

Abstract
The mechanism by which activation of the Hedgehog (Hh) pathway modulates differentiation and promotes oncogenesis in specific tissues is poorly understood. We therefore, analysed rhabdomyosarcomas from mice that were haploinsufficient for the Hh-binding protein, Hip1, or for the Hh receptor, Patched 1 (Ptch1). Transfection of the Hh-regulated transcription factor Gli1, which is expressed in a subset of mouse and human rhabdomyosarcomas, suppressed differentiation of myogenic rhabdomyosarcoma lines generated from Hip1+/− and Ptch1+/− mice. The closely related factor, Gli2, had similar effects. Gli1 and Gli2 inhibited myogenesis by repressing the capacity of MyoD to activate transcription. Deletion analysis of Gli1 indicated that multiple domains of Gli1 are required for efficient inhibition of MyoD. Gli1 reduced the ability of MyoD to heterodimerize with E12 and bind DNA, providing one mechanism whereby the Gli proteins modulate the activity of MyoD. This novel activity of Gli proteins provides new insights into how Hh signaling modulates terminal differentiation through inhibition of tissue-specific factors such as MyoD. This mechanism may contribute to the broad role of Hh signaling and the Gli proteins in differentiation decisions and cancer formation.
Keywords: Gli, MyoD, hedgehog, cancer, rhabdomyosarcoma, development
Introduction
Hedgehog (Hh) signaling is implicated in cancer formation in multiple tissues (Ruiz i Altaba et al., 2002; McMahon et al., 2003; Pasca di Magliano and Hebrok, 2003; Beachy et al., 2004). Normally, Hh signaling is initiated through binding of the Hh ligand to the 12 transmembrane domain protein Patched 1 (Ptch1). This antagonizes Ptch1 activity and allows the seven transmembrane domain protein Smoothened (Smo) to transduce the Hh signal, eventually leading to activation of the Gli family of zinc finger transcription factors whose combinatorial activities are believed to direct the entire transcriptional output of the Hh signal (Ingham and McMahon, 2001; Lum and Beachy, 2004; Briscoe and Therond, 2005; Hooper and Scott, 2005). In mammals, there are three Gli proteins, all of which contain an activation domain located at the C-terminus. Gli1 appears to function solely as a transcriptional activator and potentiates the Hh signal, while Gli2 and Gli3 also contain an N-terminal repression domain (Sasaki et al., 1997, 1999; Ruiz i Altaba, 1999). Gli2 and Gli3 can be cleaved via limited proteolysis in the absence of the Hh signal to generate truncated proteins that lack the C-terminal activation domain (Dai et al., 1999; Aza-Blanc et al., 2000; Wang et al., 2000). Consequently, Gli2 and Gli3 appear to have the capacity to both activate and repress transcription (Dai et al., 1999; Aza-Blanc et al., 2000; Pan et al., 2006), although it is not clear to which extent each function of the Gli proteins is required in vivo (Ingham and McMahon, 2001; McMahon et al., 2003; Nieuwenhuis and Hui, 2005). Hh signaling and Gli processing involves the primary cilia (Huangfu and Anderson, 2005; Liu et al., 2005; May et al., 2005), kinase-mediated phosphorylation, and the proteasome (Wang and Li, 2006) although the precise site and mechanism of downstream Hh signaling events and Gli proteolysis remains to be fully elucidated.
Hh pathway activation and increased Gli expression have been observed in different tumors. For example, haploinsufficiency of Ptch1 causes Gorlin syndrome, which is associated with very high rates of basal cell cancer, as well as increased incidences of medulloblastoma and various sarcomas including rhabdomyosarcoma (Gorlin, 1987; Watson et al., 2004). Mice that are haploinsufficient for Ptch1 are a model of Gorlin syndrome and also exhibit increased incidences of medulloblastoma (Goodrich et al., 1997) and rhabdomyosarcoma (Hahn et al., 1998), while developing basal cell carcinoma upon exposure to ultraviolet or ionizing radiation (Aszterbaum et al., 1999). Tumors that occur with increased frequency in both human and mouse Ptch1 haploinsufficiency show evidence of elevated Hh signaling and Gli1 expression (Toftgard, 2000). However, the mechanism whereby elevated Hh signaling produces tumors in specific cellular contexts is not fully established, which reflects a limited understanding of how Hh signaling regulates cell fate in normal development.
Rhabdomyosarcoma is an appealing tumor model because it has previously been used to study pathways that regulate both tumor formation and tissue-specific differentiation (Guo et al., 2003; Sirri et al., 2003). Rhabdomyosarcomas exhibit aberrant muscle differentiation while universally expressing members of the MyoD family of basic helix-loop-helix (bHLH) proteins (Hahn et al., 1998; Kappler et al., 2004), which are required for specification and differentiation of the muscle lineage during normal development (Buckingham, 2001; Buckingham et al., 2003). MyoD family members function through preferential heterodimer formation with the E proteins to activate transcription of skeletal muscle genes containing an E-box element via DNA binding (Massari and Murre, 2000). A model has emerged where inhibition of MyoD-mediated muscle gene expression and terminal differentiation is a requisite step in the generation of rhabdomyosarcoma (Merlino and Helman, 1999; Puri and Sartorelli, 2000). Hh signaling is known to regulate multiple aspects of normal myogenesis, including the initiation and maintenance of MyoD family expression, the development, survival and proliferation of the epaxial and hypaxial muscle lineages, and the selection of muscle fiber type (Munsterberg et al., 1995; Borycki et al., 1999; Kruger et al., 2001). Moreover, elevated Hh signaling has recently been observed in sporadic cases of embryonal rhabdomyosarcoma (Blandford et al., 2006; Tostar et al., 2006), in addition to the rhabdomyosarcoma formation that is associated with Ptch1 haploinsufficiency. However, a precise mechanistic understanding of how the Hh signal modulates normal skeletal muscle development and promotes rhabdomyosarcoma formation does not exist.
Here, we report that Gli1 and Gli2 have the capacity to inhibit myogenic differentiation in rhabodmyosarcoma cell lines and in C2C12 myoblasts. The mechanism of inhibition by Gli proteins includes repression of the capacity of MyoD to activate transcription, which is believed to be a critical step in the development of rhabdomyosarcomas. We propose that Hh signaling and the Gli proteins modify terminal differentiation and promote cancer formation by modulating the activity of tissue-specific factors such as MyoD.
Results
Myogenic cell lines derived from Hip1+/− and Ptch1+/− rhabdomyosarcomas
As reported previously, Ptch1+/− mice developed rhabdomyosarcomas at a frequency of 2–10% depending on the genetic backgrounds (Hahn et al., 1999). Mice haploinsufficient for Hip1, a gene whose protein product binds to Hh proteins and negatively regulates their activities (Chuang and McMahon, 1999; Chuang et al., 2003), also developed flank masses at a low frequency (Gerber and Chuang, unpublished). Histologic analysis of portions of excised tumors confirmed the presence of typical features of rhabdomyosarcoma including aberrant myogenic differentiation (Figure 1a–d). Taking advantage of the β-galactosidase gene knocked in to the mouse Ptch1 genomic locus (Goodrich et al., 1997), we used X-gal staining to determine the transcriptional activation status of the Ptch1 locus, which is a faithful marker of Hh pathway activation (Goodrich et al., 1996; Marigo et al., 1996), in one of the excised Ptch1+/− rhabdomyosarcomas. Numerous cells stained positive with X-gal (Figure 1g), in contrast to normal muscle tissue from Ptch1+/− animals (Figure 1h), indicating elevated transcriptional activity at the Ptch1 locus in tumor tissue in vivo. Semi-quantitative reverse transcription–polymerase chain reaction (RT–PCR) revealed that Gli1 message was expressed at higher levels than Gli2 in primary tumor tissue (Supplementary Figure 1). This is consistent with previous characterizations of elevated Hh pathway activity and Gli1 expression in Ptch1+/− rhabdomyosarcomas (Hahn et al., 1998; Kappler et al., 2003).
Figure 1.

Analysis of rhabdomyosarcomas from Hip1+/− and Ptch1+/− mice. (a–f) Hematoxylin-and-eosin stained sections of rhabdomyosarcoma isolated from Hip1+/− (a, b) and Ptch1+/− (c, d) mice and of normal muscle from Hip1+/− mice (e, f). (b, d, f) represent higher magnification of (a, c, e). Note the presence of the tumor capsule (indicated by grey arrows in a and c) and the disorganized, highly cellular growth pattern in both tumors (a–d)in comparison to normal muscle (e, f). Histologic features of aberrant skeletal muscle formation, the cardinal feature of rhabdomyosarcoma, were present. For example, black arrows depict multi-nucleated cells (b) and elongated, eosin-positive cells (d). (g, h) X-gal-stained sections of rhabdomyosarcoma and normal muscle isolated from Ptch1+/− mice. In this allele, the β-galactosidase gene was under the control of the Ptch1 promoter (Goodrich et al., 1997) and positive X-gal staining indicates Ptch1 expression. The tumor contained many positively staining cells (blue) not seen in normal muscle. (i–l) Immunohistochemical analysis of myogenic differentiation of rhabdomyosarcoma cell lines (RMH and RMP), derived from Hip1+/− and Ptch1+/− mice, respectively, in comparison to two human rhabdomyosarcoma cell lines, RD and Rh30. The RMP (i) and RMH (j) lines both exhibited extensive myotube formation and expressed myosin heavy chain (MyHC) (red) and sarcomeric actin (not shown), markers for terminal differentiation. In contrast, in the RD (k) and Rh30 (l) lines, only occasional mononucleated cells that expressed sarcomeric actin (red) and MyHC (not shown) were observed. (m) Northern blot analysis of gene expression in the rhabdomyosarcoma cell lines used in this study. The RMH and RMP lines both expressed high levels of MyHC, but only low levels of Gli1 message. In contrast, the Rh30 line expressed moderate levels of Gli1 while the RD line did not. Both of these human rhabdomyosarcoma lines do not exhibit terminal differentiation. The expression level of the housekeeping gene GAPDH serves as the loading control.
As rhabdomyosarcoma cell lines have been used previously to study oncogenic pathways, we generated cell lines from flank masses that occurred in Ptch1+/− and Hip1+/− mice. The masses were removed within their capsules, making contamination from surrounding tissue unlikely. We selected one rapidly growing Hip1+/− line (RMH) and one Ptch1+/− line (RMP) for subsequent analysis. Surprisingly, both lines were highly myogenic (Figure 1i and j), in contrast to previous studies of rhabdomyosarcoma lines such as the human-derived RD and Rh30 lines (Figure 1k and l) that fail to fully differentiate (Tapscott et al., 1993). Northern blot analysis confirmed ongoing transcription of high levels of myosin heavy chain (MyHC) in both the RMP and RMH lines (Figure 1m). In contrast, Gli1, which was detected previously in primary rhabdomyosarcoma tissue from Ptch1+/− mice (Kappler et al., 2003), was only weakly expressed in RMP and RMH cells, but was easily detected in the nonmyogenic Rh30 line (Figure 1m), which harbors a chromosomal amplification that spans the Gli1 locus, among other abnormalities (Fiddler et al., 1996). Ptch1 (Goodrich et al., 1996), was not detected in either the RMP or RMH lines as determined by both Northern analysis and X-gal staining (not shown). This is consistent with reports that the Hh pathway can be downregulated ex vivo in Ptch1+/− medulloblastoma (Romer et al., 2004) and in other Ptch1+/− rhabdomysarcomas (H Hahn, personal communication).
Inhibition of myogenic differentiation by activation of the Hh pathway
It was intriguing that in contrast to the primary tumors, cell lines derived from Ptch1+/− and Hip1+/− rhabdomyosarcomas exhibited both reduced Hh pathway activity and apparently increased myogenic differentiation. One possibility is that the ex vivo RMP and RMH rhabdomyosarcoma cell lines have less exposure to the Shh ligand. To test this, we cultured RMP and RMH cells in Shh-conditioned media and used immunohistochemistry to assess for myogenin (Mgn) expression (red, Figure 2a and b), which is an early marker of myogenic differentiation. Concomitantly, we assessed localization of Smo to the primary cilium (green, Figure 2a and b), which serves as a sensitive marker of mammalian Hh pathway activation in individual cells (Corbit et al., 2005). We found that localization of Smo to the primary cilium in individual cells was negatively correlated with detectable myogenin expression (Table 1). This suggests that activation of Hh signaling via exposure to exogenous Hh ligand is associated with reduced or delayed activation of the myogenic program in ex vivo Hip1+/− and Ptch1+/− rhabdomysarcoma cells.
Figure 2.

Activation of the Hh pathway inhibits terminal muscle differentiation. (a, b) Immunohistochemical analysis of Smo localization to the primary cilium and myogenin expression in RMH and RMP cells cultured in Shh-conditioned medium. Localization of Smo to the primary cilium was verified by staining with anti-acetylated tubulin, which labels the primary cilium (not shown). In both cell lines, a subset of cells (~40%, data not shown) had detectable Smo expression (green as indicated by white arrows) in the primary cilium following culture in Shh-conditioned medium. A subset of cells also exhibited Myogenin expression (red as indicated by yellow arrows). There was a negative correlation between Smo expression in the primary cilium and myogenin expression (see Table 1). (c, d) X-gal staining of myogenic RMH cells following transfection of plasmids encoding the muscle reporter MCK-lacZ and Gli1 as indicated. Note the elongated cells (arrow) with multiple blue nuclei indicative of MCK expression and myotube formation in the absence of Gli1 (c) but not in the presence of Gli1 (d), indicating that Gli1 represses muscle differentiation. Transfection efficiencies in (c) and (d) were similar based on visualization of a co-transfected GFP reporter (not shown). (e) Immunostaining of RMP cells following transfection with a plasmid encoding Gli1. Antibodies against Gli1 and myosin heavy chain (MyHC) were used. Gli1-positive cells (green) indicated by the white arrows were mononucleated and failed to express MyHC (red). Similar results were obtained in the RMH line. (f–h) Luciferase assays using RMP, RMH and C2C12 cells as indicated following transfection of a control or a Gli expression plasmid together with MCK-luc or with the Gli-specific reporter 8xGliBSδ51-luc. Gli1 and Gli2 substantially inhibited the activity of MCK-luc, while activating 8xGliBSδ51-luc.
Table 1.
RMP and RMH cells with Smo in the primary cilium express myogenin at a low frequency in comparison to the cell population as a whole
| RMP |
RMH |
|||
|---|---|---|---|---|
|
Cells with Smo in cilium |
All cells |
Cells with Smo in cilium |
All cells | |
| Myogenin+ | 16a | 45a | 14b | 48b |
| Myogenin− | 84 | 55 | 86 | 52 |
Statistically different (P<0.01).
Statistically different (P<0.005).
To further assess the consequences of reactivating the Hh pathway in cultured Hip1+/− and Ptch1+/− rhabdomysarcoma cells, we analysed the effects of expressing individual members of the Gli family in RMP and RMH cells. We determined by assaying the activity of the muscle creatine kinase (MCK) reporter, MCK-lacZ (Figure 2c and d), that transfection of a Gli1 expression plasmid strongly inhibited myogenesis. Immunohistochemical analysis showed that Gli1-positive cells (green, Figure 2e) also failed to express MyHC (red, Figure 2e), were mononucleated, and lacked typical myotube morphology. Transfection of either a Gli1 or a Gli2 expression plasmid also reduced the activity of a second MCK reporter, MCK-luciferase (luc), in RMP (Figure 2f) and RMH cells (Figure 2g). In contrast, Gli3, which is generally associated with repression, rather than activation, of Hh signaling, had no effect on MCK-luc activity. These results demonstrate that Gli1 and Gli2, which both generally potentiate Hh signaling, have the capacity to inhibit differentiation of the Hip1+/− and Ptch1+/− rhabdomyosarcoma cell lines.
The ability of Gli1 and Gli2 to inhibit muscle gene expression was not due to a generalized repression of transcription as both Gli1 and Gli2 activated the Gli reporter 8xGliBSδ51-luc, while functional Gli3 expression is indicated by repression of the Gli reporter (Figure 2f and g) (Sasaki et al., 1997). Qualitatively similar results indicating that Gli1 and Gli2 inhibit muscle differentiation were also obtained in C2C12 myoblasts (Figure 2h and Supplementary Figure 2), which are a well-established model of muscle differentiation.
Gli1 and Gli2 inhibit the ability of MyoD to activate transcription
The MyoD family of bHLH proteins plays a central role in skeletal muscle differentiation and it is believed that inhibition of MyoD activity is an important step in rhabdomyosarcoma formation. Therefore, we tested whether Gli1 limits muscle differentiation through inhibition of MyoD activity. This analysis was accomplished with C3H10T1/2 cells, an established model to assess MyoD activity (Weintraub et al., 1991). Co-transfection of C3H10T1/2 cells with Gli1 and MyoD substantially reduced the expression of several MyoD-responsive muscle reporters including Mgn-luc, MCK-luc and MyHC-luc in comparison to transfection with MyoD alone (Figure 3a). Gli1 also inhibited the activation of a synthetic MyoD reporter, 4RTK-luc (Figure 3a), which has four multimerized MyoD-binding sites placed upstream of the viral thymidine kinase (TK) promoter (Bengal et al., 1994). We also tested whether Gli2 and Gli3 can inhibit the capacity of MyoD to activate transcription of 4RTK-luc in CH310T1/2 cells (Figure 3b). In this assay, Gli2 reduced the transcriptional activation by MyoD in a comparable fashion to Gli1 (compare Figure 3b to bottom graph of Figure 3a), while Gli3 had little effect. This correlates with the capacity of Gli1 and Gli2 to inhibit differentiation of RMP, RMH and C2C12 cells and provides corroborative evidence that Gli1 and Gli2 inhibit myogenic differentiation through repression of MyoD-mediated transcriptional activation.
Figure 3.

Gli1 and Gli2 inhibit transcriptional activation by MyoD. (a) Luciferase assays of lysates of CH310T1/2 cells following transfection with muscle reporters (Mgn-luc, MCK-luc, MyHC-luc and 4RTK-luc) together with various combinations of plasmids encoding MyoD and Gli1 as indicated. In all cases, the presence of Gli1 (bottom row in all bar graphs) substantially inhibited the activity of MyoD (compare the relative luciferase activity of the middle row to bottom row for each reporter). (b) Luciferase assays of lysates of CH310T1/2 cells following transfection with the MyoD reporter 4RTK-luc, together with combinations of plasmids encoding MyoD, Gli2, and Gli3 as indicated. Gli2 substantially inhibited the activity of MyoD while Gli3 had minimal effects in this assay. (c) Luciferase assays of MyoD reporter (4RTK-luc) activity in C3H10T1/2 cells following transfection as indicated with a plasmid encoding Gli1 and various MyoD deletion constructs depicted in the top diagram (WT –wild-type, NTAD –N-terminal activation domain, C/H –cysteine/histidine rich region, bHLH –basic helix-loop-helix domain, HIII –helix three; the numbers below correspond to amino acid positions in MyoD protein). The numbers after the Δ sign indicate the corresponding amino acids of MyoD deleted in each construct. TM167 contains a stop codon introduced at amino acid position 167 of MyoD. Gli1 inhibited transcriptional activation by all deletion constructs of MyoD tested (compare the shaded bar to the solid bar), suggesting that only the bHLH domain is required for Gli1-mediated inhibition of MyoD. (d) Luciferase assays of lysates of C3H10 T1/2 cells following transfection with 4RTK-luc, Gli1, and various bHLH protein expression constructs (EMSV-MyoD, EMSV-Myf5, RSV-Mash1 and CS2-E12) as indicated. In addition to inhibiting MyoD-mediated activity, Gli1 inhibited the ability of both Myf5 and Mash1 to activate the E-box reporter (4RTK-luc); E12 was, however, refractory to inhibition by Gli1 (compare bottom row in all graphs to middle row).
To identify specific domains of MyoD that are required for Gli-mediated inhibition, we analysed constructs with deletions in regions of MyoD (Figure 3c, top panel). The overlapping deletion constructs encompassed all regions of MyoD except for the bHLH domain and included deletions of the N-terminal activation domain (NTAD) (MyoDΔ3–56) (Weintraub et al., 1991) and domains involved in the activation of genes within repressive chromatin (MyoDΔ78–91, MyoDΔ92–99 and MyoDΔ218–269) (Gerber et al., 1997). Although the deletion constructs were generally weaker activators than wild-type MyoD, their abilities to activate a simple reporter were all substantially reduced by co-transfection with Gli1 (Figure 3c, bottom panel). Therefore, the major regions of MyoD, except the segment from amino acids 99 to 167 that encompasses the bHLH domain, were shown to be dispensable for Gli1-mediated inhibition.
The mapping of the region required for inhibition of MyoD by Gli1 to the bHLH domain led us to examine the effects of Gli1 on the activity of several other bHLH proteins: the myogenic bHLH protein, Myf-5; the neural regulator, Mash1, and the broadly expressed E12, which functions as a dimerization partner for tissue-specific bHLH factors such as MyoD. Co-transfection with Gli1 inhibited all activator constructs tested except E12 (Figure 3d). Thus, Gli1 inhibits the capacity of several tissue-specific bHLH factors to activate transcription.
Multiple domains of Gli1 are required for inhibition of MyoD activity
To further understand the mechanism through which Gli1 inhibits the bHLH domain of MyoD, we constructed a series of deletion mutations that span the majority of Gli1 (Figure 4a). Expression of transfected Gli1 mutant proteins in cultured cells was verified by Western blotting (Figure 4b) and immunohistochemistry (data not shown). The capacity of these Gli1 mutations to prevent MyoD-mediated activation of the reporter Mgn-luc was assessed in transient transfections of C3H10T1/2 cells (Figure 4c). The capacity of these mutants to inhibit the endogenous myogenic program in C2C12 myoblasts was also assessed (Figure 4d). Several Gli1 mutations had a reduced capacity to inhibit myogenic gene activation in these assays including Gli1Δ1–243, which contains a deletion of the N-terminal portion of Gli1 including part of the first zinc finger; Gli1Δ764–1016, which contains a deletion of an internal region of Gli1 with no previously ascribed function; and Gli1Δ1020–1111, which contains a deletion of the C-terminal activation domain. Gli1 mutations containing deletions of other regions largely retained their capacity to inhibit myogenic gene activation.
Figure 4.

Multiple domains of Gli1 are required to inhibit transcriptional activation by MyoD. (a) Schematic diagram of Gli1 deletion constructs. Deletions of Gli1 were constructed that cover the N-terminal region (Gli1Δ1–171), the first zinc finger (Zn Fngr) (Gli1Δ1–243), the nuclear export signal (NES) (Gli1Δ404-593), and the C-terminal activation domain (AD) (Gli1Δ1020–1111), as well as two other internal regions of the protein for which no clear function has been ascribed (Gli1Δ572–707 and Gli1Δ764–1016). The numbers indicate the corresponding amino acid residues in the Gli1 protein. (b) Western blot showing comparable levels of protein expression of the various Gli1 deletion constructs depicted in (a) following transient transfection into Cos1 cells. Western blots were probed as indicated with primary antibodies against Gli1, FLAG and β-tubulin. FLAG-tagged Gli1 constructs were used as the available Gli1 antibodies that detect wild-type Gli1 in Western blots failed to detect untagged Gli1Δ1020–1111 and Gli1Δ764–1016, presumably because they recognize C-terminal motifs in wild-type Gli1 that are absent in the deletion constructs. The expression level of β-tubulin serves as the loading control. (c) Luciferase assays of muscle reporter (Mgn-luc) activity in C3H10T1/2 cells following transfection, as indicated, with a MyoD expression plasmid in conjunction with the various Gli1 deletions depicted in (a). Multiple Gli1 mutants, including those deleting the zinc finger domain (Gli1Δ1–243), the VP16-like activation domain (Gli1Δ1020–1111), and a region spanning amino acids 764–1016 of Gli1, failed to efficiently inhibit MyoD activity in this assay. (d) Luciferase assays of muscle reporter (Mgn-luc and MCK-luc) activity in C2C12 cells following transfection as indicated with various Gli1 deletions depicted in (a). Similar to (c) above, Gli1Δ1-243, Gli1Δ1020–1111, and Gli1Δ764–1016 exhibited a reduced capacity to repress myogenic gene activation. (e) Luciferase assays of Gli reporter (8xGliBSδ51-luc) activity in C3H10T1/2 cells following transfection, as indicated, with the Gli1 deletions constructs. Gli mutants without the fully intact zinc finger domain (Gli1Δ1–243) or the C-terminal activation domain (Gli1Δ1020–1111) failed to efficiently activate the Gli reporter. In contrast, the three internal deletions tested were dispensable for activation in this assay. As previously reported, deletion of the region containing the NES (GliΔ404–593) resulted in stronger transcriptional activation by Gli1. Comparison of the regions required for transcriptional activation by Gli1 and those required for inhibition of MyoD (compare c to b) revealed that the zinc finger domain and the activation domain are required both for Gli1-mediated transcriptional activation and for inhibition of MyoD. In contrast, the region of Gli1 between amino acids 764–1016 is required for antagonism of MyoD activity but not for transcriptional activation. (f) Luciferase assays of MyoD reporter (4RTK-luc) activity in C3H10T1/2 cells following transfection as indicated with various previously described Gli2 deletions. Deletions that included the C-terminal activation domain of Gli2 (Gli2Δ1183–1544 and Gli2Δ641–1544) exhibited a reduced capacity to inhibit MyoD activity in this assay.
To determine if Gli1-mediated inhibition of MyoD and muscle differentiation is correlated with the capacity of Gli1 to activate transcription, we analysed the activation of the Gli reporter 8xGliBSδ51-luc by the various Gli1 deletion mutations (Figure 4e) in C3H10T1/2 cells. As previously reported, the N-terminal region (Gli1Δ1–176) was dispensable for transcriptional activation in this assay, while the C-terminal region (Gli1Δ1020–1111) containing the Gli1 activation domain was required for activation (Yoon et al., 1998). Deletion of a small portion of the zinc finger region (Gli1Δ1–243) also greatly reduced the activation of the Gli reporter. The internal deletions largely retained the capacity to activate transcription in this assay, with deletion of the region containing the putative nuclear export signal (Gli1Δ402–593) resulting in much stronger activation than wild-type Gli1 (Kogerman et al., 1999). Intriguingly, both of the deletions that failed to activate the Gli reporter (Gli1Δ1–243 and Gli1Δ1020–1111) also failed to inhibit MyoD (compare Figures 4e to c–d). However, a third deletion, Gli1Δ764–1016, retained the capacity to activate transcription but failed to substantially inhibit MyoD. Taken together, these results indicate that several regions of Gli1 with distinct functions are required for inhibition of MyoD.
Several domains of Gli2 with distinct functions have been previously described (Sasaki et al., 1999). Like Gli1, Gli2 contains a C-terminal activation domain and a five zinc finger DNA-binding domain. Gli2 also contains an N-terminal domain that represses transcription in some contexts. We wondered whether the C- or N-terminal domains were required for inhibition of MyoD activity by Gli2. We tested the capacity of existing Gli2 mutations to prevent MyoD-mediated activation of 4RTK-luc in transient transfections of C3H10T1/2 cells (Figure 4f). Similar to Gli1, the C-terminal region that contains the Gli2 activation domain and is absent in Gli2Δ1183–1544 and Gli2Δ641–1544 was required for inhibition of MyoD. Also similar to Gli1, the region that is N-terminal to the zinc finger domain and is absent in Gli2Δ1–280 was dispensable for inhibition of MyoD in this assay.
Gli1 reduces MyoD and E protein heterodimer formation
Reduction of heterodimer formation between MyoD and E proteins (including E12 and E47) (Massari and Murre, 2000) has been implicated as a mechanism by which various transcription factors inhibit the capacity of MyoD to activate transcription (Narumi et al., 2000; Sun et al., 2001). We therefore tested if Gli1 reduces heterodimer formation between MyoD and E proteins. For this analysis, we utilized a GAL4-binding site reporter, pG5E1b-luc, and pSG424-MyoDbHLH (GAL4~MyoDbHLH), a fusion between the GAL4 DNA-binding domain and the MyoD bHLH domain previously used to assess for interactions and dimerization with the bHLH portion of MyoD in two-hybrid assays (Verzi et al., 2002). Transfection of GAL4~MyoDbHLH alone or in combination with E12ΔN, an E protein construct that lacks an activation domain, resulted in low activity of the GAL4-binding site reporter (Figure 5a, lanes 1 and 3). In comparison, co-transfection of wild-type E12 or an E47~VP16 activation domain fusion protein induced activation of the reporter (Figure 5a, compare lanes 1 and 3 to lane 5 and 8). Addition of Gli1 reduced the level of activation obtained by the addition of E12 (Figure 5a, compare lane 6 to 5), but had little effect on the activity of E47BVP16 (Figure 5a, compare lane 9 to 8). As E12 mediates activation in this assay through heterodimer formation between the E12 and the MyoD bHLH domains, these results are consistent with the conclusion that heterodimerization is reduced by Gli1. This effect appears to be overcome, however, by the VP16 activation domain present in the E47~VP16 fusion protein. This raises the possibility that the Gli-mediated reduction in GAL4~MyoDbHLH/E12 heterodimer activation of the GAL4-binding site reporter involves inhibition of the native E12 activation domain in the heterodimer context.
Figure 5.

Gli1 reduces the formation of MyoD and E protein heterodimers. (a) Luciferase assays of the GAL4-binding site reporter pG5E1b-luc in C3H10T1/2 cells following transfection. Combinations of the pSG424~MyoDbHLH plasmid encoding GAL4DBD~MyoDbHLH, a fusion between the GAL4 DNA-binding domain (DBD) and MyoD bHLH; E12ΔN, an E12 lacking the activation domain (lane 3 and 4); full-length E12 (lane 5 and 6); E47~VP16, a fusion protein between E47 and the VP16 activation domain (lane 7–9); and Gli1 were transfected as indicated. The addition of either E12 or E47~VP16 to GAL4DBD~MyoDbHLH allows MyoD bHLH to dimerize with E12 or E47 and provides an activation domain to the resultant heterodimeric DNA-binding complex. This leads to substantial activation of the pG5E1b-luc reporter (compare lanes 5 and 8 to 1). The addition of Gli1 reduced the activity associated with the full-length E12 and GAL4DBD~MyobHLH combination (compare lane 6 to 5), suggesting that Gli1 reduces the formation of MyoD and E protein heterodimers in this assay. This effect was overcome by the presence of the heterologous VP16 activation domain (compare lane 9 to 8). (b, c) The plasmids encoding MyoD~E47, a fusion protein between MyoD and E47, or wild-type MyoD were transfected into C3H10T1/2 cells with a control or Gli1 plasmid as indicated. Activation of the 4RTK-luc (b) and Mgn-luc (c) reporters was assayed as described. The addition of Gli1 resulted in less inhibition of MyoD~E47 than of MyoD (compare the shaded bars in each panel).
To further clarify whether the inhibitory effects of Gli1 on MyoD activity are attributable to reduced heterodimer formation, we utilized MyoD~E47, a fusion protein refractory to inhibition by proteins that prevent heterodimer formation of MyoD with E proteins (Neuhold and Wold, 1993). The capacity of MyoD~E47 to activate 4RTK-luc was only slightly reduced (to 82%) by the addition of Gli1, in comparison to the substantial reduction (to 8%) in wild-type MyoD activity observed with the addition of Gli1 (Figure 5b). Likewise, the activation of Mgn-luc by MyoD~E47 was reduced to 37% by the addition of Gli1, in comparison to a much stronger Gli1-mediated reduction (to 4%) of Mgn-luc activity by wild-type MyoD (Figure 5c). We conclude that the obligate MyoD~E47 heterodimer is partially resistant to the inhibitory effects of Gli1. This supports the notion that one mechanism whereby Gli1 reduces the capacity of MyoD to activate transcription involves limiting MyoD~E12 heterodimer formation.
Gli protein reduces the formation of MyoD/E12 heterodimers that bind DNA
To further explore the mechanistic basis for the inhibition of MyoD activity by Gli1, we tested the direct effect of Gli1 on the binding of MyoD/E12 heterodimers to DNA. For this analysis, we used electrophoretic mobility shift assays (EMSA) with an E-box from the Mef2c regulatory region that can be bound strongly by MyoD/E protein heterodimers (Figure 6, lane 3) (Dodou et al., 2003). The addition of increasing amounts of Gli1 modestly reduced binding of the MyoD/E12 complex to this Mef2c E-box (Figure 6, lanes 4–6); similar results were obtained using an MCK E-box (not shown). These results suggest that Gli1 inhibits the formation of MyoD/E12/DNA complexes.
Figure 6.
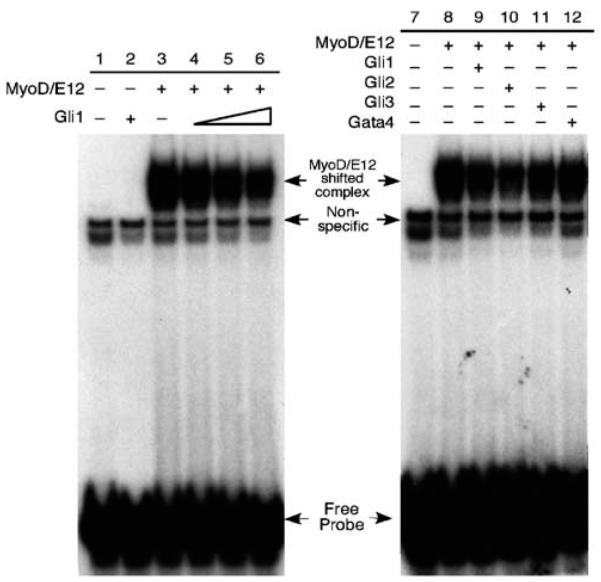
Gli proteins reduce the formation of MyoD/E12 complexes bound to DNA electrophoretic mobility shift analysis (EMSA) of the effects of Gli proteins on the capacity of MyoD and E12 to form heterodimer/DNA complexes. In lanes 1–6, mixtures of in vitro translated (IVT) MyoD, E12 and Gli1 proteins were incubated with radiolabeled, double-stranded oligonucleotides spanning the Mef2c E2 MyoD-binding site. Lane 1: lysate alone; lane 2: Gli1 alone; lane 3: MyoD/E12 alone; lane 3-6: MyoD/E12 plus increasing amounts of Gli1. In samples where a particular protein was not included (denoted by a minus sign), an equal amount of unprogrammed reticulocyte lysate was included. The amount of MyoD/E12/DNA complex (labeled as shifted complex), which runs as a doublet, was modestly reduced with increasing amounts of IVT Gli1 lysate. Experiments in lane 7–12 were performed in a similar way. Gli1 (lane 9), Gli2 (lane 10), Gli3 (lane 11) and Gata4 (lane 12-control) were mixed with MyoD/E12 in this assay. All three Gli proteins appeared to reduce the amount of the MyoD/E12/DNA complex, with Gli2 exhibiting the most dramatic effect.
We also tested whether Gli2 and Gli3 could abrogate the formation of MyoD/E12/DNA complexes using EMSA. Gli2 appeared to cause a more profound inhibition of MyoD/E12/DNA complex formation (Figure 6, lane 10) than Gli1 (lane 9), while Gli3 had an effect that was similar to that of Gli1 (Figure 6, compare lane 11 to 9). We also tested several control proteins, including Gata4, which is another zinc finger protein (Figure 6, lane 12), and an unrelated transcription factor, Xenopus Tcf7 (not shown); the addition of equal or greater amounts of control factors had only a small effect on MyoD/E12 binding to DNA. We conclude that Gli1 and Gli2 inhibit myogenesis in part by interfering with heterodimer formation and DNA binding of the MyoD family of bHLH proteins, a novel activity for the Gli protein family.
Discussion
Elevated Hh signaling and expression of the oncogene Gli1 is associated with rhabdomyosarcomas in humans and mice. As disruption of the normal MyoD family-dependent myogenic program is thought to be a requirement for preventing terminal differentiation and cell cycle withdrawal in rhabdomyosarcoma (Merlino and Helman, 1999), the novel capacity of Gli1 to inhibit transcriptional activation and DNA binding by MyoD provides a possible mechanism whereby Hh pathway activation promotes the formation of a specific tumor. Our study complements and yet contrasts prior work showing that Hh signaling activates genes such as the cyclins and N-myc, which promote proliferation in a general fashion (Duman-Scheel et al., 2002; Kenney et al., 2003; Oliver et al., 2003).
Our work supports a developmental model (Figure 7) where the Hh signal activates or maintains myogenic bHLH family expression during development (Gustafsson et al., 2002; Teboul et al., 2003), while initially repressing myogenesis through Gli-mediated antagonism of transcriptional activation by MyoD and Myf5. In an extension of this model, persistent or inappropriate activation of Hh signaling in individual cells would prevent or retard normal terminal muscle differentiation and promote rhabdomyosarcoma formation. Parallels exist between the model we propose for Hh signaling and the activities of Pax3 and Pax7, which can play a causative role in rhabdomyosarcoma formation in the context of gain of function fusion with FKHR, a forkhead protein (Barr, 2001). In normal development, similar to the combinatorial effects of the Gli proteins (McDermott et al., 2005), wild-type Pax3 and Pax7 both play roles in specifying portions of the muscle lineage (Tajbakhsh et al., 1997; Seale et al., 2000). Moreover, like Gli1 and Gli2, overexpression of either Pax3 or Pax7 can prevent muscle differentiation (Epstein et al., 1995; Olguin and Olwin, 2004). Recent reports also suggest roles for both Pax7 and Hh signaling in adult muscle satellite cells, which are undifferentiated, tissue-resident, myogenic stem cells (Koleva et al., 2005; Kuang et al., 2006; Relaix et al., 2006). It would be interesting to determine whether active Hh signaling occurs in a less differentiated subset of cells within primary rhabdomyosarcoma tissue. Whether rhabdomyosarcoma arises secondary to aberrant satellite cell programming is also a subject of ongoing investigation (Keller et al., 2004a, b; Tiffin et al., 2003).
Figure 7.

A simplified model of Hh signaling, myogenesis, and rhabdomyosarcoma. In response to Shh signaling, the Gli proteins participate in specifying myogenic precursors by promoting expression of members of the MyoD family including Myf-5 and MyoD (a). The Gli proteins also inhibit terminal differentiation of myogenic precursors by preventing the myogenic bHLH proteins from forming heterodimers with E proteins, binding DNA (E-boxes), and activating muscle gene transcription (b). Inhibition of differentiation, in combination with Shh-mediated activation of cyclins, allows for proliferation of the myogenic precursor pool (c). However, Shh signaling also activates Ptch1 and Hip1, both of which antagonize Shh signaling in a negative feedback loop (Chen and Struhl, 1996; Chuang and McMahon, 1999; Chuang and McMahon, 2003; Jeong and McMahon, 2005) (d). As a consequence of attenuated Shh signaling, Gli-mediated antagonism of myogenic bHLH proteins is reduced. This allows muscle gene transcription, terminal differentiation and myotube formation to occur. However, when Shh signaling is not appropriately attenuated due to abnormalities in Hip1 or Ptch1, persistent inhibition of MyoD activity could promote proliferation and cancer formation. The molecular mechanisms that coordinately control the switches between different states during myogenesis in vitro and in vivo need to be further investigated.
Similar to our observations, other groups have noted decreased Hh signaling in ex vivo tumor cell lines generated from mice with abnormalities in Hh signaling (Romer et al., 2004; H Hahn, personal communication). It was also recently reported that the growth of tumor allografts established from ex vivo Ptch1+/− medulloblastomas was no longer sensitive to Hh pathway antagonism (Sasai et al., 2006). We favor a model where culture conditions allow the utilization of multiple signaling pathways to support growth of Ptch1+/− tumor cells. In that regard, gene expression signatures within primary human rhabdomyosarcomas are consistent with multiple active growth factor pathways (Blandford et al., 2006) and it has been previously proposed that such heterogeneity within tumors contributes to the development of resistance to therapy (Dai et al., 2004). It would be interesting to determine whether an important role for the Hh pathway in promoting cell survival and tumor growth reemerges within Ptch1+/− tumor lines or allografts following selection with cytotoxic agents.
On the surface, our results demonstrating Gli-mediated inhibition of muscle differentiation appear opposite to previous reports that showed enhanced myogenic differentiation associated with Hh signaling (Amthor et al., 1999; Duprez et al., 1998; Kruger et al., 2001; Li et al., 2004). Interestingly, however, our findings are consistent with inhibitory effects of Shh on muscle differentiation observed in other experimental conditions (Bren-Mattison and Olwin, 2002). One explanation of these discrepancies is that the effects of the Hh signal on muscle differentiation in vivo are likely to vary with timing and level of Hh signaling (Wolff et al., 2003). For example, activation of Hh target genes may be modulated by temporal changes in the cellular environment such as the extracellular matrix or the conformation of nuclear chromatin that impact signal transduction. In that regard, histone deacetylases (HDACs) are known to regulate muscle gene expression and MyoD family activity in a temporally dependent manner (Iezzi et al., 2002). Although the MyoD family generally functions in activating muscle gene expression, certain muscle genes are actually repressed through the binding of MyoD to DNA in an HDAC1-dependent process (Puri et al., 2001; Mal and Harter, 2003). Therefore, inhibition of MyoD binding to DNA induced by Hh signaling could potentially induce upregulation of a different subset of muscle genes, explaining the enhanced myogenic differentiation observed in certain experimental contexts (Duprez et al., 1998; Amthor et al., 1999; Kruger et al., 2001; Li et al., 2004). In this fashion, elevated Hh signaling in a subset of tumor cells could also contribute to the heterogeneous partial differentiation phenotype observed in rhabdomyosar coma.
Our data suggest that Gli1 antagonizes MyoD activity through more than one mechanism. In support of this notion, multiple domains of Gli1 with distinct functions and activities were required for inhibition of MyoD activity. One mechanism appears to involve limiting MyoD/E12 heterodimer formation and DNA binding, a mechanism that is employed by many other transcription factors that inhibit MyoD activity such as Smad3 (Liu et al., 2001, 2004), Sharp-1 (Azmi et al., 2003) and CHF2 (Sun et al., 2001). It remains to be determined if this mechanism involves direct physical interaction between the Gli proteins and MyoD. A second mechanism whereby Gli1 inhibits myogenic differentiation is likely attributable to the actions of Gli target genes. Consistent with this notion, Gli2, which can function as a transcriptional activator, also inhibited myogenesis, while Gli3, which is not a strong transcriptional activator, failed to inhibit myogenesis. Gli3 did reduce the formation of MyoD/E12/DNA complexes in EMSA analysis, although direct comparisons of Gli1, 2, and 3 in this analysis are complicated by observations that Gli3 undergoes substantial post-translational modifications that may not be present within in vitro translated protein products (Dai et al., 1999; Aza-Blanc et al., 2000; Wang et al., 2000). Further experiments are required to fully determine the distinct domains, biochemical properties and expression profiles of the Gli proteins that mediate inhibition of myogenesis and promote rhabdomyosarcoma formation. Indeed, Gli1 can at least partially substitute for Gli2 in development (Bai and Joyner, 2001), while Gli2 can compensate for the absence of Gli1 in the formation of some tumors (Weiner et al., 2002), suggesting that regulation of Gli transcription plays an important role in their functional differences. Although we only observed low levels of Gli2 expression in our tumor samples, since Gli2 appears to have the intrinsic capacity to inhibit myogenesis, it would be interesting to determine whether Gli2 is expressed in human rhadomyosarcoma.
The MyoD family has served as a paradigm for understanding the functions of other bHLH proteins, which are critical determinants of cell specification and differentiation in many tissues in addition to skeletal muscle. It is therefore notable that other bHLH proteins including Mash1, whose activity we demonstrated was inhibited by Gli1, are expressed in tissues and tumors where Hh signaling is active (Ball et al., 1993; Lu et al., 2000; Berman et al., 2002; Watkins et al., 2003). Future investigations will be required to determine if inhibition of bHLH proteins such as MyoD and Mash1 is a general mechanism through which the Gli proteins modulate cell fate and promote tumor formation in different contexts. The relative contribution of the direct and indirect effects of the Gli proteins in modulating bHLH protein activity also remains to be determined.
Materials and methods
Molecular biology
Standard molecular biology techniques, including molecular cloning, genomic DNA preparation, RNA isolation, PCR, Southern analysis and histologic techniques were performed as described (Sambrook and Russell, 2001; Nagy et al., 2003).
Iimmunohistochemistry and 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-Gal) staining
Immunostaining was performed following standard procedures. Primary antibodies and dilutions used were: mouse anti-myosin heavy chain (MyHC) MF20 at 1:10 (Developmental Studies Hybridoma Bank), mouse anti-myogenin F5D at 1:10 (gift of Stephen Tapscott), mouse anti-sarcomeric actin at 1:400, mouse anti-acetylated tubulin at 1:2000 (Sigma, MO, USA), rabbit anti-Smo at 1:500, and rabbit anti-Gli1 at 1:400 (Santa Cruz). Secondary antibodies (goat anti-rabbit Alexa 488 and goat anti-mouse Alexa 594 antibodies) were used at a dilution of 1:400. Nuclear staining was performed with a 4′,6-diamidino-2-phenylindole (DAPI). The production of the anti-Smo antibody will be described elsewhere.
For X-gal staining, the RMP, RMH, and the C2C12 cells were transfected with 0.1 μg of muscle reporter (e.g., MCK-lacZ, RSV-lacZ) together with 0.3–0.4 μg of control or Gli expression plasmids. Following transfection cells were cultured in growth media for 24–48 h, switched into differentiation media and assayed after an additional 36–48 h. X-gal staining of cultured cells and tissue sections was performed following standard procedures.
Northern blot analysis
Total RNA was isolated from confluent cells grown on a 10-cm tissue culture dish using the Trizol reagent (Invitrogen) according to the manufacturer’s instructions. Total RNA (~15 μg) was used in each lane and Northern blots were hybridized with probes specific to MyHC, mouse Gli1 and human GAPDH following standard procedures.
Plasmids, cell culture, transfections and luciferase assays
Details on plasmids and the generation of rhabdomyosarcoma cell lines are contained in the supplementary material. The standard growth media used for all cell lines was DMEM (Cellgro) supplemented with glutamine, penicillin, streptomycin and 15% fetal bovine serum (FBS) (all from Invitrogen). For differentiation assays, the growth media was switched to DMEM supplemented with penicillin, streptomycin, glutamine, 1% heat inactivated horse serum (Invitrogen), insulin (10 μg/ml) (Sigma) and transferrin (10 μg/ml) (Sigma).
Transfections were performed using Lipofectamine Plus reagents (Invitrogen, CA, USA). The day before transfection, 0.5 × 105 cells were plated in 24-well dishes. For transfection of the RMP, RMH, and the C2C12 lines, 0.1 μg of muscle reporter (e.g., MCK-luciferase, Mgn-luciferase) or Gli1 reporter (8xGliBSδ51-luciferase) and 0.05 μg of CMV-Renilla luciferase were co-transfected with 0.3–0.4 μg of control or Gli expression plasmids. For transfection of C3H10T1/2 cells, 0.1 μg of muscle reporter (e.g., MCK-luciferase, Mgn-luciferase, MyHC-luciferase, 4RTK-luciferase, 8xGliBSδ51-luciferase) and 0.05 μg of CMV-Renilla luciferase were co-transfected with 0.1 or 0.2 μg of control or activator plasmid (e.g., EMSV-MyoD, MyoD~E47 and various deletion constructs of MyoD, EMSV-Myf5, RSV-Mash1 and CS2-E12) and 0.2 μg of control or Gli expression plasmid (including various Gli1 deletion constructs). Alternatively, 0.1 μg of GAL4-binding site reporter pG5E1b-luciferase and 0.05 μg of CMV-Renilla luciferase were co-transfected with 0.1 or 0.2 μg of activator plasmid (e.g., GAL4DBD~MyoDbHLH) and different combinations of 0.2 μg of expression plasmids encoding Gli, E12ΔN, E12 or E47~VP16. DMEM with 1.5 μl of plus reagent was added to each DNA mix to a total volume of 25 μl. An equal volume of DMEM with 1.5 μl of lipofectamine was also prepared for each sample. After 15 min at room temperature, the DMEM/DNA/Plus reagent was mixed with the DMEM/lipofectamine. After another 15 min, the 50 μl mix was added to 200 μl of DMEM and added to each well of cells. The cells were incubated at 37°C for 3–4 h after which the media was changed to regular growth media. Following transfection cells were cultured in growth media for 24–48 h, switched into differentiation media and assayed after an additional 36–48 h.
Luciferase assays were performed using the Promega’s Dual-Luciferase Reporter (DLR) Assay system. Briefly, cells were harvested in 125 μl of 1x Passive Lysis Buffer (Promega, WI, USA) and 10 μl of lysate was used for analysis. Luciferase assay buffer (40 μl) was added to each well and the luminescence was determined. Stop Reagent (40 μl) was added to each well and luminescence was again determined. Luminescence readings and sample processing were accompanied using a Luminescence Microplate Reader (Molecular Devices, CA, USA). Each luciferase assay was done in triplicate and normalized to a co-transfected CMV-Renilla luciferase control. All experiments were repeated multiple times. Normalized averages and standard deviations were shown. As noted in the figures, activity is depicted as a percentage relative to the activity achieved in the absence of Gli proteins, which was set at 100%.
Shh-conditioned medium was prepared from transfected Bosc23 cells as previously described (Chen et al., 2004). For immunohistochemical analysis of Smo expression in the primary cilium, cells were cultured in Shh-conditioned medium for 24 h and then in differentiation media for 12 h. The expression of myogenin in groups of cells with Smo in the primary cilium was compared to expression of myogenin in the cell population as a whole. For statistical analysis, the average percentage of cells with Smo expression in the cilium that expressed myogenin was compared to the average observed in the whole population using a t-test on five groups of 20 cells.
Western blotting
Western blots were performed following standard procedures. Dishes (3.5 cm) of Cos1 cells at 50% confluence were transfected with 1.5 μg of various Gli1 expression plasmids. Transfection efficiencies were monitored by co-transfection with a GFP expression plasmid. Cells were harvested 48 h after transfection and proteins analysed using standard procedures. Primary antibodies and dilution used were rabbit anti-Gli1 (1:1000) (Santa Cruz), mouse anti-FLAG (1:2000) (Sigma) and mouse anti-β-tubulin (1:2000) (Sigma, MO, USA).
Electrophoretic mobility shift assay
Double-stranded oligonucleotides (2 μg) representing the Mef2c E2 E-box were labeled with 32P-dCTP and purified on a nondenaturing polyacrylamide–TBE gel. Recombinant proteins were generated from pCITE2B-MyoD, pCITE2A-E12, pcDNA-Gata4, pcDNA-Myc Gli1, pcDNA-FLAG Gli2, pcDNA-FLAG Gli3 and pT7XTcf plasmids using the TNT Coupled Transcription/Translation System according to the manufacturer’s instructions (Promega). Combinations of translated products were mixed as indicated in the text and legend of Figure 6 and total lysate amount added to binding reactions was kept constant through the addition of unprogrammed lysate. Binding reactions consisting of lysate mixtures (5–5.5 μl) and 1 μg of poly dI-dC (Sigma) in 1 binding buffer (40 mM KCL, 15 mM HEPES ph 7.9, 1 m × M EDTA, 0.1 mM DTT, 5% glycerol) were incubated for 10 min prior to probe addition. Reactions were incubated for an additional 20 min at room temperature after probe addition and electrophoresed on a 6% nondenaturing polyacrylamide–TBE gel. Similar quantities of Gli1, Gli2, Gli3 and Gata4 protein generated from in vitro transcription/translation were used in EMSA as visualized on SDS–PAGE.
Supplementary Material
Acknowledgements
We are indebted to those who provided plasmids and cell lines that we used for this work, including Stephen Tapscott, Steven Swoap, Brian Black, Hiroshi Sasaki, Bert Vogelstein, Robert Hill, Kenneth Baldwin, Miao-Hsueh Chen and Hisato Kondoh. We thank Stephen Tapscott, Brian Black, and Analeah Heidt for helpful discussion and Nicholas Gerber, Brian Black, Matthias Hebrok, Zena Werb, and Analeah Heidt for critical reading of the manuscript. This work was supported by NIH grant HL067822, NHLBI-funded Program for Genomics Applications (“BayGenomics”) HL66600; and NIH grant K08HL07715 and a Parker B. Francis Fellowship (A.N.G).
Footnotes
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc).
References
- Amthor H, Christ B, Patel K. A molecular mechanism enabling continuous embryonic muscle growth –a balance between proliferation and differentiation. Development. 1999;126:1041–1053. doi: 10.1242/dev.126.5.1041. [DOI] [PubMed] [Google Scholar]
- Aszterbaum M, Epstein J, Oro A, Douglas V, LeBoit PE, Scott MP, et al. Ultraviolet and ionizing radiation enhance the growth of BCCs and trichoblastomas in patched heterozygous knockout mice. Nat Med. 1999;5:1285–1291. doi: 10.1038/15242. [DOI] [PubMed] [Google Scholar]
- Aza-Blanc P, Lin HY, Ruiz i Altaba A, Kornberg TB. Expression of the vertebrate Gli proteins in Drosophila reveals a distribution of activator and repressor activities. Development. 2000;127:4293–4301. doi: 10.1242/dev.127.19.4293. [DOI] [PubMed] [Google Scholar]
- Azmi S, Sun H, Ozog A, Taneja R. mSharp-1/DEC2, a basic helix-loop-helix protein functions as a transcriptional repressor of E box activity and Stra13 expression. J Biol Chem. 2003;278:20098–20109. doi: 10.1074/jbc.M210427200. [DOI] [PubMed] [Google Scholar]
- Bai CB, Joyner AL. Gli1 can rescue the in vivo function of Gli2. Development. 2001;128:5161–5172. doi: 10.1242/dev.128.24.5161. [DOI] [PubMed] [Google Scholar]
- Ball DW, Azzoli CG, Baylin SB, Chi D, Dou S, Donis-Keller H, et al. Identification of a human achaete-scute homolog highly expressed in neuroendocrine tumors. Proc Natl Acad Sci USA. 1993;90:5648–5652. doi: 10.1073/pnas.90.12.5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr FG. Gene fusions involving PAX and FOX family members in alveolar rhabdomyosarcoma. Oncogene. 2001;20:5736–5746. doi: 10.1038/sj.onc.1204599. [DOI] [PubMed] [Google Scholar]
- Beachy PA, Karhadkar SS, Berman DM. Tissue repair and stem cell renewal in carcinogenesis. Nature. 2004;432:324–331. doi: 10.1038/nature03100. [DOI] [PubMed] [Google Scholar]
- Bengal E, Flores O, Rangarajan PN, Chen A, Weintraub H, Verma IM. Positive control mutations in the MyoD basic region fail to show cooperative DNA binding and transcriptional activation in vitro. Proc Natl Acad Sci USA. 1994;91:6221–6225. doi: 10.1073/pnas.91.13.6221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman DM, Karhadkar SS, Hallahan AR, Pritchard JI, Eberhart CG, Watkins DN, et al. Medulloblastoma growth inhibition by hedgehog pathway blockade. Science. 2002;297:1559–1561. doi: 10.1126/science.1073733. [DOI] [PubMed] [Google Scholar]
- Blandford MC, Barr FG, Lynch JC, Randall RL, Qualman SJ, Keller C. Rhabdomyosarcomas utilize developmental, myogenic growth factors for disease advantage: a report from the Children’s Oncology Group. Pediatr Blood Cancer. 2006;46:329–338. doi: 10.1002/pbc.20466. [DOI] [PubMed] [Google Scholar]
- Borycki AG, Brunk B, Tajbakhsh S, Buckingham M, Chiang C, Emerson CP., Jr Sonic hedgehog controls epaxial muscle determination through Myf5 activation. Development. 1999;126:4053–4063. doi: 10.1242/dev.126.18.4053. [DOI] [PubMed] [Google Scholar]
- Bren-Mattison Y, Olwin BB. Sonic hedgehog inhibits the terminal differentiation of limb myoblasts committed to the slow muscle lineage. Dev Biol. 2002;242:130–148. doi: 10.1006/dbio.2001.0528. [DOI] [PubMed] [Google Scholar]
- Briscoe J, Therond P. Hedgehog signaling: from the Drosophila cuticle to anti-cancer drugs. Dev Cell. 2005;8:143–151. doi: 10.1016/j.devcel.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Buckingham M. Skeletal muscle formation in vertebrates. Curr Opin Genet Dev. 2001;11:440–448. doi: 10.1016/s0959-437x(00)00215-x. [DOI] [PubMed] [Google Scholar]
- Buckingham M, Bajard L, Chang T, Daubas P, Hadchouel J, Meilhac S, et al. The formation of skeletal muscle: from somite to limb. J Anat. 2003;202:59–68. doi: 10.1046/j.1469-7580.2003.00139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MH, Li YJ, Kawakami T, Xu SM, Chuang P-T. Palmitoylation is required for the production of a soluble multimeric Hedgehog protein complex and long-range signaling in vertebrates. Genes Dev. 2004;18:641–659. doi: 10.1101/gad.1185804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Struhl G. Dual roles for patched in sequestering and transducing Hedgehog. Cell. 1996;87:553–563. doi: 10.1016/s0092-8674(00)81374-4. [DOI] [PubMed] [Google Scholar]
- Chuang P-T, Kawcak T, McMahon AP. Feedback control of mammalian Hedgehog signaling by the Hedgehog-binding protein, Hip1, modulates Fgf signaling during branching morphogenesis of the lung. Genes Dev. 2003;17:342–347. doi: 10.1101/gad.1026303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang P-T, McMahon AP. Vertebrate Hedgehog signalling modulated by induction of a Hedgehog-binding protein. Nature. 1999;397:617–621. doi: 10.1038/17611. [DOI] [PubMed] [Google Scholar]
- Chuang P-T, McMahon AP. Branching morphogenesis of the lung: new molecular insights into an old problem. Trends Cell Biol. 2003;13:86–91. doi: 10.1016/s0962-8924(02)00031-4. [DOI] [PubMed] [Google Scholar]
- Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF. Vertebrate Smoothened functions at the primary cilium. Nature. 2005;437:1018–1021. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- Dai P, Akimaru H, Tanaka Y, Maekawa T, Nakafuku M, Ishii S. Sonic Hedgehog-induced activation of the Gli1 promoter is mediated by GLI3. J Biol Chem. 1999;274:8143–8152. doi: 10.1074/jbc.274.12.8143. [DOI] [PubMed] [Google Scholar]
- Dai Z, Huang Y, Sadee W. Growth factor signaling and resistance to cancer chemotherapy. Curr Top Med Chem. 2004;4:1347–1356. doi: 10.2174/1568026043387746. [DOI] [PubMed] [Google Scholar]
- Dodou E, Xu SM, Black BL. mef2c is activated directly by myogenic basic helix-loop-helix proteins during skeletal muscle development in vivo. Mech Dev. 2003;120:1021–1032. doi: 10.1016/s0925-4773(03)00178-3. [DOI] [PubMed] [Google Scholar]
- Duman-Scheel M, Weng L, Xin S, Du W. Hedgehog regulates cell growth and proliferation by inducing Cyclin D and Cyclin E. Nature. 2002;417:299–304. doi: 10.1038/417299a. [DOI] [PubMed] [Google Scholar]
- Duprez D, Fournier-Thibault C, Le Douarin N. Sonic Hedgehog induces proliferation of committed skeletal muscle cells in the chick limb. Development. 1998;125:495–505. doi: 10.1242/dev.125.3.495. [DOI] [PubMed] [Google Scholar]
- Epstein JA, Lam P, Jepeal L, Maas RL, Shapiro DN. Pax3 inhibits myogenic differentiation of cultured myoblast cells. J Biol Chem. 1995;270:11719–11722. doi: 10.1074/jbc.270.20.11719. [DOI] [PubMed] [Google Scholar]
- Fiddler TA, Smith L, Tapscott SJ, Thayer MJ. Amplification of MDM2 inhibits MyoD-mediated myogenesis. Mol Cell Biol. 1996;16:5048–5057. doi: 10.1128/mcb.16.9.5048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber AN, Klesert TR, Bergstrom DA, Tapscott SJ. Two domains of MyoD mediate transcriptional activation of genes in repressive chromatin: a mechanism for lineage determination in myogenesis. Genes Dev. 1997;11:436–450. doi: 10.1101/gad.11.4.436. [DOI] [PubMed] [Google Scholar]
- Goodrich LV, Johnson RL, Milenkovic L, McMahon JA, Scott MP. Conservation of the hedgehog/patched signaling pathway from flies to mice: induction of a mouse patched gene by Hedgehog. Genes Dev. 1996;10:301–312. doi: 10.1101/gad.10.3.301. [DOI] [PubMed] [Google Scholar]
- Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- Gorlin RJ. Nevoid basal-cell carcinoma syndrome. Medicine (Baltimore) 1987;66:98–113. doi: 10.1097/00005792-198703000-00002. [DOI] [PubMed] [Google Scholar]
- Guo CS, Degnin C, Fiddler TA, Stauffer D, Thayer MJ. Regulation of MyoD activity and muscle cell differentiation by MDM2, pRb, and Sp1. J Biol Chem. 2003;278:22615–22622. doi: 10.1074/jbc.M301943200. [DOI] [PubMed] [Google Scholar]
- Gustafsson MK, Pan H, Pinney DF, Liu Y, Lewandowski A, Epstein DJ, et al. Myf5 is a direct target of long-range Shh signaling and Gli regulation for muscle specification. Genes Dev. 2002;16:114–126. doi: 10.1101/gad.940702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn H, Wojnowski L, Miller G, Zimmer A. The patched signaling pathway in tumorigenesis and development: lessons from animal models. J Mol Med. 1999;77:459–468. doi: 10.1007/s001099900018. [DOI] [PubMed] [Google Scholar]
- Hahn H, Wojnowski L, Zimmer AM, Hall J, Miller G, Zimmer A. Rhabdomyosarcomas and radiation hypersensitivity in a mouse model of Gorlin syndrome. Nat Med. 1998;4:619–622. doi: 10.1038/nm0598-619. [DOI] [PubMed] [Google Scholar]
- Hooper JE, Scott MP. Communicating with Hedgehogs. Nat Rev Mol Cell Biol. 2005;6:306–317. doi: 10.1038/nrm1622. [DOI] [PubMed] [Google Scholar]
- Huangfu D, Anderson KV. Cilia and Hedgehog responsiveness in the mouse. Proc Natl Acad Sci USA. 2005;102:11325–11330. doi: 10.1073/pnas.0505328102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iezzi S, Cossu G, Nervi C, Sartorelli V, Puri PL. Stage-specific modulation of skeletal myogenesis by inhibitors of nuclear deacetylases. Proc Natl Acad Sci USA. 2002;99:7757–7762. doi: 10.1073/pnas.112218599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001;15:3059–3087. doi: 10.1101/gad.938601. [DOI] [PubMed] [Google Scholar]
- Jeong J, McMahon AP. Growth and pattern of the mammalian neural tube are governed by partially overlapping feedback activities of the hedgehog antagonists patched 1 and Hhip1. Development. 2005;132:143–154. doi: 10.1242/dev.01566. [DOI] [PubMed] [Google Scholar]
- Kappler R, Bauer R, Calzada-Wack J, Rosemann M, Hemmerlein B, Hahn H. Profiling the molecular difference between Patched- and p53-dependent rhabdomyosarcoma. Oncogene. 2004;23:8785–8795. doi: 10.1038/sj.onc.1208133. [DOI] [PubMed] [Google Scholar]
- Kappler R, Calzada-Wack J, Schnitzbauer U, Koleva M, Herwig A, Piontek G, et al. Molecular characterization of Patched-associated rhabdomyosarcoma. J Pathol. 2003;200:348–356. doi: 10.1002/path.1361. [DOI] [PubMed] [Google Scholar]
- Keller C, Arenkiel BR, Coffin CM, El-Bardeesy N, DePinho RA, Capecchi MR. Alveolar rhabdomyosarcomas in conditional Pax3: Fkhr mice: cooperativity of Ink4a/ARF and Trp53 loss of function. Genes Dev. 2004a;18:2614–2626. doi: 10.1101/gad.1244004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller C, Hansen MS, Coffin CM, Capecchi MR. Pax3:Fkhr interferes with embryonic Pax3 and Pax7 function: implications for alveolar rhabdomyosarcoma cell of origin. Genes Dev. 2004b;18:2608–2613. doi: 10.1101/gad.1243904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney AM, Cole MD, Rowitch DH. Nmyc upregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development. 2003;130:15–28. doi: 10.1242/dev.00182. [DOI] [PubMed] [Google Scholar]
- Kogerman P, Grimm T, Kogerman L, Krause D, Unden AB, Sandstedt B, et al. Mammalian suppressor-of-fused modulates nuclear-cytoplasmic shuttling of Gli-1. Nat Cell Biol. 1999;1:312–319. doi: 10.1038/13031. [DOI] [PubMed] [Google Scholar]
- Koleva M, Kappler R, Vogler M, Herwig A, Fulda S, Hahn H. Pleiotropic effects of sonic hedgehog on muscle satellite cells. Cell Mol Life Sci. 2005;62:1863–1870. doi: 10.1007/s00018-005-5072-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger M, Mennerich D, Fees S, Schafer R, Mundlos S, Braun T. Sonic hedgehog is a survival factor for hypaxial muscles during mouse development. Development. 2001;128:743–752. doi: 10.1242/dev.128.5.743. [DOI] [PubMed] [Google Scholar]
- Kuang S, Charge SB, Seale P, Huh M, Rudnicki MA. Distinct roles for Pax7 and Pax3 in adult regenerative myogenesis. J Cell Biol. 2006;172:103–113. doi: 10.1083/jcb.200508001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Blagden CS, Bildsoe H, Bonnin MA, Duprez D, Hughes SM. Hedgehog can drive terminal differentiation of amniote slow skeletal muscle. BMC Dev Biol. 2004;4:9. doi: 10.1186/1471-213X-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu A, Wang B, Niswander LA. Mouse intraflagellar transport proteins regulate both the activator and repressor functions of Gli transcription factors. Development. 2005;132:3103–3111. doi: 10.1242/dev.01894. [DOI] [PubMed] [Google Scholar]
- Liu D, Black BL, Derynck R. TGF-beta inhibits muscle differentiation through functional repression of myogenic transcription factors by Smad3. Genes Dev. 2001;15:2950–2966. doi: 10.1101/gad.925901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Kang JS, Derynck R. TGF-beta-activated Smad3 represses MEF2-dependent transcription in myogenic differentiation. Embo J. 2004;23:1557–1566. doi: 10.1038/sj.emboj.7600179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu QR, Yuk D, Alberta JA, Zhu Z, Pawlitzky I, Chan J, et al. Sonic hedgehog–regulated oligodendrocyte lineage genes encoding bHLH proteins in the mammalian central nervous system. Neuron. 2000;25:317–329. doi: 10.1016/s0896-6273(00)80897-1. [DOI] [PubMed] [Google Scholar]
- Lum L, Beachy PA. The Hedgehog response network: sensors, switches, and routers. Science. 2004;304:1755–1759. doi: 10.1126/science.1098020. [DOI] [PubMed] [Google Scholar]
- Mal A, Harter ML. MyoD is functionally linked to the silencing of a muscle-specific regulatory gene prior to skeletal myogenesis. Proc Natl Acad Sci USA. 2003;100:1735–1739. doi: 10.1073/pnas.0437843100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marigo V, Scott MP, Johnson RL, Goodrich LV, Tabin CJ. Conservation in hedgehog signaling: induction of a chicken patched homolog by Sonic hedgehog in the developing limb. Development. 1996;122:1225–1233. doi: 10.1242/dev.122.4.1225. [DOI] [PubMed] [Google Scholar]
- Massari ME, Murre C. Helix-loop-helix proteins: regulators of transcription in eucaryotic organisms. Mol Cell Biol. 2000;20:429–440. doi: 10.1128/mcb.20.2.429-440.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May SR, Ashique AM, Karlen M, Wang B, Shen Y, Zarbalis K, et al. Loss of the retrograde motor for IFT disrupts localization of Smo to cilia and prevents the expression of both activator and repressor functions of Gli. Dev Biol. 2005;287:378–389. doi: 10.1016/j.ydbio.2005.08.050. [DOI] [PubMed] [Google Scholar]
- McDermott A, Gustafsson M, Elsam T, Hui CC, Emerson CP, Jr, Borycki AG. Gli2 and Gli3 have redundant and context-dependent function in skeletal muscle formation. Development. 2005;132:345–357. doi: 10.1242/dev.01537. [DOI] [PubMed] [Google Scholar]
- McMahon AP, Ingham PW, Tabin CJ. Developmental roles and clinical significance of hedgehog signaling. Curr Top Dev Biol. 2003;53:1–114. doi: 10.1016/s0070-2153(03)53002-2. [DOI] [PubMed] [Google Scholar]
- Merlino G, Helman LJ. Rhabdomyosarcoma–working out the pathways. Oncogene. 1999;18:5340–5348. doi: 10.1038/sj.onc.1203038. [DOI] [PubMed] [Google Scholar]
- Munsterberg AE, Kitajewski J, Bumcrot DA, McMahon AP, Lassar AB. Combinatorial signaling by Sonic hedgehog and Wnt family members induces myogenic bHLH gene expression in the somite. Genes Dev. 1995;9:2911–2922. doi: 10.1101/gad.9.23.2911. [DOI] [PubMed] [Google Scholar]
- Nagy A, Gertsenstein M, Vintersten K, Behringer R. Manipulating the Mouse Embryo: A Laboratory Manual. 3rd edn Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2003. [Google Scholar]
- Narumi O, Mori S, Boku S, Tsuji Y, Hashimoto N, Nishikawa S, et al. OUT, a novel basic helix-loop-helix transcription factor with an Id-like inhibitory activity. J Biol Chem. 2000;275:3510–3521. doi: 10.1074/jbc.275.5.3510. [DOI] [PubMed] [Google Scholar]
- Neuhold LA, Wold B. HLH forced dimers: tethering MyoD to E47 generates a dominant positive myogenic factor insulated from negative regulation by Id. Cell. 1993;74:1033–1042. doi: 10.1016/0092-8674(93)90725-6. [DOI] [PubMed] [Google Scholar]
- Nieuwenhuis E, Hui CC. Hedgehog signaling and congenital malformations. Clin Genet. 2005;67:193–208. doi: 10.1111/j.1399-0004.2004.00360.x. [DOI] [PubMed] [Google Scholar]
- Olguin HC, Olwin BB. Pax-7 up-regulation inhibits myogenesis and cell cycle progression in satellite cells: a potential mechanism for self-renewal. Dev Biol. 2004;275:375–388. doi: 10.1016/j.ydbio.2004.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver TG, Grasfeder LL, Carroll AL, Kaiser C, Gillingham CL, Lin SM, et al. Transcriptional profiling of the Sonic hedgehog response: a critical role for N-myc in proliferation of neuronal precursors. Proc Natl Acad Sci USA. 2003;100:7331–7336. doi: 10.1073/pnas.0832317100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Bai CB, Joyner AL, Wang B. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol Cell Biol. 2006;26:3365–3377. doi: 10.1128/MCB.26.9.3365-3377.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasca di Magliano M, Hebrok M. Hedgehog signalling in cancer formation and maintenance. Nat Rev Cancer. 2003;3:903–911. doi: 10.1038/nrc1229. [DOI] [PubMed] [Google Scholar]
- Puri PL, Iezzi S, Stiegler P, Chen TT, Schiltz RL, Muscat GE, et al. Class I histone deacetylases sequentially interact with MyoD and pRb during skeletal myogenesis. Mol Cell. 2001;8:885–897. doi: 10.1016/s1097-2765(01)00373-2. [DOI] [PubMed] [Google Scholar]
- Puri PL, Sartorelli V. Regulation of muscle regulatory factors by DNA-binding, interacting proteins, and post-transcriptional modifications. J Cell Physiol. 2000;185:155–173. doi: 10.1002/1097-4652(200011)185:2<155::AID-JCP1>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Relaix F, Montarras D, Zaffran S, Gayraud-Morel B, Rocancourt D, Tajbakhsh S, et al. Pax3 and Pax7 have distinct and overlapping functions in adult muscle progenitor cells. J Cell Biol. 2006;172:91–102. doi: 10.1083/jcb.200508044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romer JT, Kimura H, Magdaleno S, Sasai K, Fuller C, Baines H, et al. Suppression of the Shh pathway using a small molecule inhibitor eliminates medulloblastoma in Ptc1(+/− )p53(−/−) mice. Cancer Cell. 2004;6:229–240. doi: 10.1016/j.ccr.2004.08.019. [DOI] [PubMed] [Google Scholar]
- Ruiz i Altaba A. Gli proteins encode context-dependent positive and negative functions: implications for development and disease. Development. 1999;126:3205–3216. doi: 10.1242/dev.126.14.3205. [DOI] [PubMed] [Google Scholar]
- Ruiz i Altaba A, Sanchez P, Dahmane N. Gli and hedgehog in cancer: tumours, embryos and stem cells. Nat Rev Cancer. 2002;2:361–372. doi: 10.1038/nrc796. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2001. [Google Scholar]
- Sasai K, Romer JT, Lee Y, Finkelstein D, Fuller C, McKinnon PJ, et al. Shh pathway activity is down-regulated in cultured medulloblastoma cells: implications for preclinical studies. Cancer Res. 2006;66:4215–4222. doi: 10.1158/0008-5472.CAN-05-4505. [DOI] [PubMed] [Google Scholar]
- Sasaki H, Hui C, Nakafuku M, Kondoh H. A binding site for Gli proteins is essential for HNF-3beta floor plate enhancer activity in transgenics and can respond to Shh in vitro. Development. 1997;124:1313–1322. doi: 10.1242/dev.124.7.1313. [DOI] [PubMed] [Google Scholar]
- Sasaki H, Nishizaki Y, Hui C, Nakafuku M, Kondoh H. Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development. 1999;126:3915–3924. doi: 10.1242/dev.126.17.3915. [DOI] [PubMed] [Google Scholar]
- Seale P, Sabourin LA, Girgis-Gabardo A, Mansouri A, Gruss P, Rudnicki MA. Pax7 is required for the specification of myogenic satellite cells. Cell. 2000;102:777–786. doi: 10.1016/s0092-8674(00)00066-0. [DOI] [PubMed] [Google Scholar]
- Sirri V, Leibovitch MP, Leibovitch SA. Muscle regulatory factor MRF4 activates differentiation in rhabdomyosarcoma RD cells through a positive-acting C-terminal protein domain. Oncogene. 2003;22:5658–5666. doi: 10.1038/sj.onc.1206690. [DOI] [PubMed] [Google Scholar]
- Sun J, Kamei CN, Layne MD, Jain MK, Liao JK, Lee ME, et al. Regulation of myogenic terminal differentiation by the hairy-related transcription factor CHF2. J Biol Chem. 2001;276:18591–18596. doi: 10.1074/jbc.M101163200. [DOI] [PubMed] [Google Scholar]
- Tajbakhsh S, Rocancourt D, Cossu G, Buckingham M. Redefining the genetic hierarchies controlling skeletal myogenesis: Pax-3 and Myf-5 act upstream of MyoD. Cell. 1997;89:127–138. doi: 10.1016/s0092-8674(00)80189-0. [DOI] [PubMed] [Google Scholar]
- Tapscott SJ, Thayer MJ, Weintraub H. Deficiency in rhabdomyosarcomas of a factor required for MyoD activity and myogenesis. Science. 1993;259:1450–1453. doi: 10.1126/science.8383879. [DOI] [PubMed] [Google Scholar]
- Teboul L, Summerbell D, Rigby PW. The initial somitic phase of Myf5 expression requires neither Shh signaling nor Gli regulation. Genes Dev. 2003;17:2870–2874. doi: 10.1101/gad.1117603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiffin N, Williams RD, Shipley J, Pritchard-Jones K. PAX7 expression in embryonal rhabdomyosarcoma suggests an origin in muscle satellite cells. Br J Cancer. 2003;89:327–332. doi: 10.1038/sj.bjc.6601040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toftgard R. Hedgehog signalling in cancer. Cell Mol Life Sci. 2000;57:1720–1731. doi: 10.1007/PL00000654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tostar U, Malm CJ, Meis-Kindblom JM, Kindblom LG, Toftgard R, Unden AB. Deregulation of the hedgehog signalling pathway: a possible role for the PTCH and SUFU genes in human rhabdomyoma and rhabdomyosarcoma development. J Pathol. 2006;208:17–25. doi: 10.1002/path.1882. [DOI] [PubMed] [Google Scholar]
- Verzi MP, Anderson JP, Dodou E, Kelly KK, Greene SB, North BJ, et al. N-twist, an evolutionarily conserved bHLH protein expressed in the developing CNS, functions as a transcriptional inhibitor. Dev Biol. 2002;249:174–190. doi: 10.1006/dbio.2002.0753. [DOI] [PubMed] [Google Scholar]
- Wang B, Fallon JF, Beachy PA. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell. 2000;100:423–434. doi: 10.1016/s0092-8674(00)80678-9. [DOI] [PubMed] [Google Scholar]
- Wang B, Li Y. Evidence for the direct involvement of betaTrCP in Gli3 protein processing. Proc Natl Acad Sci USA. 2006;103:33–38. doi: 10.1073/pnas.0509927103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins DN, Berman DM, Burkholder SG, Wang B, Beachy PA, Baylin SB. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature. 2003;422:313–317. doi: 10.1038/nature01493. [DOI] [PubMed] [Google Scholar]
- Watson J, Depasquale K, Ghaderi M, Zwillenberg S. Nevoid basal cell carcinoma syndrome and fetal rhabdomyoma: a case study. Ear Nose Throat J. 2004;83:716–718. [PubMed] [Google Scholar]
- Weiner HL, Bakst R, Hurlbert MS, Ruggiero J, Ahn E, Lee WS, et al. Induction of medulloblastomas in mice by sonic hedgehog, independent of Gli1. Cancer Res. 2002;62:6385–6389. [PubMed] [Google Scholar]
- Weintraub H, Dwarki VJ, Verma I, Davis R, Hollenberg S, Snider L, et al. Muscle-specific transcriptional activation by MyoD. Genes Dev. 1991;5:1377–1386. doi: 10.1101/gad.5.8.1377. [DOI] [PubMed] [Google Scholar]
- Wolff C, Roy S, Ingham PW. Multiple muscle cell identities induced by distinct levels and timing of hedgehog activity in the zebrafish embryo. Curr Biol. 2003;13:1169–1181. doi: 10.1016/s0960-9822(03)00461-5. [DOI] [PubMed] [Google Scholar]
- Yoon JW, Liu CZ, Yang JT, Swart R, Iannaccone P, Walterhouse D. GLI activates transcription through a herpes simplex viral protein 16-like activation domain. J Biol Chem. 1998;273:3496–3501. doi: 10.1074/jbc.273.6.3496. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.