

Abstract
STAT5 proteins are constitutively activated in malignant cells from many patients with leukemia, including the myeloproliferative neoplasms (MPNs) chronic myeloid leukemia (CML) and polycythemia vera (PV), but whether STAT5 is essential for the pathogenesis of these diseases is not known. In the present study, we used mice with a conditional null mutation in the Stat5a/b gene locus to determine the requirement for STAT5 in MPNs induced by BCR-ABL1 and JAK2V617F in retroviral transplantation models of CML and PV. Loss of one Stat5a/b allele resulted in a decrease in BCR-ABL1–induced CML-like MPN and the appearance of B-cell acute lymphoblastic leukemia, whereas complete deletion of Stat5a/b prevented the development of leukemia in primary recipients. However, BCR-ABL1 was expressed and active in Stat5-null leukemic stem cells, and Stat5 deletion did not prevent progression to lymphoid blast crisis or abolish established B-cell acute lymphoblastic leukemia. JAK2V617F failed to induce polycythemia in recipients after deletion of Stat5a/b, although the loss of STAT5 did not prevent the development of myelofibrosis. These results demonstrate that STAT5a/b is essential for the induction of CML-like leukemia by BCR-ABL1 and of polycythemia by JAK2V617F, and validate STAT5a/b and the genes they regulate as targets for therapy in these MPNs.
Introduction
The STAT5 proteins STAT5a and STAT5b are closely related transcription factors that are activated by JAK kinases in response to a broad range of cytokines, including IL-2, IL-3, IL-4, IL-7, and IL-15; G-CSF and GM-CSF; erythropoietin and thrombopoietin; and growth hormone and prolactin.1 Genetic studies indicate that mouse STAT5a/b has pleiotropic functions in lymphohematopoiesis,2 lactation,3 and metabolism.4 The first mice with targeted mutations in both the Stat5a and Stat5b genes,2 which are closely linked on mouse chromosome 11, produced N-terminally truncated STAT5a/b proteins (Stat5a/bΔN) that retained some DNA-binding and transactivation functions.5 Adult Stat5a/bΔN/ΔN mice have normal baseline hematopoiesis, modest lymphopenia and T-cell proliferative defects,6 and decreased hematopoietic stem cell (HSC)–repopulating activity.7,8 Subsequently, a conditional Stat5 allele with loxP sites flanking the entire 110-kb Stat5a/b locus (Stat5a/bfl) was created, and a true Stat5a/b-null allele produced by mating Stat5fl/+ mice with MMTV-Cre–transgenic mice.9 Stat5a/b-null (Stat5a/b−/−) mice lack CD8+ and γ/δ T cells, exhibit a block in B-lymphoid development at the pre–pro-B-cell stage,10–12 and the majority die in the perinatal period because of severe anemia and lung abnormalities.9,10,13 Whereas the frequency of phenotypic (c-Kit+Lin−Sca-1+CD150+) HSCs is relatively normal in Stat5a/b−/− fetal liver and BM,11,14 in transplantation assays, Stat5-null HSCs have severe defects in reconstituting lymphoid compartments, with more modest defects in the myeloerythroid repopulation.11,12,14
Constitutive activation of human STAT5 has been found in many hematologic malignancies,15 including acute leukemias and chronic myeloproliferative neoplasms (MPNs). STAT5 activation was reported more than a decade ago in BCR-ABL1–expressing myeloid and lymphoid leukemia cells,16,17 and more recently in Philadelphia chromosome–negative MPNs associated with the JAK2V617F mutation, including polycythemia vera (PV) and essential thrombocythemia.18,19 Whereas therapy with tyrosine kinase inhibitors such as imatinib mesylate has revolutionized the treatment of chronic myeloid leukemia (CML), the emergence of clinical resistance to imatinib has rekindled interest in STAT5 as a promising therapeutic target downstream of BCR-ABL1.20 In support of this strategy, inhibition of STAT5 by dominant-negative mutants impairs the survival of BCR-ABL1–expressing cell lines,21 whereas siRNA against STAT5 inhibits myeloid colony formation by primary CML progenitors.22 Moreover, constitutively active mutants of STAT5a induce MPN-like leukemia when expressed in primary mouse hematopoietic cells,5 and produce erythropoietin-independent colonies, the hallmark of PV, in human erythroid progenitors.23 When BCR-ABL1 (p210 isoform) was expressed by retroviral transduction in BM from Stat5a-deficient donors and transplanted into irradiated syngeneic hosts, recipients had a reduced incidence of CML-like MPN, accompanied by a relative increase in the development of B-cell acute lymphoblastic leukemia (B-ALL).24 In contrast, when Stat5a/b-null fetal liver progenitors were transduced by Abelson murine leukemia virus or retrovirus expressing the p185 isoform of BCR-ABL1, there were no transformed pre-B-lymphoid colonies detected in vitro, and no leukemias developed from these donor cells after transfer to immunodeficient (Rag2−/−) mice.10 Whereas these studies suggest a role for STAT5a and STAT5b in transformation and leukemogenesis by dysregulated ABL1 kinases, the role of STAT5 in myeloid and lymphoid leukemogenesis by BCR-ABL1 and in the pathogenesis of MPN associated with JAK2V617F remains unclear.
In the present study, we investigated the role of STAT5a/b in transformation and leukemogenesis by the human BCR-ABL1 and mouse JAK2V617F tyrosine kinases using a genetic approach. We demonstrate that STAT5a/b is necessary for the induction of CML-like disease by BCR-ABL1 and of polycythemia by JAK2V617F, whereas STAT5 deficiency attenuates but does not abolish JAK2V617F-induced myelofibrosis. Overall, our results demonstrate that STAT5a/b plays a critical role in the pathogenesis of BCR-ABL1– and JAK2V617F-induced MPNs, and validate the STAT5 pathway as a target for therapy in MPNs associated with dysregulated tyrosine kinases.
Methods
Mice
Stat5a/bfl/+ and Stat5a/bfl/− mice in a congenic Balb/c background were genotyped by Southern blotting and by PCR as described previously.9 Transgenic Balb/c Mx-Cre mice were the kind gift of Dr Paul Ney (St Jude Children's Research Hospital, Memphis, TN).25
BM transduction and transplantation
Replication-defective ecotropic retroviral stocks were generated by transient transfection of 293 cells using the kat packaging system and stocks matched for titer by transduction of NIH3T3 cells, as described previously.26 For the generation of CML-like leukemia and JAK2V617F-induced MPN, we collected BM from Stat5a/bfl/+ and Stat5a/bfl/− Balb/c mice 4 days after IV administration of 150 mg/kg of 5-fluorouracil (5-FU), transduced cells with retrovirus, and injected 5 × 105 cells IV into lethally irradiated (900 cGy) Balb/c recipients (The Jackson Laboratory). Secondary transplantations were performed by injecting 3 × 106 whole BM cells from primary recipients into pairs of lethally irradiated secondary recipients. For the induction of the Mx-Cre transgene, recipients were injected with 250 μg of polyinosinic-polycytidylic acid (pIpC; Invitrogen) IP every other day for 4 doses beginning day 10 after transplantation, as described previously.27 The clinical features and histopathology of BCR-ABL1–induced CML-like disease, B-lymphoid leukemia, and histiocytic sarcoma have been described previously.28 All mouse experiments were approved by the Institutional Animal Use and Care Committee of Tufts Medical Center.
B-lymphoid transformation and leukemogenesis
For analysis of transformation and leukemogenesis in primary B-lymphoid progenitors, BM from Stat5a/bfl/+ and Stat5a/bfl/− donors not pretreated with 5-FU were subjected to a single round of retroviral transduction with p210MIGFP or p210MIGFPCre retrovirus, then plated for in vitro growth at serial dilutions in Whitlock-Witte–style cultures,29 plated in soft agar for colony assays,30 or injected directly into lethally irradiated recipient mice to assess lymphoid leukemogenesis.28 In other experiments, BM from Mx-Cre;Stat5a/bfl/+ and Mx-Cre;Stat5a/bfl/− donors not pretreated with 5-FU were transduced with p201MIGFP retrovirus and expanded on autologous BM stroma in Whitlock-Witte–style cultures. Stromal-independent populations of BCR-ABL1–transformed B-lymphoid progenitors were then treated in vitro with IFN-β (100 U/mL; Pestka Biomedical Laboratories) or injected into nonirradiated congenic Balb/c Rag2−/− recipients (1 × 107 cells intravenously). After the development of B-ALL (as assessed by presence of GFP+CD19+ lymphoblasts in the peripheral blood [PB]), a subset of recipients were treated with 250 μg of pIpC intraperitoneally every other day for 4 doses.
Southern blot analysis
To analyze Stat5 recombination, we digested genomic DNA from leukemic tissues with BamHI, transferred DNA to nylon membranes, and hybridized with a radioactively labeled probe generated from the mouse Stat5 gene by PCR with primers Rec2f (5′-CCAGAAGGGGTGCAAATGAGTC-3′) and Rec3r2 (5′-TGGTGCAGTGTAGGTTGAGGCT-3′), which allows the simultaneous detection of the Stat5a/b wt, Stat5a/b fl, and Stat5a/b rec alleles in the same sample. For each sample, the ratio of intensity of the recombined Stat5 band to the sum of the recombined and floxed Stat5 bands (= x) was measured. The efficacy of recombination of the floxed Stat5 allele (as a percentage) is then equal to 100x for Stat5a/bfl/+ donors and 2(100x − 50) for Stat5a/bfl/− donors. The percentage of residual Stat5 gene is equal to 50 + 50(1 − x) for Stat5a/bfl/+ donors and 100(1 − x) for Stat5a/bfl/− donors. The percentage of total Stat5a/b deficiency is given by (100x)/2 for Stat5a/bfl/+ donors and 100x for Stat5a/bfl/− donors.
Results
Stat5a/b deficiency attenuates induction of CML-like MPN by BCR-ABL1
To address the role of STAT5a/b in MPN pathogenesis, we used donor mice with a targeted mutation that places loxP sites flanking the 110-kb Stat5a/b gene locus (Stat5a/bfl). We used 2 distinct methods to express Cre recombinase in the mutant hematopoietic cells: retroviral expression of a green fluorescent protein (GFP)–Cre fusion protein and inducible expression from an Mx-Cre transgene. For the former approach, we replaced the GFP gene in the retroviral vector pMIGR131 with a cDNA encoding GFP fused via the COOH-terminus to humanized Cre.32 A cDNA encoding the p210 (b3a2) isoform of BCR-ABL1 was then cloned 5′ of the internal ribosome entry site to yield the vector p210-MSCV-IRES-GFPCre (p210MIGFPCre; supplemental Figure 1A, available on the Blood Web site; see the Supplemental Materials link at the top of the online article). To test this vector, we transduced BM from wild-type donor mice with stocks of p210MIGFPCre or p210MIGFP virus that were matched for titer and transplanted the cells into irradiated wild-type recipient mice. Both viruses efficiently induced CML-like MPN in recipients (supplemental Figure 1B), although the overall survival was slightly longer in recipients of p210MIGFPCre-transduced BM. The malignant myeloid cells were uniformly GFP+ in both cohorts (data not shown). Although sustained expression of Cre recombinase in mammalian cells can result in growth arrest and cytotoxicity,33 these results demonstrate that coexpression of p210 and GFPCre in mouse HSCs can efficiently induce CML-like MPN in recipients.
We then transduced BM from Stat5a/bfl/+ and Stat5a/bfl/− donor mice with p210MIGFP or p210MIGFPCre retrovirus and transplanted the cells into irradiated wild-type recipients. Recipients of p210MIGFP-transduced Stat5a/bfl/+ BM developed fatal CML-like MPN within 25 days of transplantation, characterized by leukocytosis with maturing myeloid cells and hepatosplenomegaly with parenchymal pulmonary hemorrhaging (Figure 1). The latency and histopathology of this disease were identical to that reported previously using this retroviral transduction/transplantation model system.28 In contrast, recipients of p210MIGFP-transduced Stat5a/bfl/− BM or p210MIGFPCre-transduced Stat5a/bfl/+ BM had similar survival that was significantly prolonged relative to the p210MIGFP-Stat5a/bfl/+ cohort (median survival, approximately 50 days; Figure 1A). Several recipients in these cohorts developed mixed disease, with simultaneous MPN and B-ALL characterized by lymphadenopathy and a malignant pleural effusion composed of B220+BP-1+ pre-B lymphoblasts, whereas others succumbed to histiocytic sarcoma. Interestingly, mutations in BCR-ABL1 (such as deletion of the Src homology 2 domain) that decrease ABL1 tyrosine kinase activity also resulted in mixed MPN and B-ALL in this transplantation model,28 suggesting that haploinsufficiency of Stat5a/b attenuates the induction of CML-like MPN by BCR-ABL1. A final cohort of 11 recipients received transplantations with Stat5a/bfl/− BM transduced with p210MIGFPCre. Only one mouse in this cohort developed mixed MPN and B-ALL; 5 recipients succumbed to B-ALL without evidence of MPN and 5 additional recipients did not develop any hematologic disease (Figure 1) and had no circulating GFP+ cells after day 60 (data not shown).
Figure 1.

Reduction in Stat5 gene dosage attenuates BCR-ABL1–induced CML-like MPN. Kaplan-Meier survival curve for recipients of Stat5a/bfl/+ (n = 2) or Stat5a/bfl/− (n = 4) BM transduced with p210MIGFP retrovirus, and recipients of Stat5a/bfl/+ (n = 8) or Stat5a/bfl/− (n = 11) BM transduced with p210MIGFPCre retrovirus is shown. The symbols indicate individual recipient mice, with the disease phenotype of each designated by the shading: black, CML-like MPN; white, B-ALL; black and white, mixed CML/B-ALL; gray, histiocytic sarcoma (HS). Relative to the p210MIGFP→Stat5a/bfl/+ cohort, the survival of the p210MIGFP→Stat5a/bfl/− and p210MIGFPCre→Stat5a/bfl/+cohorts was significantly prolonged (P = .017 and P = .0009, respectively, by Mantel-Cox test). Relative to the p210MIGFPCre→Stat5a/bfl/+ cohort, the survival of the p210MIGFPCre→Stat5a/bfl/− cohort was also significantly prolonged (P = .008).
We assessed the efficacy of recombination mediated by GFPCre by Southern blot analysis of genomic DNA from leukemic tissues using a mouse Stat5 probe that simultaneously detects the Stat5a/b wild-type, floxed, and recombined alleles. In hematopoietic cells from recipients of p210MIGFPCre-transduced Stat5a/bfl/+ BM, recombination of the floxed Stat5a/b allele was efficient, ranging from approximately 50% to near 100% in the BM (supplemental Figure 1C). The leukemic cells were derived from 2-3 distinct retrovirally transduced HSC clones and contained the BCR-ABL1 gene. The prolonged survival and mixed hematopoietic disease in this cohort confirms that haploinsufficiency for Stat5 attenuates BCR-ABL1–induced MPN. In recipients of p210MIGFPCre-transduced Stat5a/bfl/− BM, the recombination efficiency was more variable, but in some tissues approached 90% (supplemental Figure 1D). Interestingly, the single mouse in this cohort (#14) that developed mixed CML/B-ALL disease had the lowest recombination efficiency (approximately 4% in the BM). The recipients in this cohort that did not develop hematologic disease (#9-13) did not engraft with donor-derived HSCs, as demonstrated by the absence of the floxed Stat5a/b allele. These results indicate that reduction in Stat5a/b gene dosage in HSCs significantly attenuates the CML-like MPN induced by BCR-ABL1, but has less of an effect on B-lymphoid leukemogenesis.
Absence of Stat5a/b abolishes CML-like MPN induced by BCR-ABL1
As an alternate approach to expressing Cre recombinase in HSCs, we crossed Stat5a/b mutant mice with Mx-Cre–transgenic mice in which Cre is expressed from an IFN-inducible promoter, and transduced BM from Mx-Cre;Stat5a/bfl/+ and Mx-Cre;Stat5a/bfl/− donors with p210MIGFP retrovirus. After transplantation into irradiated wild-type recipient mice, some mice in each cohort were injected with pIpC to induce Cre expression in hematopoietic stem-progenitor cells.27 Recipients of p210MIGFP-transduced Mx-Cre;Stat5a/bfl/+ BM mice that were pIpC treated (n = 7) all reconstituted with donor-derived BCR-ABL1+ HSCs and developed mixed MPN/B-ALL disease (data not shown), confirming that pIpC treatment (and induction of a systemic IFN response) does not attenuate or abolish BCR-ABL1 leukemogenic activity in irradiated recipient mice.27
When BM from Mx-Cre;Stat5a/bfl/− donors was used for transduction by p210MIGFP, recipients that were not pIpC treated developed mixed CML/B-ALL disease within 80 days after transplantation. In contrast, recipients injected with pIpC remained healthy and showed no signs of hematologic illness (Figure 2A). At approximately 2 months after transplantation, these mice had normal PB leukocyte counts (Figure 2B), in contrast to the other cohorts, which manifested the leukocytosis characteristic of MPN. However, these recipients did have significant populations of circulating GFP+ myeloid cells, indicative of engraftment with BCR-ABL1+ stem cells (Figure 2C). Analysis of genomic DNA from PB leukocytes demonstrated very efficient recombination of the floxed Stat5a/b allele in all pIpC-treated recipients (Figure 2D). Mice in the Mx-Cre;Stat5a/bfl/− + pIpC cohort were killed at day 100 and found to have normal spleen weight (Figure 2E) and organ histopathology (data not shown). However, populations of GFP+ myeloid and lymphoid cells were present in BM and spleen, and Western blot analysis of protein extracts from these tissues demonstrated the presence of phospho–BCR-ABL1 and phospho-CrkL, but the absence of phospho-STAT5 (Figure 2F), demonstrating that the BCR-ABL1 kinase was expressed and active in hematopoietic cells from these mice. Analysis of genomic DNA from the BM of pIpC-treated recipients confirmed efficient (88%-96%) recombination of the floxed Stat5a/b allele (supplemental Figure 2). These results demonstrate that BCR-ABL1 can be expressed in Stat5-deficient HSCs and activate signaling, but cannot induce the dramatic expansion of progenitors and differentiated myeloid cells that is characteristic of CML in the absence of STAT5.
Figure 2.

Mx-Cre–mediated deletion of Stat5a/b abolishes CML-like MPN induced by BCR-ABL1. (A) Kaplan-Meier survival curve for recipients of p210MIGFP-transduced BM from Mx-Cre;Stat5a/bfl/+ donors (solid line), and for recipients of p210MIGFP-transduced BM from Mx-Cre;Stat5a/bfl/− donors either untreated (dotted line) or treated (dashed line) with pIpC after transplantation as described in the “Methods.” The symbols indicate individual recipient mice, with the disease phenotype of each designated by the shading. No recipients in the Mx-Cre;Stat5a/bfl/− + pIpC cohort developed hematologic disease. (B) PB leukocyte counts at day 50 after transplantation in untreated or pIpC-treated recipients of p210MIGFP-transduced BM from Mx-Cre;Stat5a/bfl/+ (left) or Mx-Cre;Stat5a/bfl/− (right) donors. (C) Flow cytometric plot of GFP expression in PB myeloid cells from 2 representative recipients (mice #39 and #38) of p210MIGFP-transduced BM from Stat5a/bfl/− donors. (D) Southern blot analysis of the extent of recombination of the floxed Stat5a/b allele in genomic DNA from PB leukocytes at day 50 after transplantation. Nomenclature is as in supplemental Figure 1C. The small amount of wild-type Stat5a/b allele in recipients of Stat5a/bfl/− BM (mice #35-40) represents contribution from radioresistant host lymphocytes. (E) Spleen weights at autopsy of untreated or pIpC-treated recipients of p210MIGFP-transduced BM from Mx-Cre;Stat5a/bfl/+ (left) or Mx-Cre;Stat5a/bfl/− (right) donors. (F) Western blot analysis of primary myeloerythroid cell extracts from a representative untreated recipient of p210MIGFP-transduced BM from Mx-Cre;Stat5a/bfl/− donors that developed MPN and from 4 healthy pIpC-treated recipients. As controls, extracts from parental and BCR-ABL1–expressing Ba/F3 cell lines and BM from a nontransplanted Stat5 wild-type mouse were included. Proteins were immunoblotted with the indicated Abs against total or phosphorylated BCR-ABL1, STAT5, and CrkL. An anti-eIF4e immunoblot demonstrating equivalent protein loading is shown at the bottom.
Retrovirally marked normal HSCs sustain engraftment after Stat5 deletion
Several studies have shown that Stat5-deficient HSCs have prominent defects in reconstituting lymphopoiesis after transplantation, with more modest defects in the myeloerythroid repopulation.11,12,14 This raises the possibility that the lack of CML-like MPN in recipients of BCR-ABL1–transduced Mx-Cre;Stat5a/bfl/− BM after pIpC treatment is a consequence of failure of stable HSC engraftment and/or function in the absence of STAT5, although the persistence of circulating GFP+ myeloid cells (Figure 2C) in these mice argues against this. To test directly the consequences of Stat5 deletion on HSC function, we transduced BM from 5-FU–treated Stat5a/bfl/− and Mx-Cre;Stat5a/bfl/− donors with “empty” MIGFP virus that did not express BCR-ABL1, and transplanted the cells into lethally irradiated recipients. After engraftment, several recipients of transduced Mx-Cre;Stat5a/bfl/− BM were treated with pIpC and the other recipients were left untreated. Two months after transplantation, all 3 recipient cohorts had evidence of myeloid repopulation by retrovirally transduced HSCs, as assessed by the percentage of circulating GFP+Gr-1+ neutrophils (14% ± 3% for recipients of Stat5a/bfl/− recipients, 12% ± 2% for untreated Mx-Cre;Stat5a/bfl/− recipients, and 6% ± 1% for pIpC-treated Mx-Cre;Stat5a/bfl/− recipients). As observed previously, the efficiency of recombination of the floxed Stat5a/b allele in pIpC-treated recipients of Mx-Cre;Stat5a/bfl/− BM was high, and similar numbers of provirally marked HSC clones had engrafted in all 3 cohorts (Figure 3). Although analysis at longer intervals after transplantation would be necessary to make definitive statements about long-term engraftment, these results demonstrate that retrovirally transduced HSCs can efficiently engraft irradiated recipients and maintain myelopoiesis after ablation of Stat5a/b during a time period that is relevant to leukemogenesis.
Figure 3.

Retrovirally transduced normal HSCs sustain myelopoiesis after Stat5 deletion. Genomic DNA from BM of recipients of MIGFP-transduced Stat5a/bfl/− and Mx-Cre;Stat5a/bfl/− donor BM was isolated 2 months after transplantation and analyzed by Southern blot for efficiency of Stat5a/b deletion and number of engrafting proviral clones, as in supplemental Figure 1C. Top panel: Analysis of efficiency of recombination of the floxed Stat5a/b allele in recipients of MIGFP-transduced Stat5a/bfl/− BM (lanes 3-7) and in untreated (lanes 8-9) and pIpC-treated (lanes 10-12) recipients of MIGFP-transduced Mx-Cre;Stat5a/bfl/− BM using a Stat5 probe. Bottom panel: Analysis of the number of engrafted retrovirally transduced HSC clones in the 3 cohorts using a GFP probe.
Self-renewal of BCR-ABL1+ Stat5-deficient HSCs via serial transplantation
To test rigorously the self-renewal ability of BCR-ABL1–expressing Stat5a/b-deficient HSCs, we serially transplanted BM cells from 2 primary pIpC-treated recipients of p210MIGFP-transduced Mx-Cre;Stat5a/bfl/− donor BM (#35 and #38 in Figure 2) into 2 lethally irradiated secondary recipients each. HSCs from these primary recipients, which were completely deficient of Stat5 (supplemental Figure 2A), radioprotected the secondary recipients and resulted in efficient myeloid engraftment, with circulating GFP+ myeloid cells in all recipients at > 1 month after transplantation (average 45%, data not shown). In contrast to the primary recipients, which were healthy at the time of killing 100 days after transplantation, all secondary recipient mice succumbed to lymphoblastic leukemia/lymphoma between 39 and 58 days after transplantation. Three of the secondary recipients (#35-1, #38-1, and #38-2) developed B-ALL with lymphadenopathy and malignant pleural effusions, whereas the fourth recipient (#35-2) developed T-cell leukemia/lymphoma characterized by thymic enlargement and mesenteric lymphadenopathy. Molecular analysis of tumor-cell genomic DNA from these secondary recipients demonstrated that the leukemic cells contained the BCR-ABL1 gene, were derived from HSC clones present in the primary recipients, and were completely null for Stat5 (supplemental Figure 3A). Analysis of the IgH gene locus demonstrated clonal IgH rearrangements in the 3 secondary B-ALL leukemias (supplemental Figure 3B). Interestingly, these clonal IgH rearrangements were detectable in spleen DNA from the primary transplantation recipients, suggesting that BCR-ABL1–expressing Stat5-deficient HSCs were progressing to acute lymphoid leukemia (resembling lymphoid blast crisis of CML) in these primary recipients. These results demonstrate that BCR-ABL1–expressing Stat5-deficient HSC can self-renew, and that loss of STAT5 does not prevent progression to lymphoid blast crisis in this model system.
Role of STAT5 in B-lymphoid transformation and leukemogenesis by BCR-ABL1
The development of B-ALL in recipients of p210MIGFPCre-transduced Stat5a/bfl/− BM (Figure 1 and supplemental Figure 1D) and in secondary recipients of p210MIGFP-transduced Mx-Cre;Stat5a/bfl/− BM from pIpC-treated primary mice (supplemental Figure 3) suggests that STAT5 may not be absolutely required for B-lymphoid transformation and leukemogenesis by BCR-ABL1. To investigate this possibility further, we tested the ability of BCR-ABL1 to transform primary B-lymphoid progenitors from Stat5 mutant donors in 2 distinct in vitro assays. Dysregulated ABL kinases, including v-ABL and BCR-ABL1, can transform primary BM progenitors to form pre-B lymphoid colonies.30 We transduced BM from Stat5a/bfl/+ and Stat5a/bfl/− donors that were not pretreated with 5-FU with p210MIGFP or p210MIGFPCre retrovirus and plated the cells in agarose. There was no significant difference in the number of transformed B-lymphoid colonies arising from Stat5a/bfl/− BM transduced with either p210MIGFP or p210MIGFPCre (Figure 4A). In a complementary assay, we tested the ability of the transduced BM populations to proliferate in modified Whitlock-Witte–style cultures in which serial dilutions of transduced BM are plated on stroma from wild-type BM donors.29 The pre-B lymphoid progenitors that initially accumulate in such cultures are not fully transformed, require stroma, and are poorly leukemogenic in syngeneic mice.30 There was a small decrease in the efficiency of outgrowth of p210-transduced progenitors from Stat5a/bfl/− donors relative to Stat5a/bfl/+ donors, suggestive of a modest haploinsufficient effect of Stat5, but no difference in the overall efficiency of transformation of Stat5a/bfl/− BM by p210MIGFP and p210MIGFPCre virus (Figure 4B). Southern blot analysis of genomic DNA from these lymphoid cultures demonstrated that the clonality of the p210MIGFPCre-transduced Stat5a/bfl/− BM cultures was decreased relative to the other groups (supplemental Figure 4A), but several of the populations had extensive or complete recombination of the floxed Stat5 allele. We also tested the leukemogenicity of these progenitors by transplanting the cells into lethally irradiated Balb/c recipients immediately after retroviral transduction. Whereas all recipients of transduced Stat5a/bfl/+ BM succumbed to B-ALL within 10 weeks of transplantation, only 1 of 10 recipients of transduced Stat5a/bfl/− BM developed B-lymphoid leukemia (supplemental Figure 4B). Southern blot analysis demonstrated efficient recombination of the floxed Stat5 allele in leukemic cells from Stat5a/bfl/+ donors, but leukemic cells from the sole diseased recipient of transduced Stat5a/bfl/− BM did not express GFPCre and had minimal recombination of the floxed Stat5 allele (supplemental Figure 4C).
Figure 4.

STAT5 is not required for B-lymphoid transformation by BCR-ABL1 in vitro or for maintanance of established B-lymphoid leukemia in viro. (A-B) STAT5 is not required for transformation of primary BM B-lymphoid cells by BCR-ABL1 in vitro. (A) Primary BM from non–5-FU-treated Stat5a/bfl/+ and Stat5a/bfl/− donor mice was transduced with p210MIGFP or p210MIGFPCre retrovirus and plated directly in agarose. Transformed pre-B lymphoid colonies were counted on day 10. There was no significant difference in the number of colonies arising from Stat5a/bfl/− donor BM transduced with either p210MIGFP or p210MIGFPCre retrovirus (P = .88) or between any of the values assessed pairwise (t tests). (B) Serial dilutions of transduced BM were plated in triplicate on syngeneic stromal layers derived from nontransduced wild-type BM and cultured for 3 weeks, as described in the “Methods.”29 The plating density is indicated by the line color, the number of cultures that reached confluence (defined as 106 nonadherent cells) is indicated on the ordinate, and the time to confluence on the abscissa. (C) Growth of BCR-ABL1–transformed B-lymphoblasts derived from Mx-Cre;Stat5a/bfl/+ (top panel) or Mx-Cre;Stat5a/bfl/− (bottom panel) donors either untreated (blue squares) or treated with IFN-β (red circles). Each curve represents data from an independent population of transformed cells. (D) Southern blot analysis of Stat5a/b recombination status from the experiment in panel C. Lanes 1 and 2 contain tail DNA from Stat5 wild-type and Stat5fl/− mice, respectively. Note the complete deletion of the floxed Stat5a/b allele in IFN-treated lymphoblasts from Mx-Cre;Stat5a/bfl/+ donors (lanes 7-10) and from Mx-Cre;Stat5a/bfl/− donors (lanes 15-18). (E) Kaplan-Meier survival curve for unirradiated Balb/c Rag2−/− recipients injected intravenously (1 × 107 cells each, n = 16) with BCR-ABL1–expressing lymphoblasts derived from the in vitro transformation experiments in panels B and C. After engraftment of leukemia, half of each cohort (dashed lines) were treated with pIpC (arrowheads) to induce Cre recombinase. (F) Southern blot analysis of Stat5a/b recombination status in tumor tissue from 3 representative recipients of lymphoblasts from Mx-Cre;Stat5a/bfl/− donors from panel E that were treated with pIpC (lanes 4-6) or untreated (lanes 7-9). Lanes 1, 2, and 3 contain tail DNA from Stat5wt, Stat5fl/+, and Stat5fl/− mice, respectively. Note the complete deletion of the floxed Stat5a/b allele in lymphoblasts from pIpC-treated recipients.
These results suggest that STAT5 is dispensable for in vitro transformation of B-lymphoid progenitors by BCR-ABL1, but is required for initiation of B-ALL in recipients under these experimental conditions. To further investigate the role of STAT5 in B-lymphoid transformation and leukemogenesis, BM from Mx-Cre;Stat5a/bfl/+ and Mx-Cre;Stat5a/bfl/− donors not pretreated with 5-FU was transduced with p210MIGFP retrovirus and expanded on autologous stroma in Whitlock-Witte–style cultures. Stromal-independent populations of BCR-ABL1–transformed B-lymphoid progenitors were then treated in vitro with IFN-β (100 U/mL) to induce Cre recombinase. IFN-β treatment had only a minor effect on the growth of transformed progenitors from Mx-Cre;Stat5a/bfl/+ donors (Figure 4C top panel). Lymphoblasts derived from Mx-Cre;Stat5a/bfl/− donors proliferated at a lower rate in the absence of IFN-β and their growth was further delayed by 1-3 days after the addition of IFN-β, but the cells subsequently recovered exponential growth (Figure 4C bottom panel). There were no significant differences in the numbers of dead or apoptotic cells (assessed by flow cytometric measurement of annexin V/propidium iodide staining) between the IFN-β–treated Mx-Cre;Stat5a/bfl/+ and Mx-Cre;Stat5a/bfl/− populations (approximately 9% annexin V–positive/propidium iodide–negative on day 3, data not shown). Southern blot analysis demonstrated complete deletion of the floxed Stat5a/b allele under these conditions (Figure 4D). These results confirm that STAT5 is not absolutely required for B-lymphoid transformation by BCR-ABL1 in vitro.
To determine the effect of deletion of Stat5 on established B-ALL, we injected cultured Mx-Cre;Stat5a/bfl/+ and Mx-Cre;Stat5a/bfl/− lymphoblasts into unirradiated congenic Balb/c Rag2−/− recipients. In these experiments, Rag2-deficient recipients were used because we observed a large prolongation in survival of immunocompetent Balb/c mice bearing Stat5wt leukemia after pIpC treatment, suggestive of an IFN-induced immune response (data not shown). As a control, lymphoblasts from Stat5a/bfl/+ donors (lacking Mx-Cre) were also injected. After the development of clinical B-ALL (as assessed by presence of circulating GFP+CD19+ lymphoblasts), half of the recipients in each cohort were treated with pIpC to induce Cre recombinase. pIpC treatment was associated with a modest (approximately 3 days) prolongation of survival in all cohorts, which likely represents an attenuated IFN-mediated antileukemic response, but all pIpC-treated recipients quickly succumbed to B-ALL characterized by lymphadenopathy, hind-limb paralysis, and malignant pleural effusion (Figure 4E). Analysis of genomic DNA from these recipients demonstrated that the tumors were derived from blasts that had completely deleted Stat5a/b (Figure 4F). These results suggest that STAT5 is required for the initiation of BCR-ABL1–induced B-ALL in this retroviral transplantation model, but not for the maintenance of established B-ALL.
STAT5 is required for polycythemia induced by JAK2V617F in vivo
To address the role of STAT5a/b in the pathophysiology of PV, we used a model of PV induced by retroviral transduction of JAK2V617F into mouse BM, followed by transplantation into recipient mice, which develop nonfatal polycythemia and leukocytosis and eventually progress to myelofibrosis.26,34 We transduced BM from Stat5a/bfl/+ and Stat5a/bfl/− donor mice with JAK2V617FMIGFP or JAK2V617FMIGFPCre retrovirus (expressing mouse JAK2V617F) and transplanted the cells into lethally irradiated wild-type recipients. The JAK2V617FMIGFP retrovirus efficiently induced polycythemia and reticulocytosis in recipients of both Stat5a/bfl/+ and Stat5a/bfl/− donor BM (Figure 5A-B). Interestingly, the level of leukocytosis in recipients of JAK2V617F-tranduced Stat5a/bfl/− BM was less than half that of Stat5a/bfl/+ recipients (P = .019; Figure 5C), suggesting that haploinsufficiency for Stat5a/b impairs JAK2V617F-induced leukocytosis but not erythrocytosis. In both cohorts transplanted with MIGFPCre-transduced BM, we observed that some recipients did not engraft with donor-derived HSCs (supplemental Figure 5A and data not shown), suggesting that there may be a degree of stem-cell toxicity associated with the JAK2V617FMIGFPCre retrovirus that was not seen with the corresponding BCR-ABL1 virus. In those recipients that did engraft with donor BM, JAK2V617F induced polycythemia and reticulocytosis but not leukocytosis in recipients of MIGFPCre-transduced Stat5a/bfl/+ BM (Figure 5). In contrast, only 1 of 8 evaluable recipients of JAK2V617FMIGFPCre–transduced Stat5a/bfl/− BM developed polycythemia and reticulocytosis (Figure 5). Of the other recipients in this cohort, 5 engrafted with provirus-positive HSCs and demonstrated significant (30%-70%) recombination of the floxed Stat5 allele in the BM (supplemental Figure 5A), yet remained healthy with normal erythrocyte, reticulocyte, and leukocyte counts and had normal spleen weights on autopsy at day 200 after transplantation (data not shown). The 1 recipient in this cohort (#52) that developed erythrocytosis exhibited a deletion of the provirus with rearrangement of the JAK2V617F gene in spleen tissue (supplemental Figure 5B), suggesting that this event might have contributed to STAT5-independent erythrocytosis. The platelet counts in all 4 cohorts were normal, as observed previously.26 These results demonstrate that the loss of one donor Stat5 allele impairs leukocytosis but not erythrocytosis induced by JAK2V617F, whereas reduction of Stat5 gene dosage substantially below 50% prevents the development of polycythemia in recipient mice.
Figure 5.
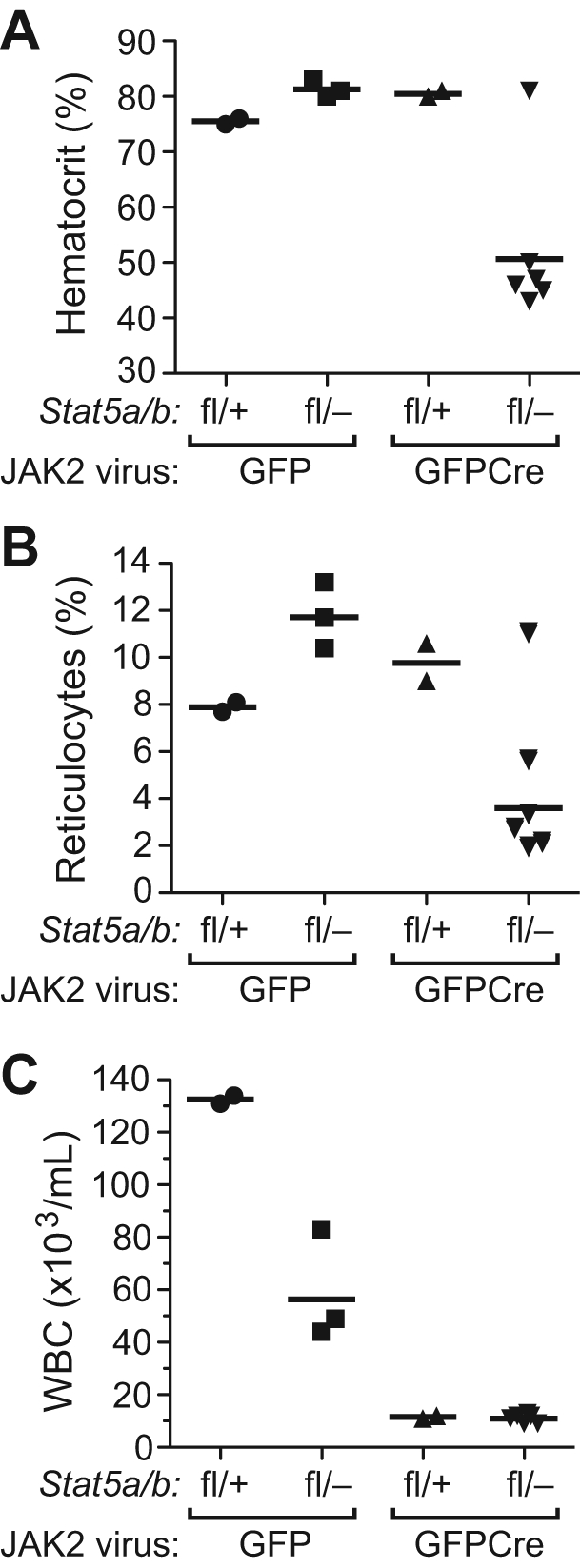
Reduction of Stat5 gene dosage impairs JAK2V617F-induced polycythemia in vivo. Hematocrit (A), reticulocyte counts (B), and leukocyte counts (C) from PB of recipients of BM from Stat5a/bfl/+ and Stat5a/bfl/− donors transduced with JAK2V617FMIGFP or MIGFPCre retrovirus analyzed on day 100 after transplantation. Hematocrit was significantly lower in the Stat5a/bfl/− JAK2V617FMIGFPCre group (P = .0114 vs Stat5a/bfl/− JAK2V617FMIGFP by t test).
As an alternative approach, we used Mx-Cre–transgenic donor mice to effect recombination of the floxed Stat5 allele. As before, we transduced BM from Mx-Cre;Stat5a/bfl/+ and Mx-Cre;Stat5a/bfl/− donors with JAK2V617FMIGFP retrovirus. After transplantation into irradiated wild-type recipient mice, some mice in each cohort were injected with pIpC to induce Cre expression and recombination. Analysis of the 4 cohorts on day 60 after transplantation demonstrated that recipients of Stat5a/bfl/+ BM and non-pIpC–treated recipients of Stat5a/bfl/− BM developed the cardinal erythroid phenotype of PV with polycythemia and reticulocytosis (Figure 6A-B). In contrast, recipients of JAK2V617F-transduced Stat5a/bfl/− BM that were treated with pIpC had normal hematocrits and reticulocyte counts, despite evidence of circulating GFP+ cells in most (Figure 6C). At autopsy on day 100 after transplantation, this cohort had normal spleen weights (Figure 6D), and Southern blot analysis of BM DNA showed that 6 of the 12 recipients had engrafted with provirus-positive, donor-derived HSCs and had evidence of efficient recombination (50%-92%) of the floxed Stat5 allele (supplemental Figure 5C). Western blotting of GFP+ BM cell extracts from these recipients demonstrated expression of JAK2 and activation of phospho-ERK signaling, but no appreciable phospho-STAT5 signaling, as expected (Figure 6E). Interestingly, although the Bcl-X gene is a known STAT5a/b target,35 we observed equivalent levels of BCL-XL protein in JAK2V617F+ cells with and without activated STAT5. These results suggest that, like BCR-ABL1 in CML, JAK2V617F cannot induce the erythropoietin-independent expansion of erythroid progenitors that is characteristic of PV in the absence of STAT5.
Figure 6.

Efficient deletion of Stat5 by Mx-Cre abolishes polycythemia and reticulocytosis induced by JAK2V617F. Hematocrit (A) and reticulocyte counts (B) of untreated (−) or pIpC-treated (+) recipients of JAK2V617FMIGFP-transduced BM from Mx-Cre;Stat5a/bfl/+ (left) or Mx-Cre;Stat5a/bfl/− (right) donors on day 60 after transplantation. Both values were significantly lower in the Stat5a/bfl/− (+pIpC) group (P < .0001 vs Stat5a/bfl/− [−pIpC] by t test). (C) Flow cytometric plots of GFP expression in PB leukocytes from 3 representative pIpC-treated recipients of JAK2V617FMIGFP-transduced BM from Stat5a/bfl/− donors, with GFP+ populations ranging from 15%-90%. (D) Spleen weights from the mice in panel A at time of autopsy on day 100. (E) Western blot analysis of primary BM myeloerythroid cell extracts from representative untreated (−) or pIpC-treated (+) recipients of JAK2V617FMIGFP-transduced BM from Mx-Cre;Stat5a/bfl/+ or Mx-Cre;Stat5a/bfl/− donors from panel A. As controls, protein extracts from parental and JAK2V617F-expressing Ba/F3 cell lines and BM from a normal mouse were used. Cell extracts were immunoblotted with the indicated Abs against total or phosphorylated STAT5 and ERK1/2 and against total JAK2 and BCL-X. An anti-eIF4e immunoblot demonstrating equivalent protein loading is shown at the bottom. Note that some STAT5 protein is detectable in pIpC-treated recipients because of contamination with cells of host origin.
JAK2V617F-induced myelofibrosis is partially independent of STAT5
As reported previously,26 polycythemic recipients of JAK2V617F-transduced BM from Stat5a/bfl/+donors and non-pIpC–treated recipients of transduced Stat5a/bfl/− donor BM all developed splenomegaly and had histopathological evidence of extramedullary erythropoiesis and myelopoiesis in spleen and liver, with disruption of the splenic follicular architecture and periportal myeloerythroid infiltrates in the liver (data not shown). Whereas the spleen weights of pIpC-treated recipients of JAK2V617F-transduced Stat5a/bfl/− BM were normal (Figure 6D), histopathological analysis revealed evidence of subclinical myeloproliferation in those recipients that engrafted with provirus-positive hematopoiesis. Whereas the spleens exhibited normal lymphoid follicles, there was significant abnormal infiltration of the red pulp with maturing myeloid and erythroid cells (Figure 7A), whereas livers demonstrated modest periportal myeloid cell infiltrates (Figure 7B). Whereas non-pIpC–treated recipients of JAK2V617F-transduced Stat5a/bfl/− BM developed moderate to heavy myelofibrosis in their BM at day 100 after transplantation, pIpC-treated recipients in this cohort that engrafted with JAK2V617F+ cells also exhibited very significant myelofibrosis, albeit slightly less dense on average (Figure 7C-D). These results suggest that myelofibrosis induced by JAK2V617F is partially independent of STAT5.
Figure 7.

Subclinical myeloproliferative disease and myelofibrosis induced by JAK2V617F in the absence of STAT5. H&E stain of spleen (A) and liver (B) from a representative pIpC-treated recipient of JAK2V617F MIGFP-transduced Mx-Cre;Stat5a/bfl/− BM that engrafted with provirus-positive donor-derived HSCs. In the spleen, a lymphoid follicle (f) and areas of erythroid (e) and myeloid (m) cell infiltration are designated. Magnification is 200×. (C-D) Reticulin stain of BM from a non-pIpC–treated recipient of JAK2V617F MIGFP-transduced Mx-Cre;Stat5a/bfl/− BM that developed polycythemia (C) and a pIpC-treated recipient with normal hematocrit (D). Magnification is 600×. All images were obtained from a BH-2 microscope using a Q-Color5 digital camera and QCapture Pro acquisition software (Olympus).
Discussion
Activation of STAT5 has been reported in a broad spectrum of hematologic malignancies,15 including the chronic MPNs. Consequently, there has been great interest in understanding the role of STAT5 in the pathogenesis of these diseases and exploring its possible utility as a target for therapy. In the present study, we used a genetic approach to define the contribution of STAT5a/b to MPNs induced by the BCR-ABL1 and JAK2V617F tyrosine kinases in a well-characterized mouse BM transduction/transplantation model system. We used a unique conditional-null allele that allows deletion of the entire Stat5a/b gene locus,9 and 2 complementary methods to express Cre recombinase in hematopoietic stem-progenitor cells: retroviral expression of a GFP-Cre fusion protein and inducible Cre expression from the IFN-responsive Mx-Cre transgene. We were able to efficiently induce CML-like MPN in recipients of Stat5+/+ BM transduced with p210MIGFPCre retrovirus (supplemental Figure 1B), but we observed less efficient engraftment of mice with Stat5a/bfl/+ HSCs transduced with JAK2V617FMIGFPCre retrovirus or with normal HSCs transduced with empty GFPCre retrovirus (data not shown). This suggests that a negative effect of Cre on HSCs33 may be partially offset by BCR-ABL1 expression. With the Mx-Cre transgene, expression of Cre is transient and leads to very efficient recombination of the floxed Stat5 allele in HSC (Figure 2D), but the ionizing radiation used to condition the transplantation recipients leads to significant recombination of floxed Stat5 in the absence of pIpC treatment (Figure 3 and data not shown). These findings reinforce the idea that studies of leukemogenesis using conditional gene targeting must be interpreted with caution.36
When either approach was used in Stat5a/bfl/+ HSCs in the present study, we observed an inhibitory effect of Stat5 haploidy on BCR-ABL1–induced CML-like leukemia, manifest as a prolongation in survival and the development of simultaneous CML-like MPN and B-ALL (Figures 1 and 2A). Single-gene deficiency for Stat5a was shown previously to attenuate MPN induced by ETV6-PDGFRβ37 and BCR-ABL124 in this model, and in the latter study, recipients also developed B-ALL alone and in combination with CML-like disease.24 When BCR-ABL1 was expressed in BM from donor mice with homozygous hypomorphic Stat5 mutations (Stat5a/bΔN/ΔN), a similar spectrum of B-lymphoid leukemia was observed in recipients.38 The BM target cells for B-ALL in this model are early B-lymphoid progenitors rather than HSCs,28,39 and previous studies have shown that the signaling requirements for myeloid and lymphoid leukemogenesis by BCR-ABL1 are different.28,40 Our findings demonstrate that reduction in Stat5 gene dosage attenuates CML-like MPN induced by BCR-ABL1, but has less of an effect on B-lymphoid leukemogenesis, defining an additional signaling difference between these distinct leukemias.
When we tested BM from Stat5a/bfl/− donors, we observed virtually complete suppression of BCR-ABL1–induced CML-like MPN with either method of Cre expression (Figures 1 and 2A). The sole recipient of p210MIGFPCre-transduced Stat5a/bfl/− BM that developed mixed MPN/B-ALL had very inefficient recombination of the floxed Stat5 allele (supplemental Figure 1D). In pIpC-treated recipients of p210MIGFP-transduced Mx-Cre;Stat5a/bfl/− BM, we demonstrated long-term engraftment with Stat5-deficient, BCR-ABL1–expressing stem cells with evidence of BCR-ABL1 kinase activity and signaling (Figure 2D and F), yet these mice remained healthy without overt hematologic disease. These observations demonstrate that STAT5a/b signaling is required for BCR-ABL1 to induce the massive expansion of myeloid progenitors that is characteristic of CML. Similar findings have been reported recently by Hoelbl et al.41 However, in the present study, the malignant stem cells persist in these recipients and show evidence of progression to lymphoid blast crisis, manifested in secondary recipients (supplemental Figure 3). In contrast, whereas Hoelbl et al transplanted CML-like leukemia stem cells (LSCs) from primary recipients of BCR-ABL1–transduced Mx-Cre;Stat5a/bfl/fl BM to lethally irradiated secondary recipients after in vitro deletion of Stat5, the resulting leukemias were derived from LSCs with unrecombined Stat5a/bfl/fl alleles, perhaps because these Stat5 wild-type LSCs outcompeted Stat5-null LSCs under these conditions.41 Our results suggest that inhibition of STAT5a/b signaling may be insufficient for eradication of LSCs in CML patients, and are consistent with recent studies demonstrating survival of malignant stem cells from CML patients despite effective pharmacologic inhibition of STAT5 with ABL1 tyrosine kinase inhibitors.42,43 We are currently investigating whether the combination of imatinib and Stat5 deficiency can eliminate LSCs in this model.
We investigated the role of STAT5a/b in B-lymphoid transformation by BCR-ABL1 directly in 2 complementary in vitro assays, pre-B-cell colony formation and growth on stroma. We observed no significant defect in transformation of Stat5a/bfl/− BM progenitors with p210MIGFPCre retrovirus in either assay (Figure 4A-B), despite evidence of efficient recombination of the floxed Stat5 allele in the transformed cells (supplemental Figure 4A). When Mx-Cre;Stat5a/bfl/− BM was used for BCR-ABL1 transduction and Cre induced by treatment with IFN-β, there was a transient delay in proliferation but an eventual outgrowth of BCR-ABL1+ lymphoblasts with complete deletion of Stat5a/b (Figure 4C-D). Our results differ from previous studies showing that transduction of Stat5-null fetal liver cells with either Abelson virus or with MIGFP retrovirus encoding the p185 isoform of BCR-ABL1 failed to yield transformed B-lymphoid colonies in vitro.10,41 However, given the profound defect in B-lymphoid development in the absence of STAT5a/b,10–12 it is plausible that the target cells for ABL1 transformation39 are significantly decreased in this donor-cell population.
For B-lymphoid leukemogenesis in vivo, our results suggest a more complex biology. Under conditions in which HSCs are transduced with BCR-ABL1 retrovirus (with 5-FU–treated BM donors) and transplanted, we have shown that B-ALL can develop in primary recipients of Stat5a/bfl/− BM when Cre recombinase is expressed and the remaining Stat5a/b allele is deleted (Figures 1 and 2A), as well as in secondary recipients of BM from such primary mice (supplemental Figure 3). It is plausible that B-lymphoid leukemia develops in these recipients as a consequence of differentiation of engrafted BCR-ABL1+ HSCs to B-lymphoid progenitors in vivo. In contrast, when B-lymphoid progenitors are transduced directly with BCR-ABL1 and transplanted (when BM donors are not treated with 5-FU), loss of STAT5 has a major negative effect on leukemogenesis (supplemental Figure 4B), but complete deletion of STAT5 does not affect established B-ALL (Figure 4E-F). We conclude that STAT5 is required for some steps in the initiation of leukemogenesis when BCR-ABL1–transduced progenitors are transplanted into irradiated recipients, but not for the maintenance of established leukemia, which is most relevant to consideration of STAT5 as a target for leukemia therapy. The precise step(s) for which STAT5 is required for leukemogenesis after IV injection of BCR-ABL1–transduced lymphoid progenitors will require further study, but we did not find a difference in the efficiency of BM homing between Stat5 wild-type and Stat5-null lymphoid progenitors (data not shown). There is a major difference between our results and the findings of Hoelbl et al,41 who concluded that STAT5 was required for the maintenance of BCR-ABL1–induced B-ALL because pIpC treatment prolonged the survival of mice bearing BCR-ABL1+ Mx-Cre;Stat5a/bfl/fl leukemias. However, mice ultimately succumbed to leukemia with unrecombined Stat5a/b alleles, suggesting that recombination of the floxed Stat5a/b locus was not completely efficient. Another important difference between the 2 studies is the genetic background of the mice: Balb/c in this study and C57Bl/6 in the work of Hoelbl et al. Further experiments, including studies in primary human leukemia cells, will be necessary to determine whether targeting STAT5 in Philadelphia chromosome–positive B-ALL will be an effective therapeutic approach.
The role of STAT5a/b in the pathogenesis of the PV-like MPN induced by JAK2V617F has important differences from its function in CML. Loss of one copy of Stat5 had no effect on polycythemia induced by JAK2V617F in Stat5a/bfl/+ BM, but did attenuate the leukocytosis in these recipients. In contrast, more extensive deficiency of Stat5 (from 66%-96%) abolished polycythemia and reticulocytosis in recipients of Stat5a/bfl/− BM. Unlike the corresponding recipients of BCR-ABL1+ Stat5-deficient BM, which had normal organ histopathology, those recipients that engrafted with JAK2V617F+ Stat5-deficient HSCs had evidence of subclinical MPN, with infiltration of spleen and liver with myeloerythroid cells and substantial myelofibrosis in the BM (Figure 7). The JAK2V617F-expressing STAT5-deficient BM cells had evidence of prominent activation of the ERK signaling pathway (Figure 6E), which represents a possible mechanism contributing to the disease process. These results suggest that inhibition of STAT5a/b signaling in PV patients may normalize their RBC mass but may also be less effective at preventing progression to myelofibrosis. Yan et al (found in this issue of Blood)44 have also demonstrated that loss of STAT5 in a transgenic model of JAK2V617F-induced MPN reversed the MPN phenotype, including the development of myelofibrosis. It is possible that retroviral expression of JAK2V617F, which results in higher levels of JAK2,45 might be responsible for the STAT5-independent subclinical MPN observed in our model.
In summary, we have demonstrated here that STAT5a/b plays a critical role in MPNs induced by both BCR-ABL1 and JAK2V617F. Our model system provides a platform for analyzing the precise molecular mechanisms through which STAT5a/b contributes to the pathogenesis of MPNs. In this regard, STAT5 has potential roles in both cytoplasmic signaling and in nuclear gene expression,46 and its activation via BCR-ABL1 and cytokine signaling have been implicated in leukemogenesis and in the response to kinase inhibitors.47 Our findings also provide a rational basis for the development of specific STAT5 inhibitors for the treatment of CML and PV.
Acknowledgments
This work was supported by the National Institutes of Health (grants CA090576 and HL089747 to R.A.V. and grant T32 CA09429 to W.A.) and the German Research Foundation Deutsche Forschungsgemeinschaft (to C.W.).
Footnotes
There is an Inside Blood commentary on this article in this issue.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: C.W., W.A., K.L., M.B., N.P., V.M.Z., and R.A.V. performed the experiments; L.H. provided essential reagents and advice; and C.W. and R.A.V. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for C.W. is Institute of Pathology, University of Munich, Munich, Germany. The current affiliation for N.P. is Department of Internal Medicine, Montefiore Medical Center, Bronx, NY. The current affiliation for V.M.Z. is Pfizer Inc, Cambridge, MA.
Correspondence: Richard A. Van Etten, Molecular Oncology Research Institute, Tufts Medical Center, 800 Washington St, Box 5609, Boston, MA 02111; e-mail: rvanetten@tuftsmedicalcenter.org.
References
- 1.Hennighausen L, Robinson GW. Interpretation of cytokine signaling through the transcription factors STAT5A and STAT5B. Genes Dev. 2008;22(6):711–721. doi: 10.1101/gad.1643908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Teglund S, McKay C, Schuetz E, et al. Stat5a and Stat5b proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell. 1998;93(5):841–850. doi: 10.1016/s0092-8674(00)81444-0. [DOI] [PubMed] [Google Scholar]
- 3.Liu X, Robinson GW, Wagner KU, Garrett L, Wynshaw-Boris A, Hennignausen L. Stat5a is mandatory for adult mammary gland development and lactogenesis. Genes Dev. 1997;11(2):179–186. doi: 10.1101/gad.11.2.179. [DOI] [PubMed] [Google Scholar]
- 4.Udy GB, Towers RP, Snell RG, et al. Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. Proc Natl Acad Sci U S A. 1997;94(14):7239–7244. doi: 10.1073/pnas.94.14.7239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moriggl R, Sexl V, Kenner L, et al. Stat5 tetramer formation is associated with leukemogenesis. Cancer Cell. 2005;7(1):87–99. doi: 10.1016/j.ccr.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 6.Moriggl R, Topham DJ, Teglund S, et al. Stat5 is required for IL-2-induced cell cycle progression of peripheral T cells. Immunity. 1999;10(2):249–259. doi: 10.1016/s1074-7613(00)80025-4. [DOI] [PubMed] [Google Scholar]
- 7.Bunting KD, Bradley HL, Hawley TS, Moriggl R, Sorrentino BP, Ihle JN. Reduced lymphomyeloid repopulating activity from adult bone marrow and fetal liver of mice lacking expression of STAT5. Blood. 2002;99(2):479–487. doi: 10.1182/blood.v99.2.479. [DOI] [PubMed] [Google Scholar]
- 8.Bradley HL, Couldrey C, Bunting KD. Hematopoietic-repopulating defects from STAT5-deficient bone marrow are not fully accounted for by loss of thrombopoietin responsiveness. Blood. 2004;103(8):2965–2972. doi: 10.1182/blood-2003-08-2963. [DOI] [PubMed] [Google Scholar]
- 9.Cui Y, Riedlinger G, Miyoshi K, et al. Inactivation of Stat5 in mouse mammary epithelium during pregnancy reveals distinct functions in cell proliferation, survival, and differentiation. Mol Cell Biol. 2004;24(18):8037–8047. doi: 10.1128/MCB.24.18.8037-8047.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoelbl A, Kovacic B, Kerenyi MA, et al. Clarifying the role of Stat5 in lymphoid development and Abelson-induced transformation. Blood. 2006;107(12):4898–4906. doi: 10.1182/blood-2005-09-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yao Z, Cui Y, Watford WT, et al. Stat5a/b are essential for normal lymphoid development and differentiation. Proc Natl Acad Sci U S A. 2006;103(4):1000–1005. doi: 10.1073/pnas.0507350103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dai X, Chen Y, Di L, et al. Stat5 is essential for early B cell development but not for B cell maturation and function. J Immunol. 2007;179(2):1068–1079. doi: 10.4049/jimmunol.179.2.1068. [DOI] [PubMed] [Google Scholar]
- 13.Zhu BM, McLaughlin SK, Na R, et al. Hematopoietic-specific Stat5-null mice display microcytic hypochromic anemia associated with reduced transferrin receptor gene expression. Blood. 2008;112(5):2071–2080. doi: 10.1182/blood-2007-12-127480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li G, Wang Z, Zhang Y, et al. STAT5 requires the N-domain to maintain hematopoietic stem cell repopulating function and appropriate lymphoid-myeloid lineage output. Exp Hematol. 2007;35(11):1684–1694. doi: 10.1016/j.exphem.2007.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benekli M, Baer MR, Baumann H, Wetzler M. Signal transducer and activator of transcription proteins in leukemias. Blood. 2003;101(8):2940–2954. doi: 10.1182/blood-2002-04-1204. [DOI] [PubMed] [Google Scholar]
- 16.Ilaria RL, Van Etten RA. P210 and P190 BCR/ABL induce the tyrosine phosphorylation and DNA binding activity of multiple specific STAT family members. J Biol Chem. 1996;271(49):31704–31710. doi: 10.1074/jbc.271.49.31704. [DOI] [PubMed] [Google Scholar]
- 17.Carlesso N, Frank DA, Griffin JD. Tyrosyl phosphorylation and DNA binding activity of signal transducers and activators of transcription (STAT) proteins in hematopoietic cell lines transformed by Bcr/Abl. J Exp Med. 1996;183(3):811–820. doi: 10.1084/jem.183.3.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aboudola S, Murugesan G, Szpurka H, et al. Bone marrow phospho-STAT5 expression in non-CML chronic myeloproliferative disorders correlates with JAK2 V617F mutation and provides evidence of in vivo JAK2 activation. Am J Surg Pathol. 2007;31(2):233–239. doi: 10.1097/01.pas.0000213338.25111.d3. [DOI] [PubMed] [Google Scholar]
- 19.Schwemmers S, Will B, Waller CF, et al. JAK2V617F-negative ET patients do not display constitutively active JAK/STAT signaling. Exp Hematol. 2007;35(11):1695–1703. doi: 10.1016/j.exphem.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walz C, Sattler M. Novel targeted therapies to overcome imatinib mesylate resistance in chronic myeloid leukemia (CML). Crit Rev Oncol Hematol. 2006;57(2):145–164. doi: 10.1016/j.critrevonc.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 21.Sillaber C, Gesbert F, Frank DA, Sattler M, Griffin JD. STAT5 activation contributes to growth and viability in Bcr/Abl-transformed cells. Blood. 2000;95(6):2118–2125. [PubMed] [Google Scholar]
- 22.Scherr M, Chaturvedi A, Battmer K, et al. Enhanced sensitivity to inhibition of SHP2, STAT5, and Gab2 expression in chronic myeloid leukemia (CML). Blood. 2006;107(8):3279–3287. doi: 10.1182/blood-2005-08-3087. [DOI] [PubMed] [Google Scholar]
- 23.Garçon L, Rivat C, James C, et al. Constitutive activation of STAT5 and Bcl-xL overexpression can induce endogenous erythroid colony formation in human primary cells. Blood. 2006;108(5):1551–1554. doi: 10.1182/blood-2005-10-009514. [DOI] [PubMed] [Google Scholar]
- 24.Ye D, Wolff N, Li L, Zhang S, Ilaria RL., Jr STAT5 signaling is required for the efficient induction and maintenance of CML in mice. Blood. 2006;107(12):4917–4925. doi: 10.1182/blood-2005-10-4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li W, Liang X, Kellendonk C, Poli V, Taub R. STAT3 contributes to the mitogenic response of hepatocytes during liver regeneration. J Biol Chem. 2002;277(32):28411–28417. doi: 10.1074/jbc.M202807200. [DOI] [PubMed] [Google Scholar]
- 26.Zaleskas VM, Krause DS, Lazarides K, et al. Molecular pathogenesis and therapy of polycythemia induced in mice by JAK2 V617F. PLoS One. 2006:1e18. doi: 10.1371/journal.pone.0000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thomas EK, Cancelas JA, Chae HD, et al. Rac guanosine triphosphatases represent integrating molecular therapeutic targets for BCR-ABL-induced myeloproliferative disease. Cancer Cell. 2007;12(5):467–478. doi: 10.1016/j.ccr.2007.10.015. [DOI] [PubMed] [Google Scholar]
- 28.Roumiantsev S, de Aos I, Varticovski L, Ilaria RL, Van Etten RA. The Src homology 2 domain of Bcr/Abl is required for efficient induction of chronic myeloid leukemia-like disease in mice but not for lymphoid leukemogenesis or activation of phosphatidylinositol 3-kinase. Blood. 2001;97(1):4–13. doi: 10.1182/blood.v97.1.4. [DOI] [PubMed] [Google Scholar]
- 29.Smith KM, Yacobi R, Van Etten RA. Autoinhibition of Bcr-Abl through its SH3 domain. Mol Cell. 2003;12(1):27–37. doi: 10.1016/S1097-2765(03)00274-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Unnikrishnan I, Radfar A, Jenab-Wolcott J, Rosenberg N. p53 mediates apoptotic crisis in primary Abelson virus-transformed pre-B cells. Mol Cell Biol. 1999;19(7):4825–4831. doi: 10.1128/mcb.19.7.4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pear WS, Miller JP, Xu L, et al. Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving P210 bcr/abl-transduced bone marrow. Blood. 1998;92(10):3780–3792. [PubMed] [Google Scholar]
- 32.Heinrich AC, Pelanda R, Klingmuller U. A mouse model for visualization and conditional mutations in the erythroid lineage. Blood. 2004;104(3):659–666. doi: 10.1182/blood-2003-05-1442. [DOI] [PubMed] [Google Scholar]
- 33.Loonstra A, Vooijs M, Beverloo HB, et al. Growth inhibition and DNA damage induced by Cre recombinase in mammalian cells. Proc Natl Acad Sci U S A. 2001;98(16):9209–9214. doi: 10.1073/pnas.161269798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lacout C, Pisani DF, Tulliez M, Gachelin FM, Vainchenker W, Villeval JL. JAK2V617F expression in murine hematopoietic cells leads to MPD mimicking human PV with secondary myelofibrosis. Blood. 2006;108(5):1652–1660. doi: 10.1182/blood-2006-02-002030. [DOI] [PubMed] [Google Scholar]
- 35.Gesbert F, Griffin JD. Bcr/Abl activates transcription of the Bcl-X gene through STAT5. Blood. 2000;96(6):2269–2276. [PubMed] [Google Scholar]
- 36.Schmidt-Supprian M, Rajewsky K. Vagaries of conditional gene targeting. Nat Immunol. 2007;8(7):665–668. doi: 10.1038/ni0707-665. [DOI] [PubMed] [Google Scholar]
- 37.Cain JA, Xiang Z, O'Neal J, et al. Myeloproliferative disease induced by TEL-PDGFRB displays dynamic range sensitivity to Stat5 gene dosage. Blood. 2007;109(9):3906–3914. doi: 10.1182/blood-2006-07-036335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sexl V, Piekorz R, Moriggl R, et al. Stat5a/b contribute to interleukin 7-induced B-cell precursor expansion, but abl- and bcr/abl-induced transformation are independent of STAT5. Blood. 2000;96(6):2277–2283. [PubMed] [Google Scholar]
- 39.Signer RA, Montecino-Rodriguez E, Witte ON, Dorshkind K. Immature B-cell progenitors survive oncogenic stress and efficiently initiate Ph+ B-acute lymphoblastic leukemia. Blood. 2010;116(14):2522–2530. doi: 10.1182/blood-2010-01-264093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu Y, Liu Y, Pelletier S, et al. Requirement of Src kinases Lyn, Hck and Fgr for BCR-ABL1-induced B-lymphoblastic leukemia but not chronic myeloid leukemia. Nat Genet. 2004;36(5):453–461. doi: 10.1038/ng1343. [DOI] [PubMed] [Google Scholar]
- 41.Hoelbl A, Schuster C, Kovacic B, et al. Stat5 is indispensable for the maintenance of bcr/abl-positive leukaemia. EMBO Mol Med. 2010;2(3):98–110. doi: 10.1002/emmm.201000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW, Druker BJ. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J Clin Invest. 2011;121(1):396–409. doi: 10.1172/JCI35721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hamilton A, Helgason GV, Schemionek M, et al. Chronic myeloid leukemia stem cells are not dependent on Bcr-Abl kinase activity for their survival. Blood. 2012;119(6):1501–1510. doi: 10.1182/blood-2010-12-326843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yan D, Hutchison RE, Mohi G. Critical requirement for Stat5 in a mouse model of polycythemia vera. Blood. 2012;119(15):3539–3549. doi: 10.1182/blood-2011-03-345215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tiedt R, Hao-Shen H, Looser R, Dirnhofer S, Schwaller J, Skoda RC. Ratio of mutant JAK2-V617F to wild type Jak2 determines the MPD phenotypes in transgenic mice. Blood. 2008;111(8):3931–3940. doi: 10.1182/blood-2007-08-107748. [DOI] [PubMed] [Google Scholar]
- 46.Kornfeld JW, Grebien F, Kerenyi MA, et al. The different functions of Stat5 and chromatin alteration through Stat5 proteins. Front Biosci. 2008;13:6237–6254. doi: 10.2741/3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Traer E, Mackenzie R, Snead J, et al. Blockade of JAK2-mediated extrinsic survival signals restores sensitivity of CML cells to ABL inhibitors [published online ahead of print November 18, 2011]. Leukemia. doi: 10.1038/leu.2011.325. doi: 10.1038/leu.2011.325. [DOI] [PMC free article] [PubMed] [Google Scholar]