of AKT that mediates its antiapoptotic effects via suppression of Fas ligand. J Biol Chem. 2010;285:20281–20290. doi: 10.1074/jbc.M110.109207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, Castoldi M, Soutschek J, Koteliansky V, Rosenwald A, Basson MA, Licht JD, Pena JT, Rouhanifard SH, Muckenthaler MU, Tuschl T, Martin GR, Bauersachs J, Engelhardt S. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980–984. doi: 10.1038/nature07511. [DOI] [PubMed] [Google Scholar]
- 48.Patrick DM, Montgomery RL, Qi X, Obad S, Kauppinen S, Hill JA, van Rooij E, Olson EN. Stress-dependent cardiac remodeling occurs in the absence of MicroRNA-21 in mice. J Clin Invest. 2010;120:3912–3916. doi: 10.1172/JCI43604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ahmed MS, Oie E, Vinge LE, Yndestad A, Oystein Andersen G, Andersson Y, Attramadal T, Attramadal H. Connective tissue growth factor--a novel mediator of angiotensin II-stimulated cardiac fibroblast activation in heart failure in rats. J Mol Cell Cardiol. 2004;36:393–404. doi: 10.1016/j.yjmcc.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 50.Chen MM, Lam A, Abraham JA, Schreiner GF, Joly AH. CTGF expression is induced by TGF- beta in cardiac fibroblasts and cardiac myocytes: a potential role in heart fibrosis. J Mol Cell Cardiol. 2000;32:1805–1819. doi: 10.1006/jmcc.2000.1215. [DOI] [PubMed] [Google Scholar]
- 51.Duisters RF, Tijsen AJ, Schroen B, Leenders JJ, Lentink V, van der Made I, Herias V, van Leeuwen RE, Schellings MW, Barenbrug P, Maessen JG, Heymans S, Pinto YM, Creemers EE. miR-133 and miR-30 regulate connective tissue growth factor: implications for a role of microRNAs in myocardial matrix remodeling. Circ Res. 2009;104:170–178. doi: 10.1161/CIRCRESAHA.108.182535. [DOI] [PubMed] [Google Scholar]
- 52.Castoldi G, di Gioia CR, Bombardi C, Catalucci D, Corradi B, Gualazzi MG, Leopizzi M, Mancini M, Zerbini G, Condorelli G, Stella A. MiR-133a regulates collagen 1A1: potential role of miR-133a in myocardial fibrosis in angiotensin II dependent hypertension. J Cell Physiol. 2011;18:22939. doi: 10.1002/jcp.22939. [DOI] [PubMed] [Google Scholar]
- 53.van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, Hill JA, Olson EN. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci U S A. 2008;105:13027–13032. doi: 10.1073/pnas.0805038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dews M, Homayouni A, Yu D, Murphy D, Sevignani C, Wentzel E, Furth EE, Lee WM, Enders GH, Mendell JT, Thomas-Tikhonenko A. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat Genet. 2006;38:1060–1065. doi: 10.1038/ng1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van Almen GC, Verhesen W, van Leeuwen RE, van de Vrie M, Eurlings C, Schellings MW, Swinnen M, Cleutjens JP, van Zandvoort MA, Heymans S, Schroen B. MicroRNA-18 and MicroRNA-19 regulate CTGF and TSP-1 expression in age-related heart failure. Aging Cell. 2011;10:769–779. doi: 10.1111/j.1474-9726.2011.00714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lin Z, Murtaza I, Wang K, Jiao J, Gao J, Li PF. miR-23a functions downstream of NFATc3 to regulate cardiac hypertrophy. Proc Natl Acad Sci U S A. 2009;106:12103–12108. doi: 10.1073/pnas.0811371106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Willis MS, Ike C, Li L, Wang DZ, Glass DJ, Patterson C. Muscle ring finger 1, but not muscle ring finger 2, regulates cardiac hypertrophy in vivo. Circ Res. 2007;100:456–459. doi: 10.1161/01.RES.0000259559.48597.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang J, Song Y, Zhang Y, Xiao H, Sun Q, Hou N, Guo S, Wang Y, Fan K, Zhan D, Zha L, Cao Y, Li Z, Cheng X, Yang X. Cardiomyocyte overexpression of miR-27b induces cardiac hypertrophy and dysfunction in mice. Cell Res. 2011;16:132. doi: 10.1038/cr.2011.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pineda Torra I, Gervois P, Staels B. Peroxisome proliferator-activated receptor alpha in metabolic disease, inflammation, atherosclerosis and aging. Curr Opin Lipidol. 1999;10:151–159. doi: 10.1097/00041433-199904000-00009. [DOI] [PubMed] [Google Scholar]
- 60.Asakawa M, Takano H, Nagai T, Uozumi H, Hasegawa H, Kubota N, Saito T, Masuda Y, Kadowaki T, Komuro I. Peroxisome proliferator-activated receptor gamma plays a critical role in inhibition of cardiac hypertrophy in vitro and in vivo. Circulation. 2002;105:1240–1246. doi: 10.1161/hc1002.105225. [DOI] [PubMed] [Google Scholar]
- 61.Nishi H, Ono K, Horie T, Nagao K, Kinoshita M, Kuwabara Y, Watanabe S, Takaya T, Tamaki Y, Takanabe-Mori R, Wada H, Hasegawa K, Iwanaga Y, Kawamura T, Kita T, Kimura T. MicroRNA-27a regulates beta cardiac myosin heavy chain gene expression by targeting thyroid hormone receptor beta1 in neonatal rat ventricular myocytes. Mol Cell Biol. 2011;31:744–755. doi: 10.1128/MCB.00581-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.da Costa Martins PA, Salic K, Gladka MM, Armand AS, Leptidis S, El Azzouzi H, Hansen A, Coenen-de Roo CJ, Bierhuizen MF, van der Nagel R, van Kuik J, de Weger R, de Bruin A, Condorelli G, Arbones ML, Eschenhagen T, De Windt LJ. MicroRNA-199b targets the nuclear kinase Dyrk1a in an auto-amplification loop promoting calcineurin/NFAT signalling. Nat Cell Biol. 1220;12:1220–1227. doi: 10.1038/ncb2126. [DOI] [PubMed] [Google Scholar]
- 63.Oka T, Maillet M, Watt AJ, Schwartz RJ, Aronow BJ, Duncan SA, Molkentin JD. Cardiac-specific deletion of Gata4 reveals its requirement for hypertrophy, compensation, and myocyte viability. Circ Res. 2006;98:837–845. doi: 10.1161/01.RES.0000215985.18538.c4. [DOI] [PubMed] [Google Scholar]
- 64.Liang Q, De Windt LJ, Witt SA, Kimball TR, Markham BE, Molkentin JD. The transcription factors GATA4 and GATA6 regulate cardiomyocyte hypertrophy in vitro and in vivo. J Biol Chem. 2001;276:30245–30253. doi: 10.1074/jbc.M102174200. [DOI] [PubMed] [Google Scholar]
- 65.Belaguli NS, Sepulveda JL, Nigam V, Charron F, Nemer M, Schwartz RJ. Cardiac tissue enriched factors serum response factor and GATA-4 are mutual coregulators. Mol Cell Biol. 2000;20:7550–7558. doi: 10.1128/mcb.20.20.7550-7558.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hautala N, Tokola H, Luodonpaa M, Puhakka J, Romppanen H, Vuolteenaho O, Ruskoaho H. Pressure overload increases GATA4 binding activity via endothelin-1. Circulation. 2001;103:730–735. doi: 10.1161/01.cir.103.5.730. [DOI] [PubMed] [Google Scholar]
- 67.van Berlo JH, Elrod JW, van den Hoogenhof MM, York AJ, Aronow BJ, Duncan SA, Molkentin JD. The transcription factor GATA-6 regulates pathological cardiac hypertrophy. Circ Res. 2010;107:1032–1040. doi: 10.1161/CIRCRESAHA.110.220764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Han M, Yang Z, Sayed D, He M, Gao S, Lin L, Yoon S, Abdellatif M. GATA4 Expression is Primarily Regulated via a miR-26b-Dependent Posttranscriptional Mechanism During Cardiac Hypertrophy. Cardiovasc Res. 2012 doi: 10.1093/cvr/cvs001. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Frangogiannis NG, Entman ML. Chemokines in myocardial ischemia. Trends Cardiovasc Med. 2005;15:163–169. doi: 10.1016/j.tcm.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 70.Xu C, Lu Y, Pan Z, Chu W, Luo X, Lin H, Xiao J, Shan H, Wang Z, Yang B. The muscle-specific microRNAs miR-1 and miR-133 produce opposing effects on apoptosis by targeting HSP60, HSP70 and caspase-9 in cardiomyocytes. J Cell Sci. 2007;120:3045–3052. doi: 10.1242/jcs.010728. [DOI] [PubMed] [Google Scholar]
- 71.Ren XP, Wu J, Wang X, Sartor MA, Qian J, Jones K, Nicolaou P, Pritchard TJ, Fan GC. MicroRNA-320 is involved in the regulation of cardiac ischemia/reperfusion injury by targeting heat-shock protein 20. Circulation. 2009;119:2357–2366. doi: 10.1161/CIRCULATIONAHA.108.814145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dong S, Cheng Y, Yang J, Li J, Liu X, Wang X, Wang D, Krall TJ, Delphin ES, Zhang C. MicroRNA expression signature and the role of MicroRNA-21 in the early phase of acute myocardial infarction. J Biol Chem. 2009;284:29514–29525. doi: 10.1074/jbc.M109.027896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cheng Y, Liu X, Zhang S, Lin Y, Yang J, Zhang C. MicroRNA-21 protects against the H(2)O(2)-induced injury on cardiac myocytes via its target gene PDCD4. J Mol Cell Cardiol. 2009;47:5–14. doi: 10.1016/j.yjmcc.2009.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Roy S, Khanna S, Hussain SR, Biswas S, Azad A, Rink C, Gnyawali S, Shilo S, Nuovo GJ, Sen CK. MicroRNA expression in response to murine myocardial infarction: miR-21 regulates fibroblast metalloprotease-2 via phosphatase and tensin homologue. Cardiovasc Res. 2009;82:21–29. doi: 10.1093/cvr/cvp015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang X, Zhang X, Ren XP, Chen J, Liu H, Yang J, Medvedovic M, Hu Z, Fan GC. MicroRNA-494 targeting both proapoptotic and antiapoptotic proteins protects against ischemia/reperfusion-induced cardiac injury. Circulation. 1308;122:1308–1318. doi: 10.1161/CIRCULATIONAHA.110.964684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Qian L, Van Laake LW, Huang Y, Liu S, Wendland MF, Srivastava D. miR-24 inhibits apoptosis and represses Bim in mouse cardiomyocytes. J Exp Med. 2011;208:549–560. doi: 10.1084/jem.20101547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hu S, Huang M, Nguyen PK, Gong Y, Li Z, Jia F, Lan F, Liu J, Nag D, Robbins RC, Wu JC. Novel MicroRNA Prosurvival Cocktail for Improving Engraftment and Function of Cardiac Progenitor Cell Transplantation. Circulation. 2011;124:S27–S34. doi: 10.1161/CIRCULATIONAHA.111.017954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hullinger TG, Montgomery RL, Seto AG, Dickinson BA, Semus HM, Lynch JM, Dalby CM, Robinson K, Stack C, Latimer PA, Hare JM, Olson EN, van Rooij E. Inhibition of miR-15 Protects Against Cardiac Ischemic Injury. Circ Res. 2011;3:3. doi: 10.1161/CIRCRESAHA.111.244442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nishi H, Ono K, Iwanaga Y, Horie T, Nagao K, Takemura G, Kinoshita M, Kuwabara Y, Mori RT, Hasegawa K, Kita T, Kimura T. MicroRNA-15b modulates cellular ATP levels and degenerates mitochondria via Arl2 in neonatal rat cardiac myocytes. J Biol Chem. 2010;285:4920–4930. doi: 10.1074/jbc.M109.082610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, Rassenti L, Alder H, Volinia S, Liu CG, Kipps TJ, Negrini M, Croce CM. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 2005;102:13944–13949. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hausenloy DJ, Tsang A, Mocanu MM, Yellon DM. Ischemic preconditioning protects by activating prosurvival kinases at reperfusion. Am J Physiol Heart Circ Physiol. 2005;288:H971–H976. doi: 10.1152/ajpheart.00374.2004. [DOI] [PubMed] [Google Scholar]
- 82.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 83.Rowland RT, Meng X, Cleveland JC, Meldrum DR, Harken AH, Brown JM. Cardioadaptation induced by cyclic ischemic preconditioning is mediated by translational regulation of de novo protein synthesis. J Surg Res. 1997;71:155–160. doi: 10.1006/jsre.1997.5142. [DOI] [PubMed] [Google Scholar]
- 84.Cai Z, Zhong H, Bosch-Marce M, Fox-Talbot K, Wang L, Wei C, Trush MA, Semenza GL. Complete loss of ischaemic preconditioning-induced cardioprotection in mice with partial deficiency of HIF-1{alpha} Cardiovasc Res. 2008;77:463–470. doi: 10.1093/cvr/cvm035. [DOI] [PubMed] [Google Scholar]
- 85.Eckle T, Kohler D, Lehmann R, El Kasmi K, Eltzschig HK. Hypoxia-inducible Factor-1 is central to cardioprotection: a new paradigm for ischemic preconditioning. Circulation. 2008;118:166–175. doi: 10.1161/CIRCULATIONAHA.107.758516. [DOI] [PubMed] [Google Scholar]
- 86.Rane S, He M, Sayed D, Vashistha H, Malhotra A, Sadoshima J, Vatner DE, Vatner SF, Abdellatif M. Downregulation of MiR-199a Derepresses Hypoxia-Inducible Factor-1{alpha} and Sirtuin 1 and Recapitulates Hypoxia Preconditioning in Cardiac Myocytes. Circ Res. 2009;104:879–886. doi: 10.1161/CIRCRESAHA.108.193102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rane S, He M, Sayed D, Yan L, Vatner D, Abdellatif M. An antagonism between the AKT and beta-adrenergic signaling pathways mediated through their reciprocal effects on miR-199a-5p. Cell Signal. 2010;22:1054–1062. doi: 10.1016/j.cellsig.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yin C, Salloum FN, Kukreja RC. A Novel Role of MicroRNA in Late Preconditioning. Upregulation of Endothelial Nitric Oxide Synthase and Heat Shock Protein 70. Circ Res. 2009;104:572–575. doi: 10.1161/CIRCRESAHA.108.193250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cheng Y, Zhu P, Yang J, Liu X, Dong S, Wang X, Chun B, Zhuang J, Zhang C. Ischaemic preconditioning-regulated miR-21 protects heart against ischaemia/reperfusion injury via anti-apoptosis through its target PDCD4. Cardiovasc Res. 2010;87:431–439. doi: 10.1093/cvr/cvq082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Harris TA, Yamakuchi M, Ferlito M, Mendell JT, Lowenstein CJ. MicroRNA-126 regulates endothelial expression of vascular cell adhesion molecule 1. Proc Natl Acad Sci U S A. 2008;105:1516–1521. doi: 10.1073/pnas.0707493105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Guo C, Sah JF, Beard L, Willson JK, Markowitz SD, Guda K. The noncoding RNA, miR-126, suppresses the growth of neoplastic cells by targeting phosphatidylinositol 3-kinase signaling and is frequently lost in colon cancers. Genes Chromosomes Cancer. 2008;47:939–946. doi: 10.1002/gcc.20596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kuhnert F, Mancuso MR, Hampton J, Stankunas K, Asano T, Chen CZ, Kuo CJ. Attribution of vascular phenotypes of the murine Egfl7 locus to the microRNA miR-126. Development. 2008;135:3989–3993. doi: 10.1242/dev.029736. [DOI] [PubMed] [Google Scholar]
- 93.Wang S, Aurora AB, Johnson BA, Qi X, McAnally J, Hill JA, Richardson JA, Bassel-Duby R, Olson EN. The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev Cell. 2008;15:261–271. doi: 10.1016/j.devcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.van Solingen C, Seghers L, Bijkerk R, Duijs JM, Roeten MK, van Oeveren-Rietdijk AM, Baelde HJ, Monge M, Vos JB, de Boer HC, Quax PH, Rabelink TJ, van Zonneveld AJ. Antagomir-mediated silencing of endothelial cell specific MicroRNA-126 impairs ischemia-induced angiogenesis. J Cell Mol Med. 2009;13:1577–1585. doi: 10.1111/j.1582-4934.2008.00613.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.van Solingen C, de Boer HC, Bijkerk R, Monge M, van Oeveren-Rietdijk AM, Seghers L, de Vries MR, van der Veer EP, Quax PH, Rabelink TJ, van Zonneveld AJ. MicroRNA-126 modulates endothelial SDF-1 expression and mobilization of Sca-1+/Lin- progenitor cells in ischaemia. Cardiovasc Res. 2011;27:27. doi: 10.1093/cvr/cvr227. [DOI] [PubMed] [Google Scholar]
- 96.Bonauer A, Carmona G, Iwasaki M, Mione M, Koyanagi M, Fischer A, Burchfield J, Fox H, Doebele C, Ohtani K, Chavakis E, Potente M, Tjwa M, Urbich C, Zeiher AM, Dimmeler S. MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science. 2009;324:1710–1713. doi: 10.1126/science.1174381. [DOI] [PubMed] [Google Scholar]
- 97.Doebele C, Bonauer A, Fischer A, Scholz A, Reiss Y, Urbich C, Hofmann WK, Zeiher AM, Dimmeler S. Members of the MicroRNA-17-92 cluster exhibit a cell intrinsic anti-angiogenic function in endothelial cells. Blood. 2010;2010:18. doi: 10.1182/blood-2010-01-264812. [DOI] [PubMed] [Google Scholar]
- 98.Janz A, Sevignani C, Kenyon K, Ngo CV, Thomas-Tikhonenko A. Activation of the myc oncoprotein leads to increased turnover of thrombospondin-1 mRNA. Nucleic Acids Res. 2000;28:2268–2275. doi: 10.1093/nar/28.11.2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ngo CV, Gee M, Akhtar N, Yu D, Volpert O, Auerbach R, Thomas-Tikhonenko A. An in vivo function for the transforming Myc protein: elicitation of the angiogenic phenotype. Cell Growth Differ. 2000;11:201–210. [PMC free article] [PubMed] [Google Scholar]
- 100.Tikhonenko AT, Black DJ, Linial ML. Viral Myc oncoproteins in infected fibroblasts down-modulate thrombospondin-1, a possible tumor suppressor gene. J Biol Chem. 1996;271:30741–30747. doi: 10.1074/jbc.271.48.30741. [DOI] [PubMed] [Google Scholar]
- 101.Fiedler J, Jazbutyte V, Kirchmaier BC, Gupta SK, Lorenzen J, Hartmann D, Galuppo P, Kneitz S, Pena JT, Sohn-Lee C, Loyer X, Soutschek J, Brand T, Tuschl T, Heineke J, Martin U, Schulte-Merker S, Ertl G, Engelhardt S, Bauersachs J, Thum T. MicroRNA-24 Regulates Vascularity After Myocardial Infarction. Circulation. 2011;2011:25. doi: 10.1161/CIRCULATIONAHA.111.039008. [DOI] [PubMed] [Google Scholar]
- 102.Lal A, Pan Y, Navarro F, Dykxhoorn DM, Moreau L, Meire E, Bentwich Z, Lieberman J, Chowdhury D. miR-24-mediated downregulation of H2AX suppresses DNA repair in terminally differentiated blood cells. Nat Struct Mol Biol. 2009;16:492–498. doi: 10.1038/nsmb.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ghosh G, Subramanian IV, Adhikari N, Zhang X, Joshi HP, Basi D, Chandrashekhar YS, Hall JL, Roy S, Zeng Y, Ramakrishnan S. Hypoxia-induced MicroRNA-424 expression in human endothelial cells regulates HIF-alpha isoforms and promotes angiogenesis. J Clin Invest. 2010;120:4141–4154. doi: 10.1172/JCI42980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pause A, Lee S, Worrell RA, Chen DY, Burgess WH, Linehan WM, Klausner RD. The von Hippel-Lindau tumor-suppressor gene product forms a stable complex with human CUL-2, a member of the Cdc53 family of proteins. Proc Natl Acad Sci U S A. 1997;94:2156–2161. doi: 10.1073/pnas.94.6.2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Camps C, Buffa FM, Colella S, Moore J, Sotiriou C, Sheldon H, Harris AL, Gleadle JM, Ragoussis J. hsa-miR-210 Is induced by hypoxia and is an independent prognostic factor in breast cancer. Clin Cancer Res. 2008;14:1340–1348. doi: 10.1158/1078-0432.CCR-07-1755. [DOI] [PubMed] [Google Scholar]
- 106.Fasanaro P, D'Alessandra Y, Di Stefano V, Melchionna R, Romani S, Pompilio G, Capogrossi MC, Martelli F. MicroRNA-210 modulates endothelial cell response to hypoxia and inhibits the receptor tyrosine kinase ligand Ephrin-A3. J Biol Chem. 2008;283:15878–15883. doi: 10.1074/jbc.M800731200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hu S, Huang M, Li Z, Jia F, Ghosh Z, Lijkwan MA, Fasanaro P, Sun N, Wang X, Martelli F, Robbins RC, Wu JC. MicroRNA-210 as a novel therapy for treatment of ischemic heart disease. Circulation. 2010;122:S124–S131. doi: 10.1161/CIRCULATIONAHA.109.928424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Katare R, Riu F, Mitchell K, Gubernator M, Campagnolo P, Cui Y, Fortunato O, Avolio E, Cesselli D, Beltrami AP, Angelini G, Emanueli C, Madeddu P. Transplantation of Human Pericyte Progenitor Cells Improves the Repair of Infarcted Heart Through Activation of an Angiogenic Program Involving Micro-RNA-132. Circ Res. 2011;109:894–906. doi: 10.1161/CIRCRESAHA.111.251546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lazzara R, El-Sherif N, Hope RR, Scherlag BJ. Ventricular arrhythmias and electrophysiological consequences of myocardial ischemia and infarction. Circ Res. 1978;42:740–749. doi: 10.1161/01.res.42.6.740. [DOI] [PubMed] [Google Scholar]
- 110.Yang B, Lin H, Xiao J, Lu Y, Luo X, Li B, Zhang Y, Xu C, Bai Y, Wang H, Chen G, Wang Z. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat Med. 2007;13:486–491. doi: 10.1038/nm1569. [DOI] [PubMed] [Google Scholar]
- 111.Lerner DL, Yamada KA, Schuessler RB, Saffitz JE. Accelerated onset and increased incidence of ventricular arrhythmias induced by ischemia in Cx43-deficient mice. Circulation. 2000;101:547–552. doi: 10.1161/01.cir.101.5.547. [DOI] [PubMed] [Google Scholar]
- 112.Plaster NM, Tawil R, Tristani-Firouzi M, Canun S, Bendahhou S, Tsunoda A, Donaldson MR, Iannaccone ST, Brunt E, Barohn R, Clark J, Deymeer F, George AL, Jr, Fish FA, Hahn A, Nitu A, Ozdemir C, Serdaroglu P, Subramony SH, Wolfe G, Fu YH, Ptacek LJ. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen's syndrome. Cell. 2001;105:511–519. doi: 10.1016/s0092-8674(01)00342-7. [DOI] [PubMed] [Google Scholar]
- 113.Terentyev D, Belevych AE, Terentyeva R, Martin MM, Malana GE, Kuhn DE, Abdellatif M, Feldman DS, Elton TS, Gyorke S. miR-1 overexpression enhances Ca(2+) release and promotes cardiac arrhythmogenesis by targeting PP2A regulatory subunit B56alpha and causing CaMKII-dependent hyperphosphorylation of RyR2. Circ Res. 2009;104:514–521. doi: 10.1161/CIRCRESAHA.108.181651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005;97:1314–1322. doi: 10.1161/01.RES.0000194329.41863.89. [DOI] [PubMed] [Google Scholar]
- 115.Lehnart SE, Wehrens XH, Laitinen PJ, Reiken SR, Deng SX, Cheng Z, Landry DW, Kontula K, Swan H, Marks AR. Sudden death in familial polymorphic ventricular tachycardia associated with calcium release channel (ryanodine receptor) leak. Circulation. 2004;109:3208–3214. doi: 10.1161/01.CIR.0000132472.98675.EC. [DOI] [PubMed] [Google Scholar]
- 116.Belevych AE, Sansom SE, Terentyeva R, Ho HT, Nishijima Y, Martin MM, Jindal HK, Rochira JA, Kunitomo Y, Abdellatif M, Carnes CA, Elton TS, Gyorke S, Terentyev D. MicroRNA-1 and-133 Increase Arrhythmogenesis in Heart Failure by Dissociating Phosphatase Activity from RyR2 Complex. PLoS One. 2011;6:6. doi: 10.1371/journal.pone.0028324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lu Y, Zhang Y, Wang N, Pan Z, Gao X, Zhang F, Shan H, Luo X, Bai Y, Sun L, Song W, Xu C, Wang Z, Yang B. MicroRNA-328 contributes to adverse electrical remodeling in atrial fibrillation. Circulation. 2010;122:2378–2387. doi: 10.1161/CIRCULATIONAHA.110.958967. [DOI] [PubMed] [Google Scholar]
- 118.Vallett SM, Sanchez HB, Rosenfeld JM, Osborne TF. A direct role for sterol regulatory element binding protein in activation of 3-hydroxy-3-methylglutaryl coenzyme A reductase gene. J Biol Chem. 1996;271:12247–12253. doi: 10.1074/jbc.271.21.12247. [DOI] [PubMed] [Google Scholar]
- 119.Najafi-Shoushtari SH, Kristo F, Li Y, Shioda T, Cohen DE, Gerszten RE, Naar AM. MicroRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science. 2010;328:1566–1569. doi: 10.1126/science.1189123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Brooks-Wilson A, Marcil M, Clee SM, Zhang LH, Roomp K, van Dam M, Yu L, Brewer C, Collins JA, Molhuizen HO, Loubser O, Ouelette BF, Fichter K, Ashbourne-Excoffon KJ, Sensen CW, Scherer S, Mott S, Denis M, Martindale D, Frohlich J, Morgan K, Koop B, Pimstone S, Kastelein JJ, Genest J, Jr, Hayden MR. Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat Genet. 1999;22:336–345. doi: 10.1038/11905. [DOI] [PubMed] [Google Scholar]
- 121.Rayner KJ, Sheedy FJ, Esau CC, Hussain FN, Temel RE, Parathath S, van Gils JM, Rayner AJ, Chang AN, Suarez Y, Fernandez-Hernando C, Fisher EA, Moore KJ. Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J Clin Invest. 2011;121:2921–2931. doi: 10.1172/JCI57275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rayner KJ, Esau CC, Hussain FN, McDaniel AL, Marshall SM, van Gils JM, Ray TD, Sheedy FJ, Goedeke L, Liu X, Khatsenko OG, Kaimal V, Lees CJ, Fernandez-Hernando C, Fisher EA, Temel RE, Moore KJ. Inhibition of miR-33a/b in non-human primates raises plasma HDL and lowers VLDL triglycerides. Nature. 2011;478:404–407. doi: 10.1038/nature10486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Xin M, Small EM, Sutherland LB, Qi X, McAnally J, Plato CF, Richardson JA, Bassel-Duby R, Olson EN. MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes Dev. 2009;23:2166–2178. doi: 10.1101/gad.1842409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Boettger T, Beetz N, Kostin S, Schneider J, Kruger M, Hein L, Braun T. Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster. J Clin Invest. 2009;119:2634–2647. doi: 10.1172/JCI38864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Elia L, Quintavalle M, Zhang J, Contu R, Cossu L, Latronico MV, Peterson KL, Indolfi C, Catalucci D, Chen J, Courtneidge SA, Condorelli G. The knockout of miR-143 and-145 alters smooth muscle cell maintenance and vascular homeostasis in mice: correlates with human disease. Cell Death Differ. 2009;16:1590–1598. doi: 10.1038/cdd.2009.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cheng Y, Liu X, Yang J, Lin Y, Xu DZ, Lu Q, Deitch EA, Huo Y, Delphin ES, Zhang C. MicroRNA-145, a novel smooth muscle cell phenotypic marker and modulator, controls vascular neointimal lesion formation. Circ Res. 2009;105:158–166. doi: 10.1161/CIRCRESAHA.109.197517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Watanabe N, Kurabayashi M, Shimomura Y, Kawai-Kowase K, Hoshino Y, Manabe I, Watanabe M, Aikawa M, Kuro-o M, Suzuki T, Yazaki Y, Nagai R. BTEB2, a Kruppel-like transcription factor, regulates expression of the SMemb/Nonmuscle myosin heavy chain B (SMemb/NMHC-B) gene. Circ Res. 1999;85:182–191. doi: 10.1161/01.res.85.2.182. [DOI] [PubMed] [Google Scholar]
- 128.Suzuki T, Sawaki D, Aizawa K, Munemasa Y, Matsumura T, Ishida J, Nagai R. Kruppel-like factor 5 shows proliferation-specific roles in vascular remodeling, direct stimulation of cell growth, and inhibition of apoptosis. J Biol Chem. 2009;284:9549–9557. doi: 10.1074/jbc.M806230200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Torella D, Iaconetti C, Catalucci D, Ellison GM, Leone A, Waring CD, Bochicchio A, Vicinanza C, Aquila I, Curcio A, Condorelli G, Indolfi C. MicroRNA-133 Controls Vascular Smooth Muscle Cell Phenotypic Switch In Vitro and Vascular Remodeling In Vivo. Circ Res. 2011;109:880–893. doi: 10.1161/CIRCRESAHA.111.240150. [DOI] [PubMed] [Google Scholar]
- 130.Davis BN, Hilyard AC, Nguyen PH, Lagna G, Hata A. Induction of MicroRNA-221 by platelet-derived growth factor signaling is critical for modulation of vascular smooth muscle phenotype. J Biol Chem. 2009;284:3728–3738. doi: 10.1074/jbc.M808788200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Liu X, Cheng Y, Zhang S, Lin Y, Yang J, Zhang C. A necessary role of miR-221 and miR-222 in vascular smooth muscle cell proliferation and neointimal hyperplasia. Circ Res. 2009;104:476–487. doi: 10.1161/CIRCRESAHA.108.185363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ji R, Cheng Y, Yue J, Yang J, Liu X, Chen H, Dean DB, Zhang C. MicroRNA expression signature and antisense-mediated depletion reveal an essential role of MicroRNA in vascular neointimal lesion formation. Circ Res. 2007;100:1579–1588. doi: 10.1161/CIRCRESAHA.106.141986. [DOI] [PubMed] [Google Scholar]
- 133.Meng F, Henson R, Lang M, Wehbe H, Maheshwari S, Mendell JT, Jiang J, Schmittgen TD, Patel T. Involvement of human micro-RNA in growth and response to chemotherapy in human cholangiocarcinoma cell lines. Gastroenterology. 2006;130:2113–2129. doi: 10.1053/j.gastro.2006.02.057. [DOI] [PubMed] [Google Scholar]
- 134.Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;133:647–658. doi: 10.1053/j.gastro.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Jones JA, Stroud RE, O'Quinn EC, Black LE, Barth JL, Elefteriades JA, Bavaria JE, Gorman JH, 3rd, Gorman RC, Spinale FG, Ikonomidis JS. Selective microRNA Suppression in Human Thoracic Aneurysms: Relationship of miR-29a to Aortic Size and Proteolytic Induction. Circ Cardiovasc Genet. 2011;18:18. doi: 10.1161/CIRCGENETICS.111.960419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Boon RA, Seeger T, Heydt S, Fischer A, Hergenreider E, Horrevoets AJ, Vinciguerra M, Rosenthal N, Sciacca S, Pilato M, van Heijningen P, Essers J, Brandes RP, Zeiher AM, Dimmeler S. MicroRNA-29 in aortic dilation: implications for aneurysm formation. Circ Res. 2011;109:1115–1119. doi: 10.1161/CIRCRESAHA.111.255737. [DOI] [PubMed] [Google Scholar]
- 137.Merk DR, Chin JT, Dake BA, Maegdefessel L, Miller MO, Kimura N, Tsao PS, Iosef C, Berry GJ, Mohr FW, Spin JM, Alvira CM, Robbins RC, Fischbein MP. miR-29b Participates in Early Aneurysm Development in Marfan Syndrome. Circ Res. 2011;23:23. doi: 10.1161/CIRCRESAHA.111.253740. [DOI] [PubMed] [Google Scholar]