

Abstract
Glycine N-methyltransferase (GNMT) is a tumor suppressor for hepatocellular carcinoma (HCC). High rates of Gnmt knockout mice developed HCC. Epigenetic alteration and dysregulation of several pathways including wingless-type MMTV integration site (Wnt), mitogen-activated protein kinase (MAPK) and Janus kinase and signal transducer and activator of transcription (JAK-STAT) are associated with HCC development in Gnmt knockout mice. We hypothesized that GNMT may regulate signal transduction through interacting with other proteins directly. In this report, we identified a mammalian target of rapamycin (mTOR) inhibitor (DEP domain containing MTOR-interacting protein [DEPDC6/DEPTOR]) as a GNMT-binding protein by using yeast two-hybrid screening. Fluorescence resonance energy transfer assay demonstrated that the C-terminal half of GNMT interact with the PSD-95/Dlg1/ZO-1 (PDZ) domain of DEPDC6/DEPTOR. Immunohistochemical staining showed that 27.5% (14/51) of HCC patients had higher expression levels of DEPDC6/DEPTOR in the tumorous tissues than in tumor-adjacent tissues, especially among HCC patients with hepatitis B viral infection (odds ratio 10.3, 95% confidence interval [CI] 1.05–11.3) or patients with poor prognosis (death hazard ratio 4.51, 95% CI 1.60–12.7). In terms of molecular mechanism, knockdown of DEPDC6/DEPTOR expression in HuH-7 cells caused S6K and 4E-BP activation, but suppressed Akt. Overexpression of DEPDC6/DEPTOR activated Akt and increased survival of HCC cells. Overexpression of GNMT caused activation of mTOR/raptor downstream signaling and delayed G2/M cell cycle progression, which altogether resulted in cellular senescence. Furthermore, GNMT reduced proliferation of HuH-7 cells and sensitized them to rapamycin treatment both in vitro and in vivo. In conclusion, GNMT regulates HCC growth in part through interacting with DEPDC6/DEPTOR and modulating mTOR/raptor signaling pathway. Both GNMT and DEPDC6/DEPTOR are potential targets for developing therapeutics for HCC.
INTRODUCTION
Hepatocellular carcinoma (HCC) is the third leading cause of cancer deaths worldwide (1,2). The pathogenesis of HCC is complex and involves many molecular pathways (2,3). Activation of the mammalian target of rapamycin (mTOR) pathway has been reported in 15–50% of human HCC (4,5), suggesting the critical role this pathway plays in hepatic tumorigenesis (5–7). The TOR proteins are evolutionarily conserved serine/threonine kinases found in nearly all eukaryotic cells (8). In response to stimulation, mTOR regulates cell growth through modulation of many processes, including protein synthesis, ribosome biogenesis and autophagy (8). Recently, Peterson et al. (9) identified that DEP domain containing MTOR-interacting protein (DEPTOR; synonym: DEP domain containing 6 [DEPDC6]) interacts with mTOR directly and serves as an mTOR inhibitor (9). Overexpression of DEPTOR activates Akt via the inhibition of a negative feedback loop from S6K to phosphatidylinositol 3-kinase (PI3K). In addition, they found that DEPTOR is overexpressed in a subset of multiple myelomas harboring cyclin D1/D3 or c-MAF/MAFB translocations. In these cells, high DEPTOR expression is necessary to maintain Akt activation, and a reduction in DEPTOR levels leads to apoptosis (9).
Glycine N-methyltransferase (GNMT) is a tumor suppressor for HCC (10,11). It regulates the ratio of S-adenosylmethionine to S-adenosylhomocysteine and serves as a folate binding protein (12,13). In addition, GNMT binds carcinogens such as polyaromatic hydrocarbons and aflatoxins and prevents the deoxyribonucleic acid (DNA) adduct formation and cytotoxicity induced by these carcinogens (14–16). Diminished levels of GNMT were observed in both human HCC cell lines and tumor tissues (17,18). Previously, we and another group reported that high rates of both sexes of Gnmt knockout mice develop HCC spontaneously (10,11). Epigenetic alteration and dysregulation of several pathways including wingless-type MMTV integration site (Wnt), mitogen-activated protein kinase (MAPK) and Janus kinase and signal transducer and activator of transcription (JAK-STAT) are associated with the HCC development in Gnmt knockout mice (10,11).
In this study, we hypothesized that GNMT may regulate signal transduction pathways through interacting with other proteins directly. Therefore, we used a yeast two-hybrid assay to screen proteins that may interact with GNMT. We identified DEPTOR as a GNMT binding protein and further mapped their interactive domains. Clinically, we showed that DEPTOR is overexpressed in hepatitis B virus (HBV)- associated HCC tissues and is associated with poor prognosis. Loss of DEPTOR in HuH-7 cells activated S6K and 4E-BP, but reduced Akt activation and cell growth. Subsequently, we revealed that GNMT affects mTOR signaling by interacting with DEPTOR. Finally, we demonstrated that GNMT can sensitize HuH-7 cells to rapamycin both in vitro and in vivo.
MATERIALS AND METHODS
HCC Patients
Pathological slides of 51 pairs of tumorous and tumor-adjacent tissues from HCC patients were obtained from the Taiwan Liver Cancer Network (TLCN). The specimens were obtained from the liver tumor tissues removed from the patients; thus, the pathology stage can represent the status of tumor progression. The mean age of the patients was 60.0 ± 13.5 years. We divided them into three groups according to types of hepatitis viral infection: 16 patients (10 males and 6 females) were hepatitis B surface antigen (HBsAg)-positive, 18 patients (9 males and 9 females) were positive for anti–hepatitis C virus (HCV) antibody (Ab), and 17 patients (7 males and 10 females) did not have any hepatitis B or C markers. Informed consent was obtained from all the patients before they had surgery. In addition, clinical and pathological data including numbers of HCC nodules, tumor size, vascular invasion of tumor cells and duration of survival were provided by TLCN. This study was approved by the Institutional Review Board of National Yang Ming University and the user committee of TLCN.
Plasmids and Lentiviral Constructs
In total, nine plasmids were constructed for the analysis of interactions between GNMT and DEPTOR. In addition, two lentiviral constructs were made to generate HuH-7 stable cells expressing GNMT or DEPTOR protein. Detailed methods are described in the Supplementary Data. Two plasmids encoding different shRNAs for DEPTOR (pLKO.1-shDEPTOR-1 and pLKO.1-shDEPTOR-2) were purchased from Addgene (http://www.addgene.org). The control plasmids for the RNA interference (pLKO.1-shLuc) and for the overexpression experiments (pLKO_AS3w.eGFP.puro) were obtained from the National RNAi Core Facility (Academia Sinica, Taiwan).
Generation of HuH-7 Stable Cells
HEK293T cells were cotransfected with a packaging plasmid-pCMV-ΔR8.91, a VSV-G envelope expressing plasmid-pMD.G and one of the following lentiviral constructs: pLKO.1-shLuc, pLKO.1-shDEPTOR-1, pLKO.1-shDEPTOR-2, pLKO_AS3w.eGFP.puro, pLV-GNMT-FLAG and pLV-HA-DEPTOR using TurboFect™ Reagent (Fermentas, Hanover, MD, USA). A supernatant containing lentiviruses was harvested according to the protocol published on the website http://rnai.genmed.sinica.edu.tw. To generate stable cell lines, HuH-7 cells were infected with pseudo-typed lentivirus in medium containing polybrene (8 μg/mL). Twenty-four hours after infection, the cells were treated with puromycin (1 μg/mL) to select stable cells.
Cell Culture and Transfection
HEK293T and HuH-7 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco BRL, Grand Island, NY) with 10% heat-inactivated fetal bovine serum (HyClone, Logan, UT, USA), penicillin (100 U/mL), streptomycin (100 μg/mL), nonessential amino acids (0.1 mmol/L), and l-glutamine (2 mmol/L) in a humidified incubator with 5% CO2. Lentivirus-infected cells including HuH-7-shLuc, HuH-7-shDEP-TOR-1, HuH-7-shDEPTOR-2, HuH-7-GFP, HuH-7-GNMT and HuH-7-DEPTOR were grown in DMEM supplemented with 1 μg/mL puromycin. Plasmid DNA was transfected by using TurboFect Reagent (Fermentas). All transfections were performed according to the manufacturer instructions.
Yeast Two-Hybrid Screening
Human GNMT cDNA was subcloned into the pGBKT7 vector (Clontech, Palo Alto, CA, USA). A human kidney cDNA library fused to the pACT2 vector (Clontech) was used as the prey. Colonies were selected under high stringency conditions according to the manufacturer instructions. After screening three times, repeatedly positive colonies were transferred onto a filter membrane and subjected to β-galactosidase assays. Plasmids retrieved from the positive clones were sequenced. The genes related to the inserts were subsequently identified by using the BLAST program and the National Center for Biotechnology Information (NCBI) GenBank database.
Immunoprecipitation and Western Blotting
Mouse liver or cultured cells were lysed by using lysis buffer supplemented with protease and phosphatase inhibitors (see Supplementary Data). Cell lysates were incubated with 10 μg anti-HA monoclonal antibody (mAb) (Covance, Berkeley, CA, USA), anti-mTOR antibody (Cell Signaling Technology, Beverly, MA, USA), anti-DEPTOR mAb (YMAC Biotech, Taipei, Taiwan) or anti-GNMT mAb (YMAC Biotech) for 1 h at 4°C, followed by the addition of 20 μL protein-A/G sepharose (Calbiochem, Merck, Darmstadt, Germany) and incubation for 4 h. The beads were washed three times with lysis buffer and resuspended in a sample buffer for sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot analyses. Similar procedures were used for immunoprecipitation of the mTOR related complex, except that for the lysis buffer was replaced by mTOR complex buffer (50 mmol/L Tris [pH 7.5], 150 mmol/L NaCl, 5 mmol/L ethylenediaminetetraacetic acid [EDTA] and 0.3% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) (19). Detailed methods for Western blotting are described in the Supplementary Data.
Indirect Immunofluorescent Antibody and Fluorescence Resonance Energy Transfer–Acceptor Bleaching Assays
Indirect immunofluorescent antibody assay was performed as described previously (15). Anti-HA mAb (1:400 dilution; Covance) and rabbit anti-GNMT antiserum (1:100 dilution; YMAC Biotech) were used as the primary antibodies, whereas fluorescein isothiocyanate (FITC)-conjugated anti-mouse IgG (1:400 dilution) and tetramethylrhodamine isothiocyanate–conjugated anti-rabbit IgG (1:400 dilution) (Chemicon, Temecula, CA, USA) were used as the secondary antibodies. Confocal microscopy was performed by using a Leica TCS SP2 inverted fluorescence microscope. A fluorescence resonance energy transfer–acceptor bleaching (FRET-AB) assay (20) was performed by using a Leica TCS SP5 Confocal Spectral Microscope Imaging System, and the acceptor photobleaching was carried out according to the manufacturer’s instructions. In brief, cells were bleached in the rhodamine channel by scanning a region of interest for 10 s with a 561-nm diode-pumped solid state (DPSS) laser line at 100% intensity. Both FITC and rhodamine images were captured before and after each bleaching. Energy-transferred efficiency was calculated by using the following formula: [(Dpost − Dpre)/Dpost] × 100%, where Dpost represents the fluorescence intensity of the donor after acceptor photobleaching and Dpre is the fluorescence intensity of the donor preceding acceptor photobleaching.
Immunohistochemical Staining
Detailed procedures for immunohisto-chemical (IHC) staining have been described previously (16). Mouse monoclonal antibodies against DEPTOR (1:50 dilution; YMAC Biotech) and Ki-67 (1:50 dilution; Becton Dickinson, Taipei, Taiwan) were used. Signals were visualized by using SuperPicTure™ Polymer Detection Kits (Zymed, Invitrogen, Carlsbad, CA, USA).
Cell Proliferation and Cytotoxicity Assays
For cell proliferation assay, HuH-7 cells were cultured in a 48-well plate in triplicate and fixed at different time points with 0.05% crystal violet in 10% formalin. Each well was then washed multiple times with water. To measure relative cell density, crystal violet was resolubilized in 10% acetic acid, and the absorbance at 595 nm was recorded by using a Varioskan Flash spectral scanning multimode reader (Thermo Scientific, Waltham, MA, USA). For cytotoxicity assay, cells were seeded in a 96-well plate in triplicate and treated with rapamycin. Then, culture medium was replaced by 100 μL fresh medium containing 10 μL of 5 mg/mL 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) stock solution (Sigma-Aldrich, St. Louis, MO, USA) (21). After 4 h of labeling the cells with MTT, the medium was replaced with 100 μL dimethyl sulfoxide for 10 min at 37°C. Samples were mixed and the absorbance was read at 540 nm by using the same reader mentioned above.
Senescence Associated–β-Gal Staining
Senescence associated (SA)-β-gal staining was performed according to the methods published previously (22). In brief, cells were fixed by using 2% formaldehyde and 0.2% glutaraldehyde for 10 min at room temperature. Then, they were incubated with SA-β-gal stain solution at 37°C (without CO2) for 12–16 h. The results were recorded by using both phase-contrast and bright-field microscopy.
Flow Cytometry
For cell cycle progression analysis, HuH-7-GFP or HuH-7-GNMT stable cells had been synchronized at G0/G1 phases by allowing them to grow to confluence (called time 0) and then reseeded the cells at subconfluent density. Both floating and adherent cells were harvested, combined and processed at different time points. Cells were fixed with ice-cold 70% ethanol at −20°C, washed and resuspended in 0.5 mL PBS containing propidium iodide (10 μg/mL) and RNase A (1 mg/mL). After incubating at 37°C for 30 min, the cells were analyzed by using a FACSCanto flow cytometer (Becton Dickinson) and the data were analyzed by using ModFit LT 2.0 software.
Xenograft and Rapamycin (RAD001) Treatment
Six-week-old athymic female NOD/SCID mice were injected with 1 × 106 HuH-7-GFP or HuH-7-GNMT stable cells in the right flank subcutaneously. Seven days later, mice were randomized into two groups and injected intraperitoneally with either RAD001 (everolimus, a soluble derivative of rapamycin; Novartis, East Hanover, NJ, USA), at a dosage of 50 μg/kg three times per week, or placebo (drug vehicle provided by Novartis). Tumor growth was monitored at least twice a week by using Vernier caliper measurement of the length (L) and width (W) of the tumor. Tumor volume (TV) was calculated as follows: TV = (L × W2)/2. The protocol was reviewed and approved by the Institutional Animal Care and Use Committee of National Yang-Ming University in compliance with the guidelines on the care and use of animals for scientific purpose.
Statistical Analysis
Statistical analysis was performed by using SPSS (version 13.0) and P < 0.05 was considered to be statistically significant. Pearson χ2 or Fisher exact tests were used to evaluate the association between DEPTOR expression and different clinico-pathological characteristics of HCC patients. Multivariate logistic regression models were used to adjust for covariate effects on the odds ratio. Comparisons between groups were made by using the Student t test. The Kaplan-Meier estimation method was used for overall survival analysis, and a log-rank test was used to compare differences. Multivariate survival analyses were conducted by using a Cox proportional hazards regression model.
All supplementary materials are available online at www.molmed.org.
RESULTS
Identification of DEPTOR as a GNMT Binding Protein and Mapping of Their Interactive Domains
To identify proteins interacting with GNMT, full-length human GNMT was used as the bait in a yeast two-hybrid screen system with a human kidney cDNA library. A positive clone containing a sequence encoding the C-terminal region of DEP domain containing 6 (DEPDC6) was identified (Figure 1A and Supplementary Figure 1). Because Peterson et al. (9) reported that DEPDC6 is an mTOR binding protein and designated it as DEPTOR, we will use DEPTOR instead of DEPDC6 in this report. The interaction between GNMT and DEPTOR was confirmed by both immunoprecipitation and FRET-AB experiments. As shown in Figures 1B and C, immunoprecipitation of either HA-tagged DEPTOR or endogenous DEPTOR coprecipitated FLAG-tagged GNMT. In addition, we detected endogenous DEPTOR in GNMT immunoprecipitants prepared from mouse liver (Figure 1D).
Figure 1.

Identification of DEPDC6/DEPTOR as a GNMT binding protein and mapping of the interactive domains. (A) Different constructs of DEPDC6/DEPTOR and GNMT used for domain mapping. The protein fragment expressed by the positive prey clone obtained from a yeast two-hybrid screening was indicated. N, amino-terminal region of GNMT; C, catalytic domains located in amino acids 37–175 and 243–295 of GNMT. (B) FLAG-tagged GNMT coimmunoprecipitated with HA-tagged DEPTOR. HEK293T cells were transfected with the indicated plasmids and harvested for immunoprecipitation analysis. (C) Interaction of endogenous DEPTOR with FLAG-tagged GNMT. FLAG-tagged GNMTs were expressed in HuH-7 cells, and cell lysates were used for immunoprecipitation analysis. (D) Interaction of endogenous DEPTOR with endogenous GNMT. Mouse liver lysates were used for immunoprecipitation analysis. Each experiment was repeated at least three times. (E) FRET-AB assay was used to assess the interaction between GNMT and DEPTOR and to map their interactive domains. Photobleaching of the GNMT rhodamine label (white dotted circle) resulted in an increase of DEPTOR fluorescent signal within the photobleached area, thus demonstrating the FRET effects (yellow bar = 10 μm; white bar = 25 μm). Positive results were also observed in HuH-7 cells coexpressed in the PDZ domain of DEPTOR and GNMT-FLAG as well as in cells coexpressed in the HA-DEPTOR and C-terminal region (amino acids 171–295) of GNMT. (F) Quantitative results of FRET efficiency in different sets of FRET-AB assays. HuH-7 cells were transfected with the indicated plasmids and were subjected to FRET-AB assay. Data are presented as means ± SD of 5–10 random fields from two independent experiments. Some image data are shown in Supplementary Figure 2. ***P < 0.001. IP, immunoprecipitation.
FRET-AB assay showed that GNMT interacted with DEPTOR directly in the cytoplasm. Compared to the prebleaching panel, the intensity of the FITC green fluorescence in the DEPTOR panel increased significantly after the rhodamine fluorophore was destroyed by laser-photobleaching (Figure 1E). To map the interactive domains between GNMT and DEPTOR, eight plasmids containing different domains of DEPTOR or GNMT were constructed (see Figure 1A). The results showed that the FRET efficiency between full-length GNMT and DEP domains of DEPTOR decreased significantly. In addition, a >50% decrease of the FRET efficiency was found between full-length DEPTOR and the N-terminal of GNMT (Figure 1F and Supplementary Figure 2). In contrast, the FRET efficiency between (a) full-length GNMT and the PSD-95/Dlg1/ZO-1 (PDZ) domain of DEPTOR and (b) full-length DEPTOR and the C-terminal 171–295 amino acid fragment of GNMT were comparable to the results observed between full-length GNMT and full-length DEPTOR (see Figures 1E, F). Therefore, the C-terminal half of GNMT interacts with the PDZ domain of DEPTOR.
Expression Levels of DEPTOR in Tumorous Tissues from HCC Patients and Its Association with Their Survival
IHC staining was used to compare the expression levels of DEPTOR between tumorous (T) and tumor-adjacent (TA) tissues obtained from HCC patients. As shown in Figure 2, DEPTOR was mainly expressed in the cytoplasm, whereas nuclear staining was also observed (Figures 2A–F). Among 51 pairs of T and TA tissue samples, 27.5% had higher expression levels of DEPTOR in tumorous tissues than in the TA tissues (Table 1). In addition, 43.8% (7/16) of patients with HBsAg-positive and 33.3% (6/18) of patients with anti-HCV antibodies had higher expression levels of DEPTOR in the tumorous tissues than in the TA tissues. Multivariate logistic-regression analysis indicated that the expression of DEPTOR significantly correlates with HBV infection (odds ratio 10.29, 95% confidence interval [CI] 1.05–11.29) (Table 2). Furthermore, higher level of DEPTOR in the tumorous tissues was associated with poorer survival (Figure 2G, log-rank test, P = 0.009). A Cox proportional hazards test was used to evaluate factors associated with prognosis of the HCC patients, and the results indicated that the association between death and DEPTOR overexpression is statistically significant (T > TA versus T ≤ TA: adjusted hazard ratio 4.51; 95% CI 1.60–12.7, P = 0.004) (Supplementary Table 1). Therefore, overexpression of DEPTOR in the tumorous tissues is frequently found in patients with HBV infection and associated with prognosis of HCC patients independent of sex, tumor type (one versus multiple tumor nodules) or tumor-node-metastasis (TNM) stages.
Figure 2.
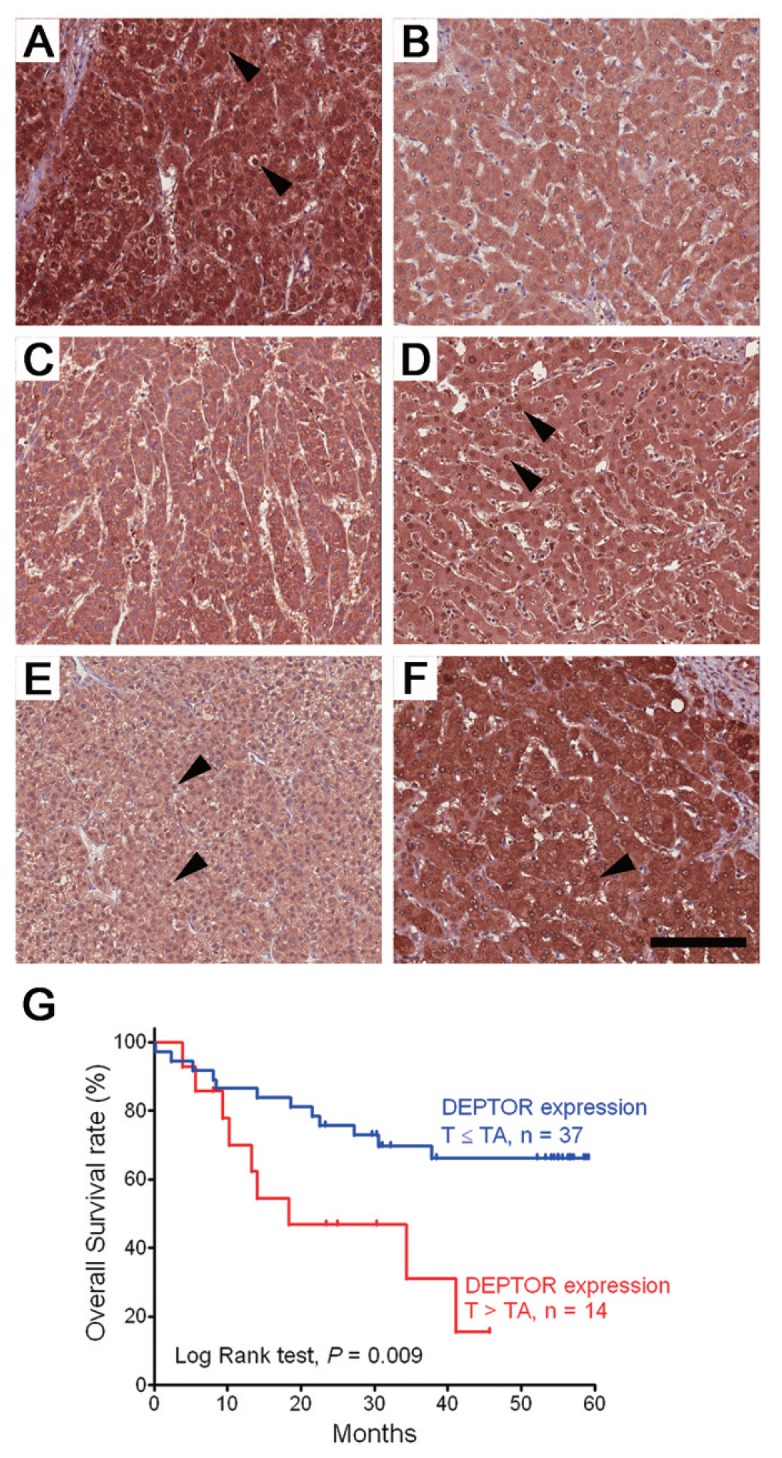
Expression profiles of DEPTOR in human HCC and the association with survival. (A–F) IHC staining of DEPTOR in three pairs of tumorous (T) and tumor-adjacent (TA) tissues from 51 HCC patients. (A, C, E) Tumorous tissues. (B, D, F) Tumor-adjacent tissues. (A, B) An example of the T > TA staining pattern. (C, D) An example of the T = TA staining pattern. (E, F) An example of the T < TA staining pattern. Arrowheads indicate nuclear staining of DEPTOR (bar = 100 μm). (G) Kaplan-Meier survival curves of two groups of HCC patients with different DEPTOR expression patterns (T > TA versus T ≤ TA).
Table 1.
Results of immunohistochemical staining of DEPTOR expression in tumorous (T) versus tumor-adjacent (TA) tissues from 51 HCC patients and the associations with their clinical and pathological characteristics.
| DEPTOR expression [n (%)] | |||||
|---|---|---|---|---|---|
|
|
|||||
| Characteristic | T < TA | T = TA | T > TA | All (%) | P |
| Patients (n) | 6 (11.8) | 31 (60.8) | 14 (27.5) | 51 | |
| Age (years), mean ± SD | 58.7 ± 18.0 | 60.0 ± 12.4 | 60.6 ± 15.0 | 60.0 ± 13.5 | |
| Sex | 0.186 | ||||
| Female | 3 (11.5) | 18 (50.0) | 4 (38.5) | 26 (100) | |
| Male | 3 (12.0) | 13 (72.0) | 10 (16.0) | 25 (100) | |
| Cirrhosis | 0.272 | ||||
| Negative | 4 (12.9) | 21 (67.7) | 6 (19.4) | 30 (100) | |
| Positive | 2 (10.0) | 11 (50.0) | 8 (40.0) | 21 (100) | |
| Viral infectiona | 0.020 | ||||
| NBNC | 5 (29.4) | 11 (64.7) | 1 (5.9) | 17 (100) | |
| HBV | 0 (0.0) | 9 (56.2) | 7 (43.8) | 16 (100) | |
| HCV | 1 (5.6) | 11 (61.1) | 6 (33.3) | 18 (100) | |
| Tumor type | 0.236 | ||||
| Solitary | 3 (8.3) | 21 (58.3) | 12 (33.4) | 36 (100) | |
| Multiple | 3 (20.0) | 10 (66.7) | 2 (13.3) | 15 (100) | |
| Vascular invasion | 0.328 | ||||
| Absent | 1 (4.3) | 15 (65.2) | 7 (30.5) | 23 (100) | |
| Vein invasion or vein tumor thrombosis | 5 (17.9) | 16 (57.1) | 7 (25.0) | 28 (100) | |
| TNM stageb (AJCC and UICC, 7th edition [46]) | 0.088 | ||||
| Early | 0 (0.0) | 14 (66.7) | 7 (33.3) | 21 (100) | |
| Late | 6 (20.0) | 17 (56.7) | 7 (23.3) | 30 (100) | |
HBV, HBV sAg (+); HCV, anti-HCV antibody (+); NBNC, HBV sAg (−) and anti-HCV antibodies (−).
Early stage, TNM stage = I; late stage, beyond TNM stage I (TNM stage = II + IIIA + IIIB + IIIC + IV).
Table 2.
Multiple logistic regression analysis of factors associated with DEPTOR overexpression in tumorous tissue.
| DEPTOR expression [n (%)] | |||||
|---|---|---|---|---|---|
|
|
|||||
| Variable | T ≤ TA | T > TA | Odds ratio | 95% CI | P |
| Sex | |||||
| Male | 21 (84.0) | 4 (16.0) | 0.32 | 0.08–1.35 | 0.121 |
| Female | 16 (61.5) | 10 (38.5) | 1.00 | ||
| TNM stagea | |||||
| Late | 23 (76.7) | 7 (23.3) | 0.64 | 0.15–2.75 | 0.552 |
| Early | 14 (66.7) | 7 (33.3) | 1.00 | ||
| Viral infectionb | |||||
| HBV | 9 (56.2) | 7 (43.8) | 10.29 | 1.05–11.29 | 0.046 |
| HCV | 12 (66.7) | 6 (33.3) | 6.65 | 0.64–68.85 | 0.112 |
| NBNC | 16 (94.1) | 1 (5.9) | 1.00 | ||
Early stage, TNM stage = I; late stage, beyond TNM stage I (TNM stage = II + IIIA + IIIB + IIIC + IV).
HBV, HBV sAg (+); HCV, anti-HCV antibody (+); NBNC, HBV sAg (−) and anti-HCV antibodies (−).
Regulation of mTOR/Raptor Signaling by DEPTOR in HuH-7 Cells
To elucidate the role of DEPTOR in the tumorigenesis of HCC, its expression was knocked down in HuH-7 cells by infection with lentiviruses carrying shRNAs targeted at DEPTOR. As shown in Figure 3A and Supplementary Figure 3, downregulation of DEPTOR resulted in activation of S6K and 4E-BP as well as in an increase in cell size. In addition, a reduction of Akt phosphorylation was also noted. Moreover, compared with the HuH-7-shLuc control cells, the proliferation rates of HuH-7-shDEPTOR-1 cells or HuH-7-shDEPTOR-2 cells decreased significantly (Figure 3B). Consistent with this observation, downregulation of DEPTOR in HA22T cells led to considerable reduction of growth rates (Supplementary Figure 4).
Figure 3.

The effects of DEPTOR on mTOR signaling in HuH-7 cells. (A) Knockdown of DEPTOR resulted in activation of S6K and a decrease of Akt phosphorylation. HuH-7 cells were infected with lentiviruses expressing shRNAs targeting DEPTOR or luciferase (shLuc). Cell lysates were analyzed by Western blotting for indicated proteins and phosphorylation states. P and T indicate phosphorylated and total protein, respectively. (B) DEPTOR knockdown in HuH-7 cells led to a decrease in proliferation. HuH-7 cells that bore shRNA against luciferase or DEPTOR were seeded on 48-well plates, and cell number at an indicated time point was evaluated by crystal violet staining. Data were normalized against OD595 values on d 1 of each treatment. Each experiment was performed in triplicate; error bars represent standard deviation (SD). **P < 0.01. (C) DEPTOR overexpression in HuH-7 cells caused an increase in the phosphorylation of Akt, whereas there was no obvious effect on the phosphorylation of S6K. (D) Transiently expressed DEPTOR in HuH-7 cells caused an increase in the phosphorylation of Akt, which could be counteracted by coexpression of GNMT or the N140S mutant GNMT. (E) DEPTOR overexpression extends survival of HuH-7 cells grown in medium containing 0.1% serum. GFP and DEPTOR stable cells were seeded on 48-well plates for 24 h (d 0); then cells were cultured in DMEM containing 0.1% serum. Cell proliferation was evaluated as described in (B). *P < 0.05; **P < 0.01. (F) Effects of DEPTOR and GNMT on apoptosis and autophagy in HuH-7 cells. Different HuH-7 stable cells were cultured in normal medium (10% serum) or in medium with 0.1% serum for 3 d and were then harvested and analyzed by Western blotting for indicated proteins. Each experiment was repeated three times. Panel B: ---○---, HuH-7-shLuc; —■—, HuH-7-shDEPTOR-1; — ▾— HuH-7-shDEPTOR-2; panel E: —○—, HuH-7-GFP; —●—, HuH-7-DEPTOR.
On the other hand, overexpression of DEPTOR in HuH-7 cells suppressed 4E-BP activation, whereas no obvious change was found in the phosphorylation of S6K (Figure 3C). However, a significant increase in Akt phosphorylation was observed in DEPTOR overexpressing HuH-7 cells (see Figure 3C). Interestingly, we showed that GNMT counteracted the effect of DEPTOR on the induction of Akt activation (Figure 3D). Furthermore, when a mutant N140S-GNMT, which possesses <0.5% enzymatic activity of wild-type GNMT (23,24), was coexpressed with DEPTOR, such a blockage effect still existed (see Figure 3D).
In addition, overexpression of DEPTOR increased the viability of HuH-7 cells significantly when they were cultured in a medium with only 0.1% fetal calf serum (FCS) (Figure 3E). Such an effect was not observed in cells cultured in a medium containing 10% FCS (Supplementary Figure 5). It is important to note that it did not matter if the cells were cultured with 10% or 0.1% FCS; there was no difference in the caspase 3 levels between HuH-7-DEPTOR and HuH-7-GFP control cells (Figure 3F). This result suggests that DEPTOR may extend cell survival through mechanism(s) other than inhibition of apoptosis. Because autophagy plays an important role for cell survival when cells are starved and it is negatively regulated by mTOR (8,25), we tested whether overexpression of DEPTOR activates autophagy in cells cultured in serum-depleted medium. The results showed that compared with the HuH-7-GFP cells, HuH-7-DEPTOR cells had significantly higher levels of both Beclin-1 and microtubule-associated protein 1 light chain 3 beta (LC3B) (see Figure 3F).
Effects of GNMT on mTOR/Raptor Downstream Signaling
Because GNMT is a DEPTOR-binding protein, we hypothesized that it is involved in the regulation of the mTOR signaling pathway. The results showed that overexpression of GNMT led to increases of both 4E-BP phosphorylation and cell size (Figure 4A and Supplementary Figure 6). In addition, overexpression of DEPTOR in HuH-7-GNMT stable cells resulted in the neutralization of the effect of GNMT on 4E-BP phosphorylation (see Figure 4A). Concerning the activation of autophagy, the amount of LC3B-I and -II in HuH-7-GNMT cells was significantly lower than in the HuH-7-GFP cells when the cells were cultured in medium containing only 0.1% FCS (Figure 3F). This result indicates that GNMT activates mTOR/raptor downstream signaling in HuH-7 cells.
Figure 4.

Effects of GNMT on mTOR signaling, cell cycle progression and proliferation. (A) Stably expression of GNMT in HuH-7 cells causes an increase in the phosphorylation of 4E-BP, which can be counteracted by transiently transfecting DEPTOR into a GNMT stable cell line. (B, C) GNMT does not interact with mTOR but interferes with the interaction between DEPTOR and mTOR. HuH-7 cells were transfected with indicated plasmids and harvested for immunoprecipitation analysis. Endogenous mTOR is not present in the anti-FLAG immunoprecipitants (B), and FLAG-tagged GNMT is not present in the precipitant pulled down by antibody against endogenous mTOR (C). In GNMT-expressing cells, the amount of DEPTOR in the mTOR precipitants is decreased and vice versa (C). (D) GNMT overexpression in HuH-7 cells led to a decrease in proliferation. GFP and GNMT stable cells were seeded on 48-well plate; cell proliferation was evaluated as described in Figure 3B. *P < 0.05. (E) GNMT overexpression in HuH-7 cells led to G2/M arrest. GFP and GNMT stable cells were synchronized at the G0/G1 phase and allowed to reenter the cell cycle. The cells were fixed at the indicated time point, and cell cycle profiles were recorded by flow cytometry. Quantification of the percentage of cells in the G2/M phase of the cell cycle is shown. Each experiment was repeated at least two times. (F) Expression of SA-β-gal in GFP and GNMT stable cells. Quantification of β-gal expression in GFP and GNMT stable cell lines are shown. Data are presented as means ± SD of 15–20 random fields with 100× magnification from two independent experiments. **P < 0.01. IP, immunoprecipitation; Ig H, IgG heavy chain; —○—, HuH-7-GFP; ----●----, HuH-7-GNMT.
Because it has been reported that DEPTOR binds to mTOR via its PDZ domain (9), we hypothesized that GNMT competes with mTOR for its binding with DEPTOR. Immunoprecipitation experiments demonstrated that GNMT and mTOR were not present in the same complex (Figure 4B, C). Moreover, in the cells overexpressing GNMT, the amount of mTOR decreased in the DEPTOR precipitants and vice versa (see Figure 4C). Therefore, GNMT activates mTOR/raptor downstream signaling via interrupting the interaction between mTOR and DEPTOR.
Effects of GNMT on Cell Cycle and Cell Proliferation
As shown in Figure 4D, the growth rate of HuH-7-GNMT cells was significantly slower than that of HuH-7-GFP cells. Similar results were also observed in HepG2 cells overexpressing GNMT (Supplementary Figure 7). Because it was reported that activation of mTOR causes cellular senescence in nonproliferating cells (26,27), we hypothesized that GNMT not only activates mTOR signaling, but also affects cell cycle progression, which results in cellular senescence and growth suppression. Cell cycle analysis showed that, 36 h after the cells entered the cell cycle, the percentages of cells in the G2/M phase for HuH-7-GNMT cells and HuH-7-GFP cells were 23.8% and 10.4%, respectively (Figure 4E). Furthermore, SA-β-gal assay demonstrated that HuH-7-GNMT cells had a significantly higher rate of positive staining than HuH-7-GFP cells (Figure 4F and Supplementary Figure 8).
GNMT Sensitizes HuH-7 Cells to Rapamycin Treatment
Because overexpression of GNMT delayed cell cycle progression, we decided to test whether overexpression of GNMT has any effect on HCC cells treated with the mTOR inhibitor rapamycin. The results of MTT assay revealed dose- responsive effects of rapamycin treatment in both HuH-7-GFP and HuH-7-GNMT cells (Figure 5A). In addition, compared with HuH-7-GFP cells treated with 4, 20 or 100 nmol/L rapamycin, the HuH-7-GNMT cells consistently had slower growth rates (see Figure 5A). In the presence of 4 nmol/L rapamycin, the viability of HuH-7-GNMT cells was significantly lower than that of HuH-7-GFP cells (Figure 5B).
Figure 5.

GNMT sensitizes HuH-7 cells to rapamycin treatment. (A, B) The effect of rapamycin on the growth of HuH-7-GFP and HuH-7-GNMT cells. Viable cells were measured by using an MTT assay. The cell viability curves were drawn by using data from d 5. The y axis represents the percentages of viable cells compared with the solvent control. **P < 0.01. Each experiment was repeated three times. (C) Effects of RAD001 on growth rates of HuH-7-GNMT and HuH-7-GFP cells in xenografts. Mean tumor volume ± standard error of the mean at a given time is shown. Treatment with RAD001 of tumors formed by inoculation of GNMT stable cells (HuH-7-GNMT + RAD001, n = 10) resulted in a significant reduction of tumor growth in comparison with the other three groups (HuH-7-GFP + RAD001, n = 8; HuH-7-GFP + placebo, n = 8; and HuH-7-GNMT + placebo, n = 5). *P < 0.05. (D) Representative photomicrographs of IHC analysis for proliferation marker Ki-67 in xenografts from (C) and quantification of Ki-67+ cells in different groups. Data are expressed as means ± SD. Means not sharing the same lowercase letters (a and c) are significantly different (P < 0.05) (bar = 100 μm). Panel A: ---○---, HuH-7-GFP + 0 (nmol/L of rapamycin);
 , HuH-7-GFP + 4;
, HuH-7-GFP + 4;
 , HuH-7-GFP + 20;
, HuH-7-GFP + 20;
 , HuH-7-GFP + 100; ●, HuH-7-GNMT + 0;
, HuH-7-GFP + 100; ●, HuH-7-GNMT + 0;
 , HuH-7-GNMT + 4;
, HuH-7-GNMT + 4;
 , HuH-7-GNMT + 20;
, HuH-7-GNMT + 20;
 , HuH-7-GNMT + 100; panel B: —○—, HuH-7-GFP; ---●---, HuH-7-GNMT; panel C: —○—, HuH-7-GFP + placebo;
, HuH-7-GNMT + 100; panel B: —○—, HuH-7-GFP; ---●---, HuH-7-GNMT; panel C: —○—, HuH-7-GFP + placebo;
 , HuH-7-GFP + RAD001;
, HuH-7-GFP + RAD001;
 , HuH-7-GNMT + placebo;
, HuH-7-GNMT + RAD001.
, HuH-7-GNMT + placebo;
, HuH-7-GNMT + RAD001.
The additive effect of GNMT to the rapamycin treatment was further tested in vivo by using a xenograft model. After being inoculated with either HuH-7-GNMT or HuH-7-GFP cells for 1 wk, the mice were treated with either RAD001 (a soluble derivative of rapamycin) or the drug vehicle (the placebo group). The results showed that compared with the tumors formed from HuH-7-GFP cells, overexpression of GNMT reduced 23% of tumor growth (Figure 5C). Compared to HuH-7-GFP tumors that received placebo, treated HuH-7-GFP tumors with RAD001 resulted in 37% reduction of tumor growth. Importantly, RAD001 treatment of HuH-7-GNMT tumors achieved better tumor shrinkage (53% reduction; see Figure 5C). Furthermore, IHC staining with anti–Ki-67 antibody showed that both GNMT overexpression and RAD001 treatment could lead to the downregulation of Ki-67 expression in the xenograft tumors, and it seems that they have additive effects to such down-regulation (Figure 5D).
DISCUSSION
In this study, we identified DEPTOR as a GNMT binding protein and confirmed that they interact with each other directly by using different methods, including coimmunoprecipitation and FRET-AB assays (Figure 1). It is important to note that GNMT uses its C-terminal domain to bind the PDZ domain of DEPTOR. Because GNMT forms dimmers or tetramers via its N-terminal domain (28), the interaction with DEPTOR should not hinder its dimmer/tetramer formation. In contrast, the interaction between GNMT and DEPTOR may interfere with DEPTOR-mTOR interaction, since the DEPTOR uses its PDZ domain to bind mTOR (9).
Previously, Peterson et al. (9) reported that DEPTOR was overexpressed abundantly in a subset of multiple myelomas with cyclin D1/D3 or c-MAF/MAFB translocations, whereas it was downregulated in most of the cancer that they tested. However, these authors did not study the expression of DEPTOR in HCC. In the present study, we found that 27.5% of tumorous tissues from HCC patients have overexpression of DEPTOR (see Table 1). Moreover, HBV infection is significantly associated with the overexpression of DEPTOR in HCC. It was reported that HBV DNA is integrated into chromosomes of the host cells, which results in a wide range of genetic alterations. Such mechanism has been proposed to play an important role in the hepatocarcinogenesis (29). The integration of viral DNA was observed within genes that are important for cell growth, such as the cyclin A gene (30), the retinoic acid receptor β gene (31) and the human telomerase reverse transcriptase gene (32). In addition, the hepatitis B virus X protein (HBx) was shown to function as a transcriptional transactivator of various cellular genes associated with growth control (29). HBx also decreases proteasomal degradation of β-catenin, which in turn increases the expression of its downstream targets c-myc and cyclin D1 (33). Therefore, it is possible that HBV DNA integration and the regulatory protein HBx are involved in the upregulation of DEPTOR in HBV-associated HCC.
In this study, the loss-of-function experiment indicated that the function of DEPTOR in the mTOR pathway in the HCC cells is similar to that in multiple myeloma cells (9). It was reported that in multiple myeloma, a plasma cell malignancy, high-level synthesis of secretary proteins makes them more susceptible to endoplasmic reticulum (ER) stress than other types of cells (34). The suppression of mTOR/raptor signaling by overexpression of DEPTOR resulted in an inhibition of protein synthesis and, subsequently, the reduction of ER stress (9). Because liver plays the dominate role in plasma proteins production (35), the mechanism mentioned above may be applicable to hepatocytes as well. In addition, HBV infection was reported to induce ER stress in hepatocytes (36,37), and it remains to be determined whether viral infection has a direct effect on DEPTOR activation or whether DEPTOR activation is a cellular defense mechanisms against HBV infection.
Chronic HBV infection was shown to increase the risk of liver cirrhosis (38). However, we did not found any correlation between DEPTOR overexpression and liver cirrhosis in this study. This result may be because of the limitation of the sample size. Further study with a larger sample size is needed to elucidate the association between DEPTOR overexpression and different clinical features of HCC.
It is important to note that despite increased Akt phosphorylation was observed when DEPTOR was overexpressed in HuH-7 cells, S6K phosphorylation was not suppressed significantly. This result suggests that activation of Akt by DEPTOR may be through a pathway other than the feedback inhibitory loop from S6K to PI3K in HuH-7 cells. This outcome is distinctively different from the phenomenon found in multiple myeloma cells. Recently, we found that DEPTOR interacts with phosphatidylinositol-3,4,5-trisphosphate-dependent Rac exchange factor 2 (P-Rex2), which was reported to be an inhibitor of phosphatase and tensin homolog (PTEN) (39). In addition, knocking down of P-Rex2 expression in HuH-7 cells abrogated Akt activation induced by DEPTOR (C-H Li, personal communication). Therefore, DEPTOR activates Akt through other mechanisms.
In addition, our results also indicate that, besides mTOR, there might be other kinases that are capable of phosphorylating S6K in hepatocytes. Consistent with this observation, it was reported that rapamycin dramatically decreases the phosphorylation of 4E-BP, but it has little effect on the phosphorylation of S6K in HuH-7 cells (40). Previously, Belham et al. (41) identified NIMA (never in mitosis gene a)–related kinase 6 (NEK6) and NEK7 as the major kinases responsible for the phosphorylation of hydrophobic regulatory sites of S6K in rat liver (41). They demonstrated that NEK6 phosphorylates and activates S6K in vitro and in vivo. Although there was some controversy (42), these results do not rule out the possibility that activation of S6K could be regulated by multiple mechanisms, especially in a major secretary organ such as the liver.
In this study, we demonstrated that in addition to participating in the mTOR signaling pathway through interacting with DEPTOR, GNMT counteracts DEPTOR-induced Akt activation in HuH-7 cells. Moreover, the N140S mutant of GNMT also possesses such a blockage effect (Figure 3D). It was reported that an N140S mutant of GNMT lost 99.5% of enzyme activity, while still possessing almost identical quaternary, tertiary and secondary structures as the wild-type GNMT (24). Therefore, the regulatory function of GNMT on these signaling cascades is not associated with its enzyme activity. In addition, we demonstrated that overexpression of GNMT leads to G2/M arrest of the cell cycle. It is plausible that GNMT participates in various biological functions through interacting with different proteins. Studies on the role that GNMT plays in cell cycle control are currently under investigation.
CONCLUSION
The application of the multitargeted kinase inhibitor sorafenib in the clinical management of patients with HCC represents a breakthrough in translational medicine (2,3). However, its benefits are modest and only occur in select patients (2). Currently, many clinical trials by using mTOR inhibitors alone or in combination with other molecular targeting agents are taking place (2,43,44). To optimize these treatments, more studies are needed to understand the network of mTOR signaling. In this study, we demonstrate that GNMT overexpression reduces tumor growth in vivo, which is consisting with the in vitro data. Importantly, combination of rapamycin and GNMT overexpression showed an additive anticancer effect. Because the haplotypes and phenotypes of GNMT have been characterized (45), such information may serve as a predictive marker for the responsiveness of HCC patients to rapamycin treatment. Further studies are needed to elucidate whether GNMT could serve as a therapeutic agent or as a sensitizer for other molecular targeting agents.
Supplemental Data
ACKNOWLEDGMENTS
We thank TLCN for providing the HCC tissue samples and related clinical data. This network currently includes five major medical centers (National Taiwan University Hospital, Chang-Gung Memorial Hospital-Linko, Veteran General Hospital-Taichung, Chang-Gung Memorial Hospital-Kaohsiung and Veteran General Hospital-Kaohsiung). TLCN was supported by grants from the National Science Council since 2005 (NSC 100-2325-B-182-006) and the National Health Research Institutes, Taiwan. In addition, we thank the Sequencing Core Facility of the National Yang-Ming University Genome Research Center and the Confocal Microscope Lab of National Taiwan University for technical support and for assistance in the FRET experiments. This work was supported in part by grants from the National Science Council of the Republic of China (National Research Program for Biopharmaceuticals [NRPB], grant NSC100-2325-B-010-008) and the Ministry of Education of the Republic of China (Aim for the Top University Plan).
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
REFERENCES
- 1.El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–76. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 2.Finn RS. Development of molecularly targeted therapies in hepatocellular carcinoma: where do we go now? Clin Cancer Res. 2010;16:390–7. doi: 10.1158/1078-0432.CCR-09-2084. [DOI] [PubMed] [Google Scholar]
- 3.Llovet JM, Bruix J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology. 2008;48:1312–27. doi: 10.1002/hep.22506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sahin F, et al. mTOR and P70 S6 kinase expression in primary liver neoplasms. Clin Cancer Res. 2004;10:8421–5. doi: 10.1158/1078-0432.CCR-04-0941. [DOI] [PubMed] [Google Scholar]
- 5.Villanueva A, et al. Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology. 2008;135:1972–83. doi: 10.1053/j.gastro.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lane HA, Breuleux M. Optimal targeting of the mTORC1 kinase in human cancer. Curr Opin Cell Biol. 2009;21:219–29. doi: 10.1016/j.ceb.2009.01.016. [DOI] [PubMed] [Google Scholar]
- 7.Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729–34. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- 8.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–84. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 9.Peterson TR, et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137:873–86. doi: 10.1016/j.cell.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liao YJ, et al. Characterization of a glycine N-methyltransferase gene knockout mouse model for hepatocellular carcinoma: implications of the gender disparity in liver cancer susceptibility. Int J Cancer. 2009;124:816–26. doi: 10.1002/ijc.23979. [DOI] [PubMed] [Google Scholar]
- 11.Martinez-Chantar ML, et al. Loss of the glycine N-methyltransferase gene leads to steatosis and hepatocellular carcinoma in mice. Hepatology. 2008;47:1191–9. doi: 10.1002/hep.22159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ogawa H, Fujioka M. Purification and properties of glycine N-methyltransferase from rat liver. J Biol Chem. 1982;257:3447–52. [PubMed] [Google Scholar]
- 13.Cook RJ, Wagner C. Glycine N-methyltransferase is a folate binding protein of rat liver cytosol. Proc Natl Acad Sci U S A. 1984;81:3631–4. doi: 10.1073/pnas.81.12.3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhat R, Bresnick E. Glycine N-methyltransferase is an example of functional diversity: role as a polycyclic aromatic hydrocarbon-binding receptor. J Biol Chem. 1997;272:21221–6. doi: 10.1074/jbc.272.34.21221. [DOI] [PubMed] [Google Scholar]
- 15.Chen SY, et al. Glycine N-methyltransferase tumor susceptibility gene in the benzo(a)pyrene-detoxification pathway. Cancer Res. 2004;64:3617–23. doi: 10.1158/0008-5472.CAN-03-3726. [DOI] [PubMed] [Google Scholar]
- 16.Yen CH, et al. Glycine N-methyltransferase affects the metabolism of aflatoxin B1 and blocks its carcinogenic effect. Toxicol Appl Pharmacol. 2009;235:296–304. doi: 10.1016/j.taap.2008.12.013. [DOI] [PubMed] [Google Scholar]
- 17.Chen YM, et al. Genomic structure, expression, and chromosomal localization of the human glycine N-methyltransferase gene. Genomics. 2000;66:43–7. doi: 10.1006/geno.2000.6188. [DOI] [PubMed] [Google Scholar]
- 18.Liu HH, et al. Characterization of reduced expression of glycine N-methyltransferase in cancerous hepatic tissues using two newly developed monoclonal antibodies. J Biomed Sci. 2003;10:87–97. doi: 10.1007/BF02256001. [DOI] [PubMed] [Google Scholar]
- 19.Jacinto E, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122–8. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- 20.Yan Y, Marriott G. Analysis of protein interactions using fluorescence technologies. Curr Opin Chem Biol. 2003;7:635–40. doi: 10.1016/j.cbpa.2003.08.017. [DOI] [PubMed] [Google Scholar]
- 21.Carmichael J, DeGraff WG, Gazdar AF, Minna JD, Mitchell JB. Evaluation of a tetrazolium-based semiautomated colorimetric assay: assessment of radiosensitivity. Cancer Res. 1987;47:943–6. [PubMed] [Google Scholar]
- 22.Dimri GP, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92:9363–7. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Augoustides-Savvopoulou P, et al. Glycine N-methyltransferase deficiency: a new patient with a novel mutation. J Inherit Metab Dis. 2003;26:745–59. doi: 10.1023/B:BOLI.0000009978.17777.33. [DOI] [PubMed] [Google Scholar]
- 24.Luka Z, Wagner C. Effect of naturally occurring mutations in human glycine N-methyltransferase on activity and conformation. Biochem Biophys Res Commun. 2003;312:1067–72. doi: 10.1016/j.bbrc.2003.11.037. [DOI] [PubMed] [Google Scholar]
- 25.Baehrecke EH. Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol. 2005;6:505–10. doi: 10.1038/nrm1666. [DOI] [PubMed] [Google Scholar]
- 26.Demidenko ZN, Blagosklonny MV. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle. 2008;7:3355–61. doi: 10.4161/cc.7.21.6919. [DOI] [PubMed] [Google Scholar]
- 27.Ruggero D, et al. The translation factor eIF-4E promotes tumor formation and cooperates with c-Myc in lymphomagenesis. Nat Med. 2004;10:484–6. doi: 10.1038/nm1042. [DOI] [PubMed] [Google Scholar]
- 28.Fu Z, et al. Crystal structure of glycine N-methyltransferase from rat liver. Biochemistry (Mosc) 1996;35:11985–93. doi: 10.1021/bi961068n. [DOI] [PubMed] [Google Scholar]
- 29.Tsai WL, Chung RT. Viral hepatocarcinogenesis. Oncogene. 2010;29:2309–24. doi: 10.1038/onc.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang J, Chenivesse X, Henglein B, Brechot C. Hepatitis B virus integration in a cyclin A gene in a hepatocellular carcinoma. Nature. 1990;343:555–7. doi: 10.1038/343555a0. [DOI] [PubMed] [Google Scholar]
- 31.Yaginuma K, et al. Multiple integration site of hepatitis B virus DNA in hepatocellular carcinoma and chronic active hepatitis tissues from children. J Virol. 1987;61:1808–13. doi: 10.1128/jvi.61.6.1808-1813.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Horikawa I, Barrett JC. cis-Activation of the human telomerase gene (hTERT) by the hepatitis B virus genome. J Natl Cancer Inst. 2001;93:1171–3. doi: 10.1093/jnci/93.15.1171. [DOI] [PubMed] [Google Scholar]
- 33.Ding Q, et al. Erk associates with and primes GSK-3beta for its inactivation resulting in upregulation of beta-catenin. Mol Cell. 2005;19:159–70. doi: 10.1016/j.molcel.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 34.Lee AH, Iwakoshi NN, Anderson KC, Glimcher LH. Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc Natl Acad Sci U S A. 2003;100:9946–51. doi: 10.1073/pnas.1334037100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miller LL, Bly CG, Watson ML, Bale WF. The dominant role of the liver in plasma protein synthesis; a direct study of the isolated perfused rat liver with the aid of lysine-epsilon-C14. J Exp Med. 1951;94:431–53. doi: 10.1084/jem.94.5.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ji C, Kaplowitz N. ER stress: can the liver cope? J Hepatol. 2006;45:321–33. doi: 10.1016/j.jhep.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 37.Wang HC, et al. Different types of ground glass hepatocytes in chronic hepatitis B virus infection contain specific pre-S mutants that may induce endoplasmic reticulum stress. Am J Pathol. 2003;163:2441–9. doi: 10.1016/S0002-9440(10)63599-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liaw YF, Chu CM. Hepatitis B virus infection. Lancet. 2009;373:582–92. doi: 10.1016/S0140-6736(09)60207-5. [DOI] [PubMed] [Google Scholar]
- 39.Fine B, et al. Activation of the PI3K pathway in cancer through inhibition of PTEN by exchange factor P-REX2a. Science. 2009;325:1261–5. doi: 10.1126/science.1173569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jasinghe VJ, et al. J Hepatol. Vol. 49. ABT; 2008. -869, a multi-targeted tyrosine kinase inhibitor, in combination with rapamycin is effective for subcutaneous hepatocellular carcinoma xenograft; pp. 985–97. [DOI] [PubMed] [Google Scholar]
- 41.Belham C, Comb MJ, Avruch J. Identification of the NIMA family kinases NEK6/7 as regulators of the p70 ribosomal S6 kinase. Curr Biol. 2001;11:1155–67. doi: 10.1016/s0960-9822(01)00369-4. [DOI] [PubMed] [Google Scholar]
- 42.Lizcano JM, et al. Molecular basis for the substrate specificity of NIMA-related kinase-6 (NEK6): evidence that NEK6 does not phosphorylate the hydrophobic motif of ribosomal S6 protein kinase and serum-and glucocorticoid-induced protein kinase in vivo. J Biol Chem. 2002;277:27839–49. doi: 10.1074/jbc.M202042200. [DOI] [PubMed] [Google Scholar]
- 43.Rao RD, Buckner JC, Sarkaria JN. Mammalian target of rapamycin (mTOR) inhibitors as anti-cancer agents. Curr Cancer Drug Targets. 2004;4:621–35. doi: 10.2174/1568009043332718. [DOI] [PubMed] [Google Scholar]
- 44.Carr BI, Kralian S. Molecular Targeted Therapies for HCC. In: Carr BI, editor. Hepatocellular Carcinoma. Humana Press; New York: 2010. pp. 589–614. [Google Scholar]
- 45.Huang YC, et al. Haplotypes, loss of heterozygosity, and expression levels of glycine N-methyltransferase in prostate cancer. Clin Cancer Res. 2007;13:1412–20. doi: 10.1158/1078-0432.CCR-06-1551. [DOI] [PubMed] [Google Scholar]
- 46.Edge SB, Compton CC. The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol. 2010;17:1471–4. doi: 10.1245/s10434-010-0985-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.