

Abstract
In a genetic screen for regulators of synaptic morphology, we identified the single Caenorhabditis elegans flamingo-like cadherin fmi-1. The fmi-1 mutants exhibit defective axon pathfinding, reduced synapse number, aberrant synapse size and morphology, as well as an abnormal accumulation of synaptic vesicles at nonsynaptic regions. Although FMI-1 is primarily expressed in the nervous system, it is not expressed in the ventral D-type (VD) GABAergic motorneurons, which are defective in fmi-1 mutants. The axon and synaptic defects of VD neurons could be rescued when fmi-1 was expressed exclusively in non-VD neighboring neurons, suggesting a cell nonautonomous action of FMI-1. FMI-1 protein that lacked its intracellular domain still retained its ability to rescue the vesicle accumulation defects of GABAergic motorneurons, indicating that the extracellular domain was sufficient for this function of FMI-1 in GABAergic neuromuscular junction development. Mutations in cdh-4, a Fat-like cadherin, cause similar defects in GABAergic motorneurons. The cdh-4 is expressed by the VD neurons and seems to function in the same genetic pathway as fmi-1 to regulate GABAergic neuron development. Thus, fmi-1 and cdh-4 cadherins might act together to regulate synapse development and axon pathfinding.
Introduction
Patterning of the nervous system requires interactions between neurons and their environment. Multiple secreted factors and cell adhesion molecules have been demonstrated to coordinate axon guidance, target selection, and synapse formation (Shen et al., 2004; Cólon-Ramos et al., 2007; Inaki et al., 2007). The Caenorhabditis elegans neuromuscular system has a simple architecture with stereotyped synaptic positioning (White, 1986), making it attractive to elucidate the molecular mechanisms that underlie neural circuit formation (Jin, 2005; Richmond, 2005). Body wall muscles are innervated by both excitatory (cholinergic) and inhibitory (GABAergic) motorneurons, with neuromuscular junctions (NMJs) formed coordinately with two cholinergic NMJs per one GABAergic NMJ. The cholinergic neurons make synapses onto the muscles (NMJs) and on the GABAergic neurons, but the GABAergic neurons predominantly form NMJs (White, 1986; Hall and Russell, 1991).
In a screen for GABAergic motorneuron synapse-defective mutants, we recovered two loss-of-function alleles in fmi-1, the single C. elegans flamingo-like nonclassical cadherin. Flamingo was isolated as a component of the planar cell polarity (PCP) pathway in Drosophila, where it functions with Frizzled, in parallel to Fat and Dachsous (Lu et al., 1999; Usui et al., 1999; Casal et al., 2006). Several studies have shown roles for flamingo and the vertebrate orthologs (Celsr1–3) in nervous system formation, indicating that these molecules are functionally conserved. Flamingo- and Celsr-disrupted animals have developmental defects in target selection, dendrite extension, axon outgrowth, synapse development, and circuit formation (Gao et al., 2000; Curtin et al., 2003; Lee et al., 2003; Senti et al., 2003; Shima et al., 2004; Formstone and Mason, 2005; Tissir et al., 2005; Chen and Clandinin, 2008; Lewis et al., 2011). In the Drosophila neuromuscular system, Flamingo functions after development to prevent the formation of inappropriate ectopic synapses and inhibit axonal degradation (Bao et al., 2007).
Flamingo belongs to the class-B2 family of G-protein-coupled receptors (GPCRs), which includes other molecules that function at synapses, e.g., latrophilins (Südhof, 2001). Unlike classical GPCRs, however, there is no evidence that Flamingo binds directly to G-proteins, although there is evidence for indirect interactions with G-proteins (Shima et al., 2007). Overall, the downstream signaling pathways by which Flamingo/Celsr molecules exert their influence is unclear. In multiple contexts, flamingo mutants are not phenocopied by PCP mutations, implying that Flamingo may use different signaling pathways in these events (Gao et al., 2000; Lee et al., 2003). Flamingo functions cell nonautonomously in retinal neurons to regulate level-specific target selection (Chen and Clandinin, 2008).
Here we show that fmi-1 loss of function causes specific patterning defects in the GABAergic ventral D-type (VD) neurons. Interestingly, FMI-1 is not expressed in the VD neurons themselves; rather, it is found in multiple adjacent neurons and is present along the ventral nerve cord. Expression of FMI-1 in the cholinergic neurons can specifically rescue the synaptic patterning defects in VD neurons. We provide additional evidence suggesting that fmi-1 interacts genetically with the Fat-like cadherin, cdh-4, which is expressed in the VD neurons. Thus, the fmi-1 and cdh-4 cadherins might function together in VD neuron development.
Materials and Methods
C. elegans strains.
All C. elegans used were hermaphrodites, and the strains were maintained at 20–22.5°C as described (Brenner, 1974), with the exception of eri-1(mg366) animals, which were maintained at 18°C. The Pfmi-1::FMI-1venusGFP integrants (lhIs9 and lhIs10), Pfmi-1:: FMI-1Δint::GFP (lhIs27-lhIs30), and Punc-55::GFP (lhIs35) integrants were isolated using trimethyl psoralen/UV mutagenesis, and all were out-crossed to wild-type six times before analyses. The following alleles were used in this work: fmi-1(ju43), fmi-1(ju58), fmi-1(tm306), fmi-1(rh308), fmi-1(gm188), cdh-4(lq83), cdh-4(lq56), eri-1(mg366), juIs1 [Punc-25:: SNB-1::GFP], nuIs94 [Pacr-2::SNB-1::GFP], juIs76 [Punc-25::GFP], juIs14 [Pacr-2::GFP], juIs145 [Pflp-13::GFP], lhIs9 and lhIs10 [Pfmi-1:: FMI-1::venusGFP], lhIs27-lhIs30 [Pfmi-1::FMI-1Δint::GFP+Punc-25:: SNB-1RFP], lhIs35 [Punc-55::GFP], and hdIs46 [Pcdh-4::GFP].
Molecular biology.
The fmi-1 cDNA was prepared by reverse transcription (RT)-PCR using RNA made from wild-type (N2) animals. RNA was isolated using the Trizol reagent (Invitrogen), and RT-PCR was conducted using the dT23VN primer and the ProtoScript II RT-PCR kit (NEB). Amplification of the cDNA was conducted using Phusion polymerase (NEB). The resulting fmi-1 7.7 kb PCR product was cloned into the pCR8/GW/TOPO vector to generate pEVL89. The plasmid was completely sequenced and determined to be free of PCR-induced mutations. For the fmi-1 promoter driving GFP clone pBA202, an ∼2.6 kb region upstream of the ATG was amplified by PCR and cloned into pCR8/GW/TOPO (Invitrogen). The plasmid (pBA202) was then cut with Sph1 and ClaI, and the fmi-1 promoter was cloned upstream of the GFP cassette from pPD114.108 in place of the mec-7 promoter cut with the same enzymes. The fmi-1 cDNA was independently recombined into destination expression clones containing the unc-25 promoter (pBA153) and acr-2 promoter (pBA223) using LR clonase (Invitrogen). To rescue the synaptic phenotypes associated with fmi-1(ju58), we injected a fosmid (WRM066DB07) (GeneService Ltd.) that covered the fmi-1 genomic region, including ∼1 kb upstream. The fosmid was injected into fmi-1(ju58);juIs1 animals at a concentration of 2 ng/μl, along with pBA182 (5 ng/μl) as a coinjection marker. The sequence of all primers and plasmids used in this work are available on request.
FMI-1 antisera.
Antisera were raised against the intracellular domain of FMI-1 corresponding to the entire intracellular domain (amino acids 2455–2596). We purified the antisera using a maltose-binding protein–FMI-1 fusion produced in Escherichia coli and purified on an amylose-resin (NEB) by column chromatography using the manufacturer's instructions.
Whole-mount immunostaining.
Animals were fixed as described (Ackley et al., 2003), with the following modification. The animals were fixed for 2 h on ice, and the reduction step was performed using 1× borate buffer for 2 h at 37°C. The following primary antisera were used in this study: mouse anti-GFP (Roche) used at 1:1000, rabbit anti-UNC-10 (Koushika et al., 2001) used at 1:1000, mouse anti-UNC-10 (Developmental Studies Hybridoma Bank, http://dshb.biology.uiowa.edu/) used at 1:1000, rabbit anti-synaptotagmin (SNT-1) (Nonet et al., 1993) used at 1:2000, and rat anti-FMI-1 used at 1:1000. The following secondary antibodies were purchased from Invitrogen and were used at 1:2000: Alexa 488-labeled anti-mouse, Alexa 594-labeled anti-rabbit, Alexa 488 anti-rabbit, and Alexa 594 anti-rat. All images were collected on either a Pascal confocal microscope (Zeiss) or an FV1000 confocal microscope (Olympus) equipped with Fluoview software. Images were acquired using multitrack parameters, with a 63× Plan-apochromat objective. Deconvolution was done using the “Iterative_Deconvolve_3D” plug-in for ImageJ (Dougherty, 2005).
Colocalization analysis.
We used the “Just Another Colocalization Plug-in” available for ImageJ (Bolte and Cordelières, 2006). We used this program to calculate the Pearson's coefficient and the percentage of pixels that colocalize from each channel.
GFP analysis.
For axon morphology, animals were scored blind to genotype by examining cell-type-specific GFP markers (GABAergic, juIs76[Punc-25-gfp] and juIs145[pflp-13-gfp]; cholinergic, juIs14[Pacr-2-gfp]) with an Axioplan 2 microscope using a 63× Plan-apochromat objective and a GFP long-pass filter set (Chroma Technology) as described (Ackley et al., 2005). Animals that had any differences from the expected wild-type pattern were scored as axon guidance defective. A defasciculation event was counted as a region of the nerve cord where two or more processes appeared to become split from one fascicle and were visible along a distance that was greater than the length of two neuronal cell bodies, which is ∼10–20 μm. Synapse morphology of D-type neurons was visualized by juIs1 [Punc-25 SNB-1::GFP], and the A/B-type neurons were visualized using nuIs94 [Pacr-2 GFP-SNB-1] and using either a Pascal LSM confocal microscope (Zeiss) or an FV1000 confocal microscope (Olympus). Animals were anesthetized using 0.5% phenoxy-propanol (TCI America) in M9 and mounted on 2% agarose pads. Images were acquired using a 63× Plan-apochromat objective.
Image quantification.
To quantify the area and density of each SNB-1::GFP punctum, we generated two-dimensional projections of image stacks acquired by confocal microscopy. The images were converted to thresholded binary images, and the size and location (x–y coordinates) of single GFP puncta were acquired automatically using the Analyze Particle command in ImageJ. Measurements were performed with the experimenter blind to genotype. The procedure was done as described (Ackley et al., 2003) with minor modifications. In ImageJ, the files were thresholded, and a region of interest was drawn around the nerve cord. The following measurement options were selected: Area, Center of Mass, Circularity, Perimeter, Fit Ellipse, and Limit to Threshold. The Analyze Particle command was used with a minimum of 4 pixels and no maximum size. The following options were selected: Outline Particles, Ignore Particles Touching Edge, Include Interior Holes, and Reset Counter. The resulting measurements were exported to Excel (Microsoft) for statistical analysis. Significance was determined by a two-tailed Student's t test or F test as indicated.
Sequence alignments.
Alignments were performed pairwise using the EMBOSS::needle method (http://www.ebi.ac.uk/Tools/emboss/align/) with the following options selected: Gap Open (10.0), Gap Extend (0.5), and Matrix (Blosum62). The GenBank accession numbers for the non-nematode proteins used are as follows: Mus musculus Celsr2 (NP_001004177.2) and Drosophila melanogaster Flamingo/starry night (AAF58763.5).
RNAi.
Animals with each marker (juIs1, juIs76, or lhIs9) were tested in the eri-1(mg366) background to increase RNAi penetrance in the nervous system (Kennedy et al., 2004). Bacteria from the Ahringer RNAi library (Gene Service Ltd.) were grown overnight in Luria broth supplemented with 100 μm ampicillin and 25 μm tetracycline, and 100 μl of the saturated culture was placed on Nematode Growth Media plates supplemented with 1 mm isopropyl β-d-1-thiogalactopyranoside, 30 μm carbenecillin, and 1 μg/ml nystatin. The plates were incubated at 37°C overnight. Three to five L4 animals of each genotype were placed on the RNAi lawn and allowed to have self-fertilized offspring. The F1 offspring were analyzed for phenotypes. All RNAi experiments were replicated at least two times, each with freshly prepared medium.
Electron microscopy.
Young adult hermaphrodites of genotypes CZ333 juIs1, CZ1677 fmi-1(ju58);juIs1, and CZ460 fmi-1(ju43);juIs1 were fixed in parallel using glutaraldehyde and osmium fixative as described (Hallam et al., 2002). Three hundred to 500 serial 45 nm ultrathin sections were collected from two wild-type animals (juIs1) and one each of the fmi-1(ju58) and fmi-1(ju43) mutant worms. Nerve cords were photographed with a Gatan digital camera on a JEOL 1200 electron microscope. NMJs were identified as synapses with muscles as postsynaptic partners (White et al., 1986). GABAergic NMJs have muscles as sole postsynaptic partners, whereas cholinergic NMJs usually have two postsynaptic partners, a muscle and a neuronal process. Small, round, clear vesicles of 30–35 nm diameter were counted as synaptic vesicles. Measurement of presynaptic densities was taken as the number of sections that contained recognizable electron density, multiplied by 45 nm. Statistical analysis was conducted using the standard t test (Excel).
Results
fmi-1 mutations cause synapse defects
We recovered two alleles, ju43 and ju58, in a visual screen for mutants with altered synaptic vesicles clustering at GABAergic NMJs using the juIs1[Punc-25::SNB-1::GFP] reporter. Compared with the uniformly sized and distributed fluorescent puncta in the wild-type animals (puncta area, 0.8 μm2; 25.7 puncta per 100 μm), these mutants showed enlarged, aberrantly shaped accumulations of GFP-labeled vesicles (Table 1, Fig. 1), with extended regions lacking fluorescent puncta (gaps) (ju43: 1.2 μm2, 22.6 puncta per 100 μm; ju58: 1.5 μm2, 19.2 puncta per 100 μm). Overall, the NMJs formed on the ventral body wall muscles were more severely disrupted in ju43 (1.1 μm2, 21.7 puncta per 100 μm) and ju58 (1.8 μm2, 17.6 puncta per 100 μm) than those along the dorsal muscles, although defects were noticeable in the dorsal cord (ju43: 1.3 μm2, 23.4 puncta per 100 μm; ju58: 1.2 μm2, 21.7 puncta per 100 μm). Because the VD and dorsal D-type neuron (DD) class of GABAergic neurons innervate the ventral and dorsal muscles, respectively, these observations suggest that the DD and VD neurons may be differentially affected by the ju43 and ju58 mutations.
Table 1.
Measurements of synaptic vesicle accumulation
| Ventral cord |
Dorsal cord |
|||||
|---|---|---|---|---|---|---|
| Area ± SEM (n)a | Maximum (μm2) | Density ± SEM (n)b | Area ± SEM (n)a | Maximum (μm2) | Density ± SEM (n)b | |
| GABAergic | ||||||
| Wild type | 0.80 ± 0.02 (530) | 3.9 | 25.7 ± 1.0 (16) | 0.83 ± 0.03 (344) | 3.0 | 22.9 ± 0.9 (10) |
| fmi-1(ju43) | 1.10 ± 0.13 (102)* | 8.0 | 21.7 ± 0.9 (4)* | 1.27 ± 0.08 (157)* | 4.8 | 23.4 ± 0.7 (4) |
| fmi-1(ju58) | 1.80 ± 0.14 (328)* | 17.4 | 17.6 ± 2.4 (9)* | 1.15 ± 0.06 (233)* | 7.0 | 21.7 ± 3.9 (6) |
| fmi-1(tm306) | 1.43 ± 0.15 (199)* | 19.7 | 19.2 ± 1.9 (5)* | 0.89 ± 0.05 (147) | 3.4 | 18.4 ± 3.0 (4)* |
| fmi-1(rh308) | 1.74 ± 0.20 (118)* | 18.9 | 16.3 ± 1.2 (4)* | 0.91 ± 0.05 (147) | 3.8 | 19.0 ± 1.5 (3) |
| fmi-1(ju58)+[WRM066DB07] | 1.15 ± 0.10 (167)*‡ | 9.0 | 19.9 ± 1.7 (7)*‡ | 1.24 ± 0.10 (119)* | 7.7 | 17.5 ± 1.8 (4)* |
| juIs1+[WRM0066DB07) | 0.86 ± 0.06 (203) | 6.0 | 26.5 ± 2.0 (6) | 0.92 ± 0.06 (184) | 6.0 | 23.1 ± 1.5 (5) |
| fmi-1(tm306)+[Pacr-2::fmi-1] | 1.06 ± 0.07 (129)*‡ | 4.6 | 15.5 ± 2.4 (4)*‡ | 1.22 ± 0.09 (110)* | 3.8 | 17.2 ± 2.2 (4) |
| fmi-1(tm306)+[Punc-25::fmi-1] | 2.11 ± 0.22 (135)* | 16.8 | 12.3 ± 2.4 (5)* | 1.14 ± 0.11 (77)* | 4.4 | 20.0 ± 4.4 (4) |
| cdh-4(lq83) | 1.82 ± 0.11 (259)* | 14.7 | 16.6 ± 1.6 (7)* | 1.24 ± 0.09 (169)* | 7.2 | 20.3 ± 2.0 (4)* |
| fmi-1(tm306);cdh-4(lq83) | 1.45 ± 0.13 (171)*† | 13.2 | 16.6 ± 1.8 (5)*† | 0.97 ± 0.07 (134)* | 6.0 | 18.1 ± 2.6 (4)* |
| cdh-4(lq56) | 1.40 ± 0.10 (155)* | 6.7 | 16.7 ± 3.6 (5)* | N/D | ||
| fmi-1(tm306);cdh-4(lq56) | 1.48 ± 0.18 (113)*† | 12.8 | 18.1 ± 1.0 (3)*† | N/D | ||
| eri-1(mg366)juIs1;CDH-4(RNAi) | 1.56 ± 0.15 (137)* | 11.3 | 16.7 ± 1.8 (4)* | N/D | ||
| fmi-1(tm306); eri-1(mg366)juIs1;CDH-4(RNAi) | 1.57 ± 0.24 (101)*† | 18.9 | 17.8 ± 3.1 (4)*† | N/D | ||
| Cholinergic | ||||||
| Wild type | 0.90 ± 0.06 (281) | 6.2 | 26.7 ± 2.0 (5) | 1.03 ± 0.07 (208) | 6.0 | 26.1 ± 2.8 (4) |
| fmi-1(ju58) | 0.98 ± 0.06 (459) | 10.9 | 23.8 ± 2.1 (9) | 1.17 ± 0.09 (323)* | 16.6 | 26.9 ± 2.5 (7) |
N/D, Not determined.
aMeasurements are in square micrometers.
bAverage number of puncta observed per 100 μm.
*p < 0.01, significantly different from wild type;
‡p < 0.01, significantly different than fmi-1;
†not significantly different from fmi-1(tm306).
Figure 1.
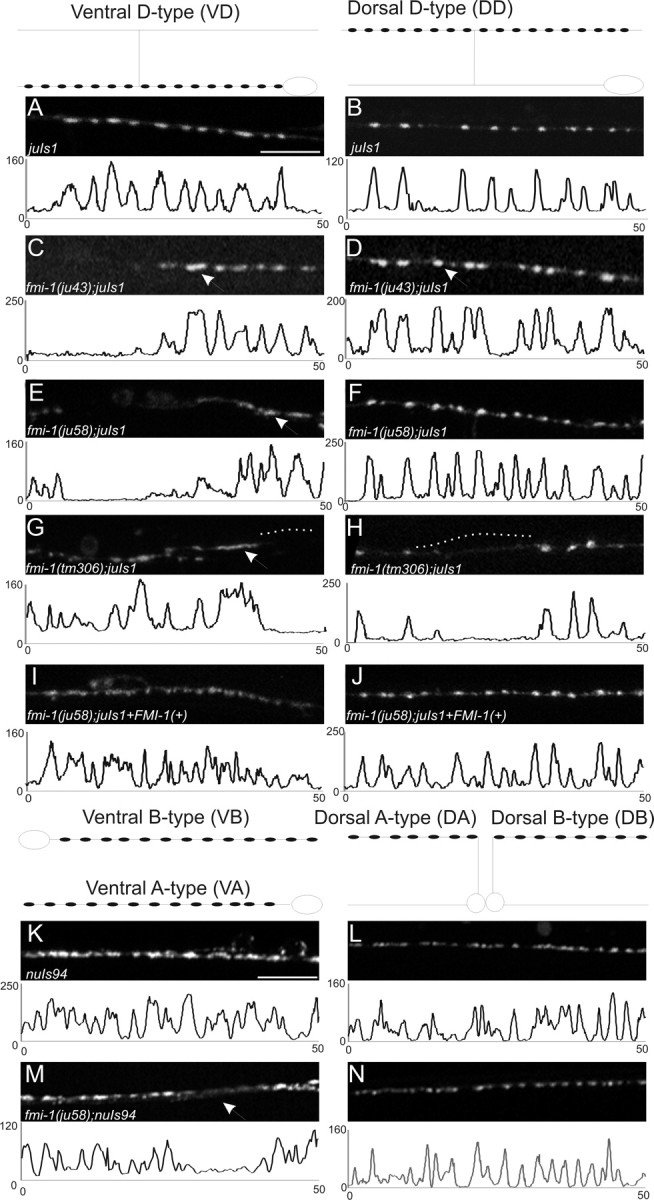
GABAergic NMJs are malformed in fmi-1 mutants. GABAergic (A–J) and cholinergic (K–N) NMJs were examined using SNB-1::GFP to visualize synaptic vesicle clusters. The shape of the VD and DD motorneurons is depicted above A and B, respectively, and the location of the synapses within those cells is illustrated (black circles). In all panels, left is anterior. A–J, Images are a 50 μm section of the ventral (A, C, E, G, I) or dorsal (B, D, F, H, J) nerve cords, and below each image is a plot profile for each image to demonstrate the approximately similar-sized puncta in wild-type and how they are divergent in fmi-1 mutants. Wild-type animals (A, B) had consistently sized puncta that were evenly spaced along the nerve cords. C–H, In the GABAergic motorneurons of fmi-1 mutants, we observed larger synapses (C, D, arrows) and misshapened (E, G, arrows) puncta. Frequently, there were long regions of the nerve cord lacking synapses (gaps) (G, H, dotted lines). I, J, We partially rescued the ju58 defects using a fosmid containing fmi-1. These animals had a significant reduction in synaptic size relative to ju58 alone, and fewer long gaps lacking GFP were found in the nerve cords. K–N, SNB-1::GFP expressed in the cholinergic neurons appear less defective than in the GABAergic neurons. Schematics of the shape and location of synapses within these neurons are shown above K and L. Note that the VA and VB synapses, as well as those in the DA and DB neurons, would overlap in vivo, which is not illustrated here for simplicity. In wild-type animals, SNB-1::GFP puncta were normally sized and tightly clustered. M, N, In the fmi-1(ju58) animals, the puncta were approximately the same size, although we observed a few gaps where puncta are weaker or absent (M, arrow). Scale bars, 10 μm.
We mapped ju43 and ju58 to the right arm of linkage group V close to the locus encoding the single flamingo cadherin-domain-containing protein (F15B9.7/cdh-6/fmi-1) (Pettitt, 2005). We also identified molecular changes in both alleles (see below). We analyzed additional alleles of fmi-1, including a deletion allele, tm306 (Fig. 2), isolated by the National Bioresource Project (http://www.shigen.nig.ac.jp/c.elegans/index.jsp); gm188, isolated in a screen for Hermaphrodite Specific Neuron (HSN) migration defects; and rh308, isolated in a screen for pioneer axon guidance defects (Steimel et al., 2010). We observed that all fmi-1 mutants exhibited grossly similar patterns of synaptic defects (Table 1).
Figure 2.

Lesions identified in the fmi-1 gene and protein. A, Schematic presentation of the fmi-1 gene and locations of lesions. Nucleotide numbering is with respect to the start of translation. The promoter regions used for GFP expression (Pfmi-1L) and the segment present in the rescuing fosmid (Pfmi-1S) are indicated. Exons and their corresponding protein domains are color coded. B, The FMI-1 protein structure was determined using SMART (Simple Modular Architecture Research Tool). The extracellular region of the protein is predicted to have nine cadherin repeats (green ovals), five EGF-like domains (orange diamonds), two LamG domains (yellow ovals), as well as a hormone receptor-related domain (HormR; blue hexagon) and a proteolytic cleavage site that is present in G-coupled protein receptors, e.g., latrophilins [G-protein-coupled receptor proteolytic site domain (GPS); blue pentagon]. FMI-1 has seven predicted transmembrane domains and an intracellular domain of ∼142 amino acids. C, The gm188 mutation in the seventh Cadherin repeat affects a highly conserved glycine residue [alignment of C. elegans (C.e.) FMI-1, Drosophila (D.m.) Flamingo, and M. musculus (M.m.) Celsr2]. D, Partial alignment of the first LamG domain showing the glycine affected by ju43.
The fmi-1 coding region spans ∼17 kb and is predicted to contain 25 exons (Fig. 2). We generated a full-length cDNA for fmi-1 (∼7.7 kb), using primers directed at the predicted translational start and stop sites (see Materials and Methods); sequence analyses of this cDNA confirmed the exon–intron junctions of fmi-1. We determined the molecular nature of each of the mutations. The gm188 and ju43 lesions are missense mutations that result in amino acid changes in highly conserved glycines present in the seventh Cadherin domain and first Laminin G domain (LamG) domain, respectively, in the extracellular domain (ECD) (Fig. 2). The ju58 mutation causes a premature stop, and tm306 deletes a portion of the coding region and introduces a premature stop codon (Fig. 2). We partially rescued the defects using a fosmid genomic clone covering the fmi-1 locus (WRM066DB07) (Fig. 1). In fmi-1(ju58) animals, the ventral cord SNB-1::GFP puncta, which are formed by the VD neurons, had an apparent area of 1.80 μm2 with 17.6 puncta per 100 μm, whereas ju58 animals containing the fmi-1 fosmid formed puncta of 1.15 μm2, with 19.9 puncta per 100 μm, which was a significant improvement (p < 0.05). To determine whether the fmi-1 fosmid caused defects in wild-type animals, we crossed the rescuing extrachromosomal array away from ju58 and found the animals had normally sized puncta (0.9 μm2), with a small increase in the number of puncta formed (juIs1, 25.7 ventral cord puncta per 100 μm vs juIs1 plus fmi-1, 26.5 puncta/100 μm), but the effect was not statistically significant (p > 0.05). The rescue of ju58 by transgenic expression suggests that the altered SNB-1::GFP patterning was likely caused by a loss of fmi-1 function.
The cholinergic motorneurons also make NMJs onto body wall muscles. A synaptic vesicle marker for the cholinergic motorneurons (nuIs94) displayed differences in fmi-1 mutants (Fig. 1, Table 1). In wild-type nuIs94 animals, SNB-1::GFP were clustered in a relatively even sized and shaped pattern along the dorsal and ventral cords. In ju58 animals, the dorsal cord SNB-1::GFP puncta were slightly larger (WT, 1.03 μm2 vs ju58, 1.17 μm2; p = 0.002), whereas the puncta in the ventral cord were approximately the same sizes (WT, 0.90 μm2 vs ju58, 0.98 μm2; p > 0.05). In ju58 animals, there was a slight reduction in the total puncta formed (WT, 26.7 puncta per 100 μm vs ju58, 23.8/100 μm; p < 0.05). Overall, the SNB-1::GFP marker in GABAergic neurons appeared to be more severely affected by fmi-1 mutations than in cholinergic neurons.
fmi-1 synaptic defects occur during development
In our screen and subsequent analyses, we primarily scored synapses in young adults. For the regions of the nerve cord lacking synaptic puncta, we asked whether these gaps could be attributable to a degeneration of synapses that were formed earlier in development, as has been reported for Drosophila Flamingo mutants (Bao et al., 2007). The 13 VD motorneurons form and begin to establish NMJs late in the first larval stage (L1). From L2 until adult, the animals grow approximately threefold to fivefold in length, and because no new VD neurons are born, the axons must elongate and add synapses during development. We therefore examined wild-type and fmi-1 mutant animals in stages L2, L3, and L4. We found that the fmi-1 mutants had regions of the ventral cord lacking synaptic puncta as early as L2 (Fig. 3). We did not see a significant change in the frequency of regions in the nerve cord lacking vesicles in younger versus older animals (L2, 1.2 gaps per ventral cord; L3, 1.5; L4, 1.5; adult, 1.7; F = 1.02; p = 0.4; n = 25 animals per stage). Therefore, we conclude that the absent synapses are more likely attributable to developmental defects in these neurons.
Figure 3.
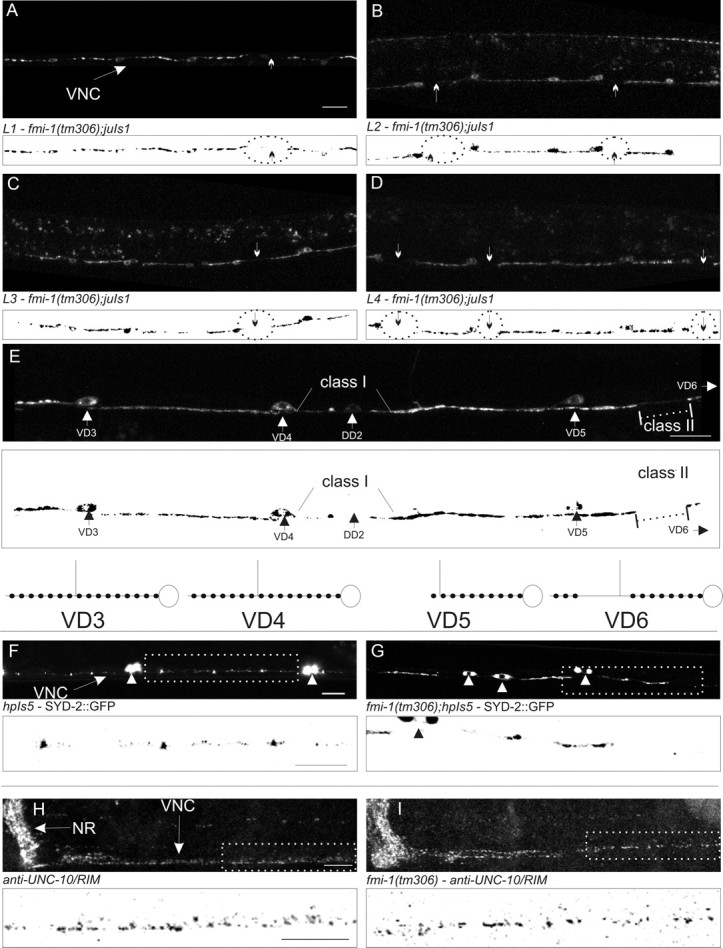
FMI-1 causes synaptic defects during development. A–D, GABAergic neurons in the ventral nerve cord (VNC) in tm306;juIs1 (Punc-25::SNB-1::GFP) were imaged at each larval stage (L1–L4). Regions lacking SNB-1::GFP (circles and arrows) were present in all stages examined. Below each panel is a thresholded mask of the image (black/white inverted). E, An adult animal with both class I and class II gaps is shown. VD5 appears to have a class I gap because the gap extends from end of the synaptic puncta (large arrowhead) to the point of VD4. The small amount of fluorescent material in that region seems to come from the DD2, which is more weakly fluorescent. In comparison, the puncta formed in VD4 extend all the way to VD3. VD6 (cell body not shown) has a class II gap (dotted line) because VD6 forms puncta distal to the gap before VD5. A diagram illustrating these cells is drawn above and to the right. F, G, SYD-2::GFP accumulates in small puncta in the nerve cord (F, arrow) and motorneuron cell bodies (F, G, arrowheads). In fmi-1 mutants (G), SYD-2::GFP is clustered in larger puncta with bigger gaps between puncta. H, I, UNC-10 localized to discretely sized and spaced puncta in the nerve ring (NR) and VNC. I, UNC-10 localization was grossly wild type in fmi-1(tm306). Boxed regions in F–I are depicted with a black/white inverted image below each panel. Scale bars, 10 μm.
We also examined the failure of SNB-1::GFP to accumulate evenly along the axons in VD neurons (SNB-1::GFP gaps). In fmi-1 mutants, we identified two types of gaps in the SNB-1::GFP pattern in VD neurons: class I, where the gap occurred from a point in the line of synapses and then extends to the next cell body, and class II, where synaptic puncta are both anterior and posterior of the SNB-1::GFP gap (Fig. 3A–F). The transport of synaptic material distal to the gap from the cell body suggests that either the axon is intact between those two points, or that the distal puncta arose from another neuron. In fmi-1(tm306);juIs1 (SNB-1::GFP) animals (n = 30), we found 1.4 gaps per animal. Of those, 42 of 57 gaps were class I, whereas 15 of 57 appeared to be class II. To determine whether the VD neurons have axon extension defects, we examined the VD axons in tm306 animals using a Punc-55::gfp reporter. Eighty-eight percent of fmi-1(tm306) mutants (1.26 ± 0.85 gaps per animal; n = 24) exhibited defects in the VD neurons, resulting in regions of the ventral cord lacking VD axons. This suggested that axon guidance and/or extension underlies the majority of the SNB-1::GFP gaps found in VD neurons, whereas a minority could arise from synaptic patterning defects within axons.
Subsequently, we asked whether the vesicle accumulation morphology changes in VD SNB-1::GFP puncta (enlarged size) were caused by the underlying axon failures that caused gaps. Qualitatively, we saw no difference in SNB-1::GFP puncta in axons that appeared to have gaps versus those that did not. We also calculated the average area of fluorescent SNB-1::GFP puncta within 10 μm of a gap versus those that were further than 10 μm. We observed no significant effect of being near a gap on the size of the puncta [tm306 (1.75 ± 0.40 near a gap vs 1.42 ± 0.15 distal; p = 0.49) and rh308 (1.59 ± 0.60 near a gap vs 2.03 ± 0.42 distal; p = 0.59)]. Thus, we concluded that the SNB-1::GFP accumulation morphological aberrations in fmi-1 mutants are developmental events that occur independent of, or in addition to, axon guidance and extension defects.
fmi-1 contributes to VD active zone development
To determine whether other synaptic proteins were affected by loss of fmi-1, we examined the active zone proteins SYD-2 (Zhen and Jin, 1999) and UNC-10 (Koushika et al., 2001) in fmi-1 mutants. We visualized the GABAergic active zones using SYD-2::GFP (Yeh et al., 2005) and found disruptions in the pattern of accumulation, including enlarged, misshapen puncta and areas lacking SYD-2 (Fig. 3G,H). By whole-mount immunostaining using anti-UNC-10 antisera, we looked at endogenous UNC-10/Rim throughout the animal and found that global UNC-10 accumulation within the nervous system was relatively normal (Fig. 3I,J). These observations do not rule out a role for FMI-1 in the delivery of synaptic material but may suggest that fmi-1 has a broader role in GABAergic NMJ development, not limited to how synaptic vesicles are clustered.
Synaptic components accumulate in nonsynaptic sites in fmi-1 mutants
We examined the nervous system of fmi-1(ju43) and fmi-1(ju58) animals using ultrastructural analysis and compared dorsal cord motorneuron synapses to similar regions of a wild-type animal (Fig. 4). We calculated the length of the active zones and vesicle-clustering domains, as well as the number of synaptic vesicles per synapse in the dorsal cord (Table 2). We found that in both fmi-1 mutant alleles (n = 1 per genotype), the length of the GABAergic synapses, defined as a continuous region containing clusters of synaptic vesicles surrounding the active zone, was approximately half that of the wild type and that there was an ∼50% reduction in the total number of vesicles present at the presynaptic terminals. The discrepancy between the ultrastructural results (smaller synapses) and fluorescent measurement of SNB-1::GFP (enlarged puncta) suggests that the GFP-labeled material may not be present at synapses (defined by the presence of an active zone), or that the enlarged puncta, which appeared contiguous at the light microscopy level, may include closely positioned but separate synapses. The former phenotype has been observed previously, in unc-16 mutants where synaptic material becomes trapped in enlarged cisternae within the axons (Brown et al., 2009).
Figure 4.
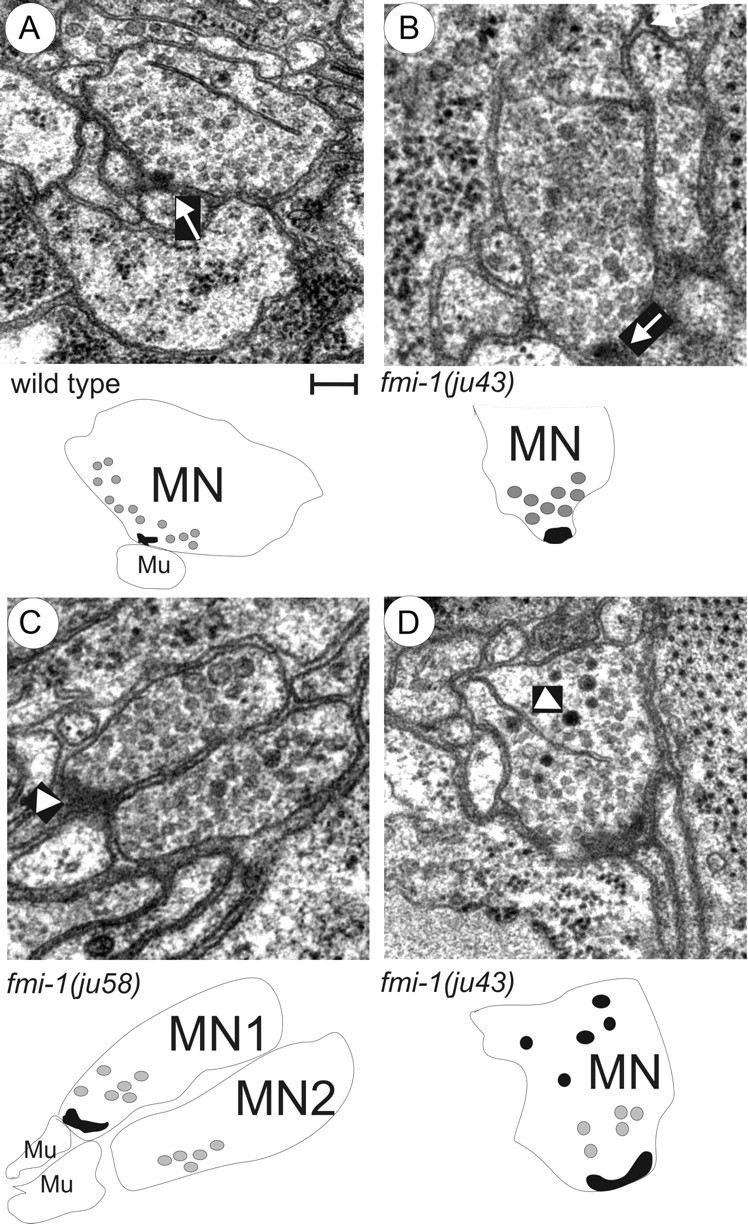
Ultrastructural analysis of fmi-1 synaptic defects. A, Wild-type animal NMJs formed as a single active zone (arrow) surrounded by evenly sized, clear synaptic vesicles. Below, a schematic is provided to illustrate the motorneuron (MN) and muscle (Mu). The active zone (black object) was surrounded by synaptic vesicles (gray circles), only some of the vesicles are illustrated for simplicity. B–D, The fmi-1 mutants did have some normal NMJs (B). However, many fmi-1 animals also exhibited abnormal junctions (C, D). C, In this junction, two neurons had vesicle-filled swellings at the point of contact with the muscle; however, only one had an active zone (arrowhead). This is illustrated below where motorneuron 1 (MN1) has an active zone, but motorneuron 2 (MN2) does not, despite both having an accumulation of synaptic vesicles. D, Electron-dense vesicles were present at ju43 NMJs (arrowhead), but not in wild-type animals (A). In the schematic, the electron-dense structures are illustrated as black circles. Scale bar, 100 nm.
Table 2.
Comparison of neuromuscular junctions by genotype
| Type of NMJ | Number of NMJs | Active zone length (nm) | Vesicles per active zone | Synapse length (nm)a | Vesicles per profile | |
|---|---|---|---|---|---|---|
| Wild type | GABAergic | 11 | 122.7 ± 15.0 | 40.7 ± 2.7 | 1006.2 ± 77.5 | 26.1 ± 0.9 |
| fmi-1(ju43) | GABAergic | 3 | 75.0 ± 15.0 | 60.7 ± 11.8 | 465.0 ± 150.0* | 23.4 ± 7.2 |
| fmi-1(ju58) | GABAergic | 6 | 120.0 ± 19.0 | 21.5 ± 2.6* | 472.5 ± 25.3* | 16.6 ± 1.3* |
| wild type | Cholinergic | 23 | 184.0 ± 14.1 | 26.5 ± 2.8 | 637.8 ± 56.4 | 20.4 ± 1.9 |
| fmi-1(ju43) | Cholinergic | 19 | 165.8 ± 23.1 | 21.4 ± 1.1 | 746.1 ± 67.0 | 21.4 ± 1.1 |
| fmi-1(ju58) | Cholinergic | 21 | 182.1 ± 24.0 | 27.8 ± 2.5 | 615.0 ± 55.5 | 22.2 ± 1.9 |
aDefined as consecutive sections with synaptic vesicles.
*p < 0.01, significantly different from wild type.
We also noticed differences in the morphology of synaptic vesicles at the GABAergic NMJs in the fmi-1 mutants. In wild type, the synaptic vesicles at GABAergic NMJs were of mostly equal size (30–50 nm) and were internally clear. In contrast, the vesicles in the fmi-1 mutants were enlarged and often had electron-dense material within the vesicles (Fig. 4). The observation of electron-dense material within vesicles could also reflect a failure in the assembly of active zone components at the plasma membrane, consistent with the SYD-2::GFP results.
Within the region of the animals we examined, the cholinergic synapses were more affected in the ju43 than ju58 background. In ju43 animals, the cholinergic synapses were longer than wild type (WT, 618 nm vs ju43, 922.5 nm) but had approximately the same number of synaptic vesicles as wild type. In ju43 mutants, cholinergic synapses also had a disrupted morphology whereby they formed NMJs containing multiple presynaptic densities, and some synapses appeared to be misplaced away from muscle targets (Fig. 4). Overall, we found that fmi-1 mutations caused abnormalities in the ultrastructure of synapses.
fmi-1 mutant axon guidance and fasciculation defects
We examined the axonal morphology of the GABAergic motorneurons with two GFP markers, juIs145 and juIs76 (Huang et al., 2002; Wu et al., 2007). These mark the DD neurons and DD/VD neurons, respectively (Fig. 5, Table 3). We observed multiple defects in the outgrowth of the VD and DD neurons, including errors in the left (L) or right (R) side of the ventral cord on which the commissural axon exits (L/R decision, 2% of the animals had defects in WT vs 48% in ju43 and 67% in tm306) and the ability of the neurons to remain tightly bundled (defasciculations, 0% of WT animals vs 50% of ju43 and 36% of tm306) (Fig. 5, Table 3). The fmi-1 animals also exhibited failures in longitudinal axon extension (Fig. 5) in both DDs (juIs76 observed in L1 animals) and VDs (see above) (Table 4). Only 4% of wild-type animals exhibited a gap between the cell body of a DD and the process from the next DD posterior (i.e., cell body of DD2 and axon of DD3). In fmi-1 mutants, we found multiple gaps (approximately two per animal) in >70% of animals examined. Also, similar defects were observed for the cholinergic neurons using the juIs14 GFP marker (Fig. 5) (Huang et al., 2003). Transgenic expression of a functional FMI-1::GFP rescued the axon extension phenotypes observed in fmi-1(tm306);juIs76 L1 animals (tm306lhIs9: 30% gaps, 0.36 ± 0.60 per animal, n = 33; vs tm306: 73% of animals, 1.72 ± 1.17 gaps per animal, n = 30) (Table 4).
Figure 5.

Axon guidance defects in fmi-1. GABAergic and cholinergic motorneurons were visualized using GFP reporters juIs76-Punc-25::GFP (A, B, D–H), juIs145-Pflp-13::GFP (C), and juIs14-Pacr-2::GFP DA, DB, VA, and VB (I, J). A–D, The six DD motorneurons are the only GABAergic motorneurons present during L1s [VDs form at the end of the L1 stage; 4 RME neurons are in the nerve ring region (NR)]. The process of each neuron extends anteriorly to the next cell body, i.e., DD3 extends to DD2. B, C, DDs in the fmi-1 mutants often exhibit incomplete axon extension (arrows). C, DD axons often defasciculated into two nerve bundles (arrowhead). D, DD axon extension defects were rescued by FMI-1::GFP using its endogenous promoter (lhIs9, *Pmyo-2::RFP, which is the coinjection marker). E, In wild type, GABAergic axons form as a single contiguous fascicle in the ventral cord (arrow). Commissures (arrowhead) posterior to DD1 and VD1 grow toward the right side, which is up in this ventral view. F, In fmi-1 mutants, GABAergic motorneurons exited the ventral cord on the incorrect side and often had defects in the extension of the axons (arrow). The gap is likely attributable to the premature termination of both the VD and DD axons. G, In wild-type animals (not shown) and fmi-1(tm306), commissures extend along the dorsoventral axis (indicated by arrow) fully to the dorsal cord. H, Overexpression of FMI-1::GFP (lhIs9) caused aberrant guidance and premature turning and termination of GABAergic commissures (arrow), with some having ectopic neurites (arrowhead). The position of the dorsal cord is indicated by the dotted line. I, Cholinergic processes within the ventral cord (arrow) appear as a single, cohesive fascicle in wild-type animals. J, In the ju58 animals, cholinergic axons of defasciculated (arrow). Scale bar, 10 μm.
Table 3.
Axon guidance defects in fmi-1 and cdh-4 mutants
| Percentage of wild typea | L/R defects | Defasciculation | Number | |
|---|---|---|---|---|
| GABAergic (DD and VD) | ||||
| Wild type | 98% | 2% | 0% | 50 |
| fmi-1(ju43) | 12% | 48% | 50% | 70 |
| fmi-1(tm306) | 10% | 67% | 36% | 70 |
| fmi-1(tm306) lhIs9 | 24% | 27% | N/D | 76 |
| lhIs9 | 12% | 68% | N/D | 70 |
| fmi-1(tm306);FMI-1GFP (high) | 16% | 64% | N/D | 25 |
| cdh-4(lq83) | 15% | 30% | 29% | 79 |
| cdh-4(lq83);fmi-1(tm306) | 6% | 35% | 22% | 49 |
| L1 GABAergic (DD only) | ||||
| Wild type | 98% | 0% | 2% | 100 |
| fmi-1(ju43) | 92% | 5% | 3% | 100 |
| Cholinergic (DA and DB) | ||||
| Wild type | 90% | 8% | 2% | 60 |
| fmi-1(ju58) | 5% | 5% | 95% | 20 |
N/D, Not determined.
aPercentage of animals without any L/R or defasciculation events.
Table 4.
Axon extension defects by genotype
| Percentage of gaps | Gaps per animal | Number | |
|---|---|---|---|
| GABAergic (DD only) (juIs76, L1 stage) | |||
| Wild type | 4% | 0.08 ± 0.40 | 25 |
| fmi-1(ju43) | 76% | 1.76 ± 1.09 | 25 |
| fmi-1(tm306) | 73% | 1.72 ± 1.17 | 30 |
| fmi-1(tm306)lhIs9 | 30% | 0.36 ± 0.60 | 33 |
| cdh-4(lq83) | 41% | 0.55 ± 0.78 | 29 |
| cdh-4(lq83);fmi-1(tm306) | 50% | 0.75 ± 0.91 | 20 |
| GABAergic (VD only) (lhIs35, L3 stage) | |||
| Wild type | 0% | 0.00 ± 0.00 | 25 |
| fmi-1(tm306) | 88% | 1.26 ± 0.85 | 24 |
Axon guidance phenotypes were also observed in the ultrastructural analysis. Wild-type animals had, on average, 12.6 processes in the dorsal cord and 52.4 processes in the ventral cord (right bundle). The ju43 animals had a significant increase in the number of processes in the dorsal cord, 15.6, and a significant decrease in the ventral cord, 47.6 (p < 0.05). In ju58 animals, the dorsal cord was normal, 12.4 processes, but the ventral cord had a significant decrease, 44.4 (p < 0.05). These differences could be attributable to premature termination, to inappropriate posterior extension, or to axons crossing into the contralateral bundle, as has been described here and previously (Steimel et al., 2010). Overall, we found evidence that mutations in fmi-1 can cause defects in the patterning of both cholinergic and GABAergic motorneurons. However, defects in the morphology of synapses and accumulation of synaptic material were present in intact axons that appear to develop normally (Figs. 3, 4), suggesting that fmi-1 may affect both axon outgrowth and synaptogenesis.
Axon guidance defects are FMI-1 dose dependent
We also found that overexpression of Flamingo induced axon outgrowth errors, which has also been reported in Drosophila (Chen and Clandinin, 2008). Our FMI-1::GFP integrant, lhIs9, when present in the wild-type background, induced axon guidance defects, including L/R errors (68% of animals) and neurites forming on VD commissures (Fig. 5). When the total amount of FMI-1 was reduced, by crossing the integrant to fmi-1(tm306), many of the defects associated with the integrated chimera were improved. For example, in lhIs9;juIs76, animals (8 of 30) had commissural processes that deviated along the anteroposterior axis, and some of these had ectopic neurites growing out of them (6 of 30) (Fig. 5). This phenotype was not present in wild type or in tm306 alone. The fmi-1(tm306)lhIs9;juIs76 animals rarely (2 of 36) had misrouted axons along the anteroposterior axis, and none (0 of 36) had ectopic neurites (p < 0.05). Overall, the percentage of animals not displaying any L/R or ectopic neurite outgrowth phenotypes (percentage of wild type) was higher in the lhIs9tm306 (24%) animals than in either single mutant background (lhIs9, 12%; tm306, 10%). We interpret these results, along with our loss-of-function data, as an indication that the GABAergic motorneurons were sensitive to the levels of FMI-1 present in the animal. Either too much or too little FMI-1 resulted in axon patterning defects in the GABAergic motorneurons.
FMI-1 protein localization is nonoverlapping with synaptic vesicle proteins
To examine the subcellular localization of FMI-1, we generated antisera to the intracellular domain of FMI-1 as well as a full-length GFP-tagged genomic clone (see Materials and Methods). Using both methods, we found that FMI-1 accumulated to a region of neuropil around the nerve ring and the ventral cord (Fig. 6). FMI-1 was also present in the dorsal cord in young animals but was rarely observed in neuronal cell bodies. We observed no staining in the candidate null alleles, ju58 (Fig. 6), rh308 (Steimel et al., 2010), or tm306 (data not shown), indicating that our antisera is specific. Because deletion of extracellular FMI-1 domains affected protein localization (Steimel et al., 2010), we examined the effects of the missense mutations, ju43 and gm188, which affect the extracellular domain. These animals retain immunoreactivity, and the protein was still localized to axon segments, indicating that the phenotypes in these animals are not caused by gross mislocalization of the protein (Fig. 6).
Figure 6.
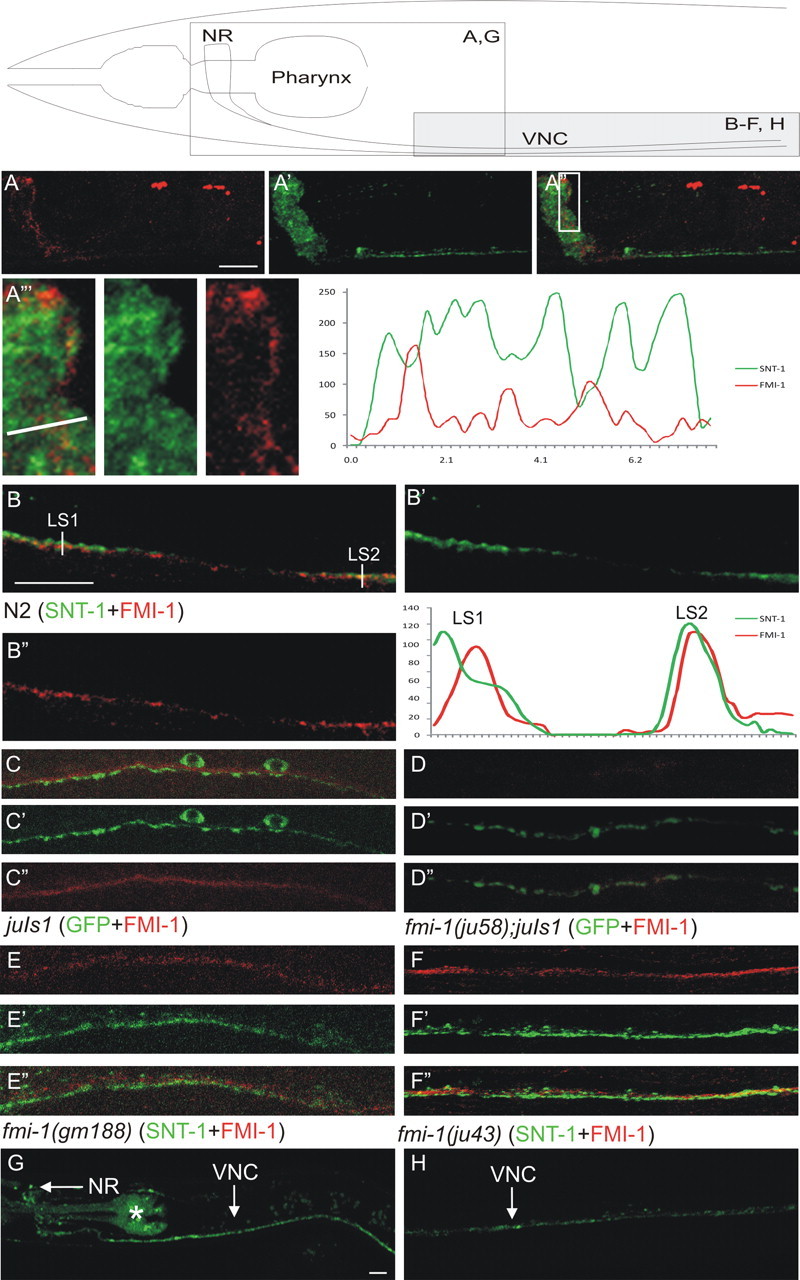
FMI-1 is concentrated at nonsynaptic regions of the nervous system. Using specific antisera, we visualized FMI-1 (A–F, red) relative to the synaptic vesicle protein SNT-1 (A, B, E, F, green) or juIs1 (SNB-1::GFP) (C, D, green). Above the panels is a schematic to orient the region being examined. A, B, In wild-type animals, FMI-1 localized to the nerve ring (NR; A) and ventral nerve cord (VNC; B). FMI-1 was predominantly adjacent to, but not overlapping with, SNT-1. A‴, Enlarged view of the boxed region in A″. FMI-1- and SNT-1-containing regions have little overlap. An intensity profile corresponding to the line in A‴ is on the right. B, In the ventral nerve cord, FMI-1 was adjacent to SNT-1 [line scan 1 (LS1)] but occasionally overlapped [e.g., line scan 2 (LS2)]. C, FMI-1 was adjacent to, but not within, GABAergic NMJs labeled by SNB-1::GFP. D, The ju58 allele lacked FMI-1 immunoreactivity, demonstrating the antisera specificity. Results were similar for tm306 and rh308 (data not shown). E, F, The missense mutations gm188 (E) and ju43 (F) did not grossly affect protein localization, although there is a slight increase in the overlap of the FMI-1 and SNT-1 (colocalization coefficient, 0.38). G, H, FMI-1::GFP accumulated in the NR and VNC in a pattern similar to the antisera staining. The pharyngeal staining (asterisk) is from the coinjection marker (Pmyo-2-mCherry). Scale bars, 10 μm.
We costained animals with antibodies to FMI-1 and to the synaptic vesicle protein SNT-1 (Fig. 6). In wild-type animals, FMI-1 was concentrated in the nervous system but did not overlap with a marker of synaptic vesicles (SNT-1). We calculated the intensity colocalization coefficient (see Materials and Methods) for FMI-1 and SNT-1 throughout the nervous system and found that r = 0.27 ± 0.08 (n = 6) in wild-type animals, indicating that FMI-1 was not strongly localized to regions containing synaptic vesicles. We found that fmi-1 is expressed by neurons. Of those, several do form synapses in the ventral nerve cord, e.g., cholinergic motorneurons, as well as many neurons that do not, e.g., HSN, PVP, PVQ, etc. (White et al., 1976). Thus, our data suggest that FMI-1 is primarily present in nonsynaptic regions of the ventral cord. However, because of the en passant organization of synapses formed in the ventral nerve cord, it is not possible to determine where FMI-1 is localized with single axon/synapse resolution.
Flamingo is expressed in a subset of motorneurons
To determine which motorneurons express fmi-1, we fused a 2.6 kb fragment of DNA upstream of the fmi-1 start codon to GFP. Our expression pattern was consistent with what has been previously reported for fmi-1 (Steimel et al., 2010). To specifically identify which motorneurons express flamingo, we coexpressed an RFP (mCherry) reporter in GABAergic (Punc-25) or cholinergic (Pacr-2) neurons (Fig. 7). We observed consistent colocalization of GFP and RFP between Pfmi-1-gfp and Pacr-2-rfp in the DA, VA, and VB and weakly in the DB cholinergic neurons (Fig. 7). We occasionally observed a limited coexpression of Punc-25-rfp and Pfmi-1-gfp in the DDs of some L1 animals (<10%) (Fig. 7), but never in the VDs (>L2). Thus, we find no evidence that the fmi-1 promoter is active in the VD neurons, even though they are affected by mutations in the fmi-1 gene.
Figure 7.

fmi-1 is expressed in the nervous system, but not VD motorneurons. A, A Pfmi-1::GFP was used to determine the spatial expression of fmi-1. GFP was observed in a subset of neurons near the nerve ring (NR) along the ventral nerve cord (VNC) and in the lumbar ganglion (LG). B–E, To identify which motorneurons were expressing flamingo, we coexpressed Pfmi-1-gfp with either Pacr-2-rfp (B) or Punc-25-rfp (C–E). B, C, In larva and adults, the GFP-positive motorneurons also expressed Pacr-2-rfp, but not Punc-25::rfp. D, In contrast, none of the older larvae or adults coexpressed GFP and RFP when we used Punc-25-rfp. D–D″, In L1 larvae, there was limited coexpression of Punc-25-rfp and Pfmi-1-gfp in a subset of the DD motorneurons (arrows). The GFP intensity was much reduced relative to other FMI-1(+) cells. The remaining GFP-positive cells in the ventral cord are likely the DA motorneurons. E–E″, By the adult stage, DD1 (arrow) no longer expresses GFP. Scale bars, 10 μm.
fmi-1 might regulate VD development cell nonautonomously
We used the FMI-1::GFP (Steimel et al., 2010) expressed under its endogenous promoter to rescue the fmi-1 synaptic defects. We injected the Pfmi-1::fmi-1gfp (0.5 ng/μl) plasmid into fmi-1(tm306) along with a Punc-25::snb-1dsRed to visualize GABAergic NMJs. The expression of full-length FMI-1 protein efficiently rescued the synaptic spacing (gaps) (tm306, 1.72 ± 0.92 gaps per cord vs tm306 plus fmi-1gfp, 0.72 ± 0.6; p = 3 × 10−5) and partially rescued the morphology defects (two of two lines). Interestingly, we found that the intracellular domain deletion FMI-1ΔInt::GFP could also rescue the synaptic clustering defect in the GABAergic neurons (two of three lines, 0.77 ± 0.71 gaps per cord; p = 2.1 × 10−4) (Fig. 8). Importantly, we found that in the rescued animals using either full length (data not shown) or the FMI-1ΔInt, the GFP-labeled FMI-1 proteins were not detectable in VD neurons (Fig. 8). To ensure that this was not attributable to transgene mosaicism, we integrated the array carrying the Pfmi-1::FMI-1ΔInt::GFP and Punc-25::SNB-1::RFP and recovered four independent integrated lines. None (zero of four) of the integrated lines exhibited any coexpression of the GFP and RFP proteins (Fig. 8). This analysis further supports that FMI-1 is not present in VD neurons but is required for the proper development of these cells.
Figure 8.

FMI-1 functions cell nonautonomously in the VD neurons. A, VD SNB-1::GFP puncta in fmi-1(tm306 + Punc-25::fmi-1 were enlarged and still had gaps (between arrowheads). A′, A″, RFP was used to mark cells with the transgene. Below is a diagram illustrating the VD neuron (red) expressing FMI-1 [F(+)], but long gaps were present lacking SNB-1GFP. B, When fmi-1 is specifically expressed in the cholinergic cells, SNB-1::GFP puncta were evenly sized and spaced puncta. B′, B″, RFPs, the transgenically positive neurons. C–C″, In some cases, the cholinergic (red) and GABAergic (green) axons appeared defasciculated. Here, even though the fmi-1 transgene is present in these cholinergic neurons, the GABAergic synaptic defects are not rescued. D–D″, Defects in SNB-1::GFP (green) were present when cholinergic axons (red) were properly fasciculated with the GABA neurons. E–E″, FMI-1ΔInt::GFP expressed under its endogenous promoter partially rescued the synaptic defects in GABAergic neurons (as visualized by SNB-1::RFP; E′). GFP is not observed in the GABAergic neurons. F–F″, We integrated the FMI-1ΔInt::GFP and still found no evidence of expression with the GABA marker (Punc-25::SNB-1::dsRED), confirming that FMI-1 expressed from non-GABAergic neurons is sufficient to rescue the SNB-1 accumulation defects. Scale bars, 10 μm.
To test the site of action of FMI-1 more rigorously, we attempted to rescue the defects by expression of FMI-1 in specific cells. We were unable to rescue the synaptic defects in tm306 when we expressed the fmi-1 cDNA specifically in the GABAergic neurons (zero of three lines). In fact, even at the lowest concentration tested (0.5 ng/μl), we saw what appear to be more grossly distended synapses in the ventral nerve cord (tm306, 1.43 μm2 vs tm306 plus Punc-25::fmi-1, 2.11 μm2) (Fig. 7). In contrast, we could partially rescue the synaptic morphology defects in the VD neurons of fmi-1(−) animals when the cDNA was specifically expressed from a cholinergic promoter (Pacr-2) (four of six lines) (tm306, 1.43 μm2 vs tm306 plus Pacr-2::fmi-1, 1.06 μm2; p < 0.05). Thus, whereas we cannot formally rule out that a small amount of FMI-1 is produced in the VDs, expression of FMI-1 in non-VD neurons was able to either fully (Pfmi-1) or partially (Pacr-2) rescue the VD defects.
In the cell-type-specific rescue experiments, we used a cytoplasmic RFP to mark the cells that expressed the FMI-1 transgene. In fmi-1 mutants with Punc-25::fmi-1, we still observed that some axonal regions appeared to lack SNB-1::GFP (Fig. 8A) but did have RFP, although these processes could also come from the DDs, which also express unc-25. In animals expressing fmi-1 under the cholinergic promoter, we occasionally ob served defasciculation of the SNB-1::GFP-labeled GABAergic axons and the RFP-labeled cholinergic processes (Fig. 8C). This suggested that for FMI-1 to influence SNB-1::GFP patterning, the protein needs to be proximal to, perhaps even in direct contact with, VD neurons.
Another possibility that could explain the defects is that FMI-1 functions within cholinergic neurons to maintain axon fasciculation and that the synaptic defects in GABAergic neurons are secondary to disruptions in the cholinergic fascicle. To address this, we examined fmi-1(tm306) mutants with the cholinergic neurons marked by RFP, but lacking the fmi-1 rescuing transgene. VD neurons exhibited NMJ defects in regions where cholinergic axons were properly fasciculated (Fig. 8D), and we found no correlation between cholinergic axon defasciculation and VD synaptic defects (r = 0.16; n = 50). Altogether, these results suggest that FMI-1 likely directly affects VD development, rather than an indirect effect of modulating cholinergic axon bundling.
Mutations in CDH-4, a Fat-like cadherin, exhibit fmi-1-like synaptic defects
Cadherin proteins typically interact homophilically, although there are instances where cadherins exhibit heterophilic interactions (Prakasam et al., 2006). We found that the VD defects in fmi-1 mutants were rescued when FMI-1 was expressed in neighboring neurons, including the cholinergic neurons, suggesting that FMI-1 likely functions through other proteins in GABAergic neurons. Thus, we examined the other cadherin-domain-containing proteins to determine whether any were functioning in the same genetic pathway with fmi-1.
There are 11 genes in the C. elegans genome that encode for 12 proteins with cadherin repeats (Yamashita et al., 2004). Of those, six are evolutionarily conserved (fmi-1/flamingo, hmr-1B/N-cadherin, cdh-1/Dachsous, cdh-3/Fat, cdh-4/Fat, and cdh-11/calsyntenin), whereas the remainder (cdh-5, -7, -9, -10, -12) appear to be nematode specific. We tested whether systemic RNAi of the conserved cadherin genes phenocopied the fmi-1 loss-of-function alleles. Using the eri-1 mutation to potentiate RNAi in the neurons (Kennedy et al., 2004), we found that only the knockdown of cdh-4(RNAi) closely resembled the vesicle accumulation defects in fmi-1 mutants (Fig. 9, Table 1) (eri-1;cdh-4RNAi: 1.56 μm2, 16.7 puncta per 100 μm). The knockdown of cdh-4 in fmi-1 mutants did not significantly enhance the penetrance of defects (Fig. 9) (tm306;eri-1;cdh-4RNAi: 1.57 μm2, 17.8 puncta per 100 μm).
Figure 9.
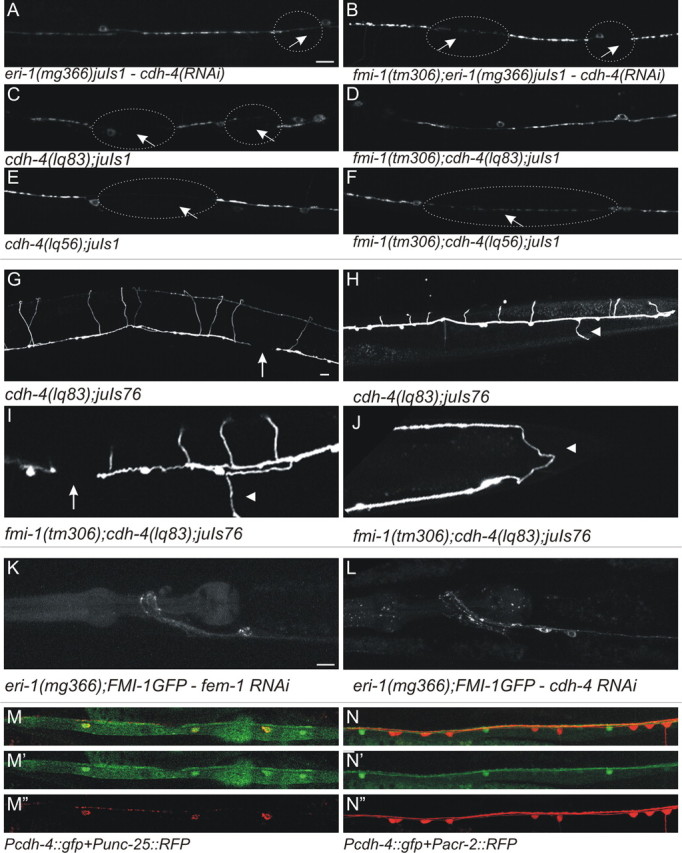
fmi-1 and cdh-4 mutations cause similar phenotypes in VD patterning. A, The eri-1(mg366)juIs1 animals treated with cdh-4 RNAi had elongated SNB-1::GFP puncta interrupted by large gaps (circles and arrows). B, The cdh-4 RNAi caused no enhancement of the phenotype present in eri-1(mg366)juIs1;fmi-1(tm306) animals. C–F, The cdh-4 loss-of-function mutations, lq83 (C, D) and lq56 (E, F), caused fmi-1-like phenotypes, but neither enhanced defects present in fmi-1(tm306). G–J, The cdh-4(lq83) caused GABAergic axon pathfinding defects, including axon extension failure (G, I, arrows) and left/right defects (H, I, arrowheads). I, J, Animals lacking both cdh-4 and fmi-1 have pathfinding defects that are similar to the single mutants. J, In this animal, the VD13 axon (arrowhead) is extended toward the posterior rather than the anterior. K, L, The cdh-4(RNAi) does not grossly disrupt FMI-1::GFP localization relative to control, fem-1(RNAi). M, N, In larva and adults, Pcdh-4::gfp (hdIs46) expression overlaps with a marker for the GABAergic neurons (red, Punc-25::rfp; M) but not the cholinergic neurons (red, Pacr-2::rfp; N). Scale bars, 10 μm.
Two cdh-4 loss-of-function alleles, lq56 and lq83, were identified in a screen for Q-cell descendant migration defects (E. Lundquist, unpublished observations). Both alleles result in premature stop codons (lq83-R126Amber and lq56-L838Amber). We found that the pattern of SNB-1::GFP in GABAergic motorneurons was similar to those observed in the RNAi experiments for cdh-4 and fmi-1 single mutants (Fig. 9, Table 1) [cdh-4(lq56): 1.40 μm2, 16.7 puncta per 100 μm; cdh-4(lq83): 1.82 μm2, 16.6 puncta per 100 μm]. NMJs in double mutants of cdh-4 and fmi-1 were similar to the single mutants [fmi-1(tm306);cdh-4(lq56): 1.48 μm2, 18.1 puncta per 100 μm; fmi-1(tm306);cdh-4(lq83): 1.45 μm2, 16.6 puncta per 100 μm], consistent with these two molecules functioning in a linear genetic pathway.
Previously, Schmitz et al. (2008) described embryonic development, cell migration, and axon guidance phenotypes associated with mutations in the cdh-4 locus. Therefore, we examined the GABAergic axon patterning in cdh-4(lq83) and cdh-4(lq83);fmi-1(tm306). As had been reported previously, we found multiple types of defects in cdh-4 mutant animals, including animals with L/R defects (cdh-4 30%, fmi-1;cdh-4 35%) and defasciculations (cdh-4 29%, fmi-1;cdh-4 22%) (Fig. 9; Tables 3, 4). Although the percentage of animals with axon guidance defects in the double mutants was similar to the cdh-4(lq83) single mutants, in some animals we observed a slight increase in the severity of some phenotypes (Fig. 9). Thus, these proteins may function in overlapping pathways for axon guidance.
The cadherin domains of FMI-1 are partially required for their localization to axons. When the cadherin domains are specifically deleted, more of the FMI-1::GFP is localized to the cell bodies (Steimel et al., 2010). Therefore, we asked whether CDH-4 might be required for the efficient localization of FMI-1 to axons. However, we observed no gross changes in the localization of FMI-1::GFP when cdh-4 was removed by mutation or knocked down by RNAi (Fig. 9). Thus, our experiments suggest that CDH-4 is not required to localize FMI-1 to axons.
We tested whether cdh-4 is expressed in the VD motorneurons by examining the expression of Pcdh-4::gfp (hdIs46) relative to our motorneuron markers (Fig. 9). As has been described, cdh-4 is broadly expressed, including in motorneurons, starting early in development (Schmitz et al., 2008). However, we found that by the L3 stage, the broad expression of Pcdh-4::gfp throughout the nervous system had ceased but expression continues, specifically in the GABAergic neurons. That the VD neurons continued expressing cdh-4 suggests that this protein could be required for events, such synaptogenesis, that occur in later developmental stages, i.e., after axon outgrowth/guidance, which happens in early development.
Discussion
Flamingo cadherins regulate multiple facets of neuronal development
Here we have shown that the single C. elegans Flamingo protein regulates multiple aspects of motorneuron development. Mutations in fmi-1 caused axon outgrowth and synaptic defects in the cholinergic and GABAergic motorneurons, although the penetrance of phenotypes in these cell types was different. The fmi-1 and cdh-4, two conserved cadherin-domain-containing proteins, both affected GABAergic development and seemed to function in a common genetic pathway. FMI-1 influences GABAergic development nonautonomously, as we find no evidence that fmi-1 is expressed in the VD GABAergic neurons. In contrast, cdh-4 does seem to be expressed within these neurons, but where it is required for GABAergic neuron development remains to be determined. Whether FMI-1 and CDH-4 physically interact is unknown. However, it remains possible that these two molecules function in receptor-ligand-like fashion to facilitate VD development.
Planar cell polarity and synaptogenesis share a common goal
Flamingo was initially isolated as a component of the planar cell polarity pathway, where it functions to establish asymmetric protein localization within a cell. This asymmetry helps break a field of equivalent epithelia into cells polarized to a body axis. It is interesting to speculate on how a PCP-like activity may have become specialized to ensure patterning of synaptic versus nonsynaptic specializations. For example, these proteins could mark regions of the axon where synapses can or cannot form. Additional evidence for this comes from the identification that Wnt signaling regulates the accumulation of Frizzled receptors to the appropriate segment of the DA9 axon, to ensure the proper delivery of synaptic components (Klassen and Shen, 2007). Our ultrastructural analysis suggests FMI-1 functions to ensure the proper delivery or assembly of synaptic components, but the precise mechanism by which this occurs remains to be elucidated.
FMI-1 affects both axon extension and synaptic development
We have found that fmi-1 mutations result in multiple neuronal defects, including altered axon outgrowth and synaptic patterning. Drosophila Flamingo/fmi mutants also exhibit both axon pathfinding and synaptogenesis defects in motorneurons (Bao et al., 2007). Interestingly, Flamingo mutant motor axons form ectopic synapses en passant, a type of synapse that wild-type motorneurons do not form at all (Bao et al., 2007). These types of synaptic contacts are also seen in misrouted axons in the brains of fmi mutants (Lee et al., 2003; Senti et al., 2003), suggesting that these synapses may occur due to the axon being placed in an unfamiliar context. Similar to our ultrastructural studies, Bao et al. (2007) found that synaptic components can be mislocalized to regions of the axon when it is not contacting its normal target. Although it is unclear whether this reflects a failure in synaptogenesis or target recognition, it would seem that Flamingo proteins are involved in how synaptic components are directed appropriately within axons.
Axon guidance and synaptogenesis defects occur in many backgrounds where cell adhesion molecules are mutated, such as cle-1, nid-1 and ptp-3, unc-40, unc-5, and unc-6. Where it has been examined, many have concluded that these defects can be separated (Ackley et al., 2003, 2005; Gally and Bessereau, 2003; Cólon-Ramos et al., 2007; Poon et al., 2008; Killeen, 2009). The synaptic phenotypes in these cell adhesion mutants are distinct from each other as well as from those in fmi-1. Therefore, although some of the disruptions in synaptic vesicle accumulation in fmi-1 mutants can be attributed to axon guidance problems, it is unlikely that the altered axon guidance nonspecifically results in synaptic defects. Rather, our results indicate that fmi-1 functions in multiple developmental events in neurons, both in axon guidance and synaptogenesis. Whether these occur via distinct mechanisms remains to be elucidated.
Other genes involved in epithelial polarity have been found to also regulate axon growth and synapse formation in recent years. Vertebrate polarity proteins such as Par3 and Par6 interact with atypical protein kinase C, Inscuteable, and/or Partner of Inscuteable (PINS) to link signaling from extracellular ligands (e.g., NGF or netrin-1) to changes in epithelial polarity (Zhang and Macara, 2006; McCaffrey and Macara, 2009). In some cases, these signals indicate to a migrating axon to transition from a migratory, nonsynapse-forming growth cone to an adherent synapse-forming axon. Synapse formation and axon extension are also linked in C. elegans (Grill et al., 2007; Yan et al., 2009; Trujillo et al., 2010). VD neurons are born at the late L1 larval stage, and their neurites increase approximately threefold to fivefold in length by the time the animal reaches adulthood. We find that FMI-1 expression continues throughout development and that the protein is present into adulthood. This may indicate that FMI-1 functions in the continued development of the nervous system that occurs in those stages, such as axon extension and synaptogenesis.
Cell nonautonomous role of fmi-1
In Drosophila, Flamingo has been found to function via self-interactions (Usui et al., 1999; Chen and Clandinin, 2008; Chen et al., 2008). In our study, we find that the VD neurons do not produce detectable levels of Flamingo, yet they are among the most sensitive cells to the absence of FMI-1. The defects in VD neurons were rescued when we use promoters that are not active in the VD neurons (Pfmi-1 and Pacr-2). FMI-1 lacking the intracellular domain is capable of rescuing the vesicle accumulation defects, even though it is not expressed in the VD neurons. All together, these results indicate that FMI-1 is likely functioning cell nonautonomously to regulate the axon and synapse development of the VD neurons.
Flamingo has previously been shown to act cell nonautonomously, most elegantly in the Drosophila retinal axons (Chen and Clandinin, 2008). They demonstrated that mosaics, in which only one of six R neurons was fmi(+), the neurons contacting the FMI-containing neuron exhibit more defects than adjacent neurons in the same bundle. They also demonstrated that overexpression of the FMI-1 ECD was sufficient to induce targeting defects. They concluded that cells are effectively gauging the amount of Flamingo and that asymmetric levels lead to functional consequences. Similarly, we find that, in rescue and overexpression experiments, phenotypes are quite dose dependent, suggesting that changes in protein levels are important for cellular responses to Flamingo.
FMI-1 interacts genetically with a Fat-like cadherin protein
We find that knockdown of cdh-4 by RNAi or mutation phenocopies fmi-1 in synaptic development. In planar cell polarity, Fat and Dachsous have been shown to function together in parallel to Flamingo (Casal et al., 2006). Here we find that during C. elegans development, Flamingo and CDH-4 seem to have overlapping functions. We see no obvious enhancement of the embryonic or larval lethality caused by cdh-4 mutations when fmi-1 is removed from those animals, suggesting no involvement of Flamingo in this process. Our data suggest that fmi-1 and cdh-4 function in a linear genetic pathway with regard to the development of the VD motorneurons, which would be the first indication of such a relationship for these two conserved cadherins.
We find that CDH-4 has a limited, if any, role in the gross localization of FMI-1, although subtle changes in the localization of FMI-1 could be important for its activity. We also find that the promoter of CDH-4 is active in the VD neurons and that the expression of this gene continues throughout VD development. Because synaptogenesis continues after the axons have become patterned, the continued expression of cdh-4 in the VDs is consistent with a role for this protein in the formation of VD synapses. The relationship between these proteins could be key to understanding how cholinergic neurons may pattern the GABAergic synapses in the neuromuscular system of C. elegans.
Footnotes
Y.J. was supported by NIH Grant NS035546. E.A.L. was supported by NIH Grants R01 NS040945 and R21 NS070417. B.D.A. was supported in part by NCRR IDeA Networks of Biomedical Research Excellence Program Grant P20 RR016475 and NIH Grant RC1 GM091086. M.Z. was supported by a Human Frontier Long-term Fellowship and a grant from Natural Sciences and Engineering Research Council of Canada. A.G. is an associate and Y.J. is an investigator of HHMI. We thank M. Razak for assistance in isolating the lq56 and lq83 alleles of cdh-4, H. Hutter for comments on this manuscript as well as strains and reagents, S. Mitani for the tm306 allele and the CGC for strains.
References
- Ackley BD, Kang SH, Crew JR, Suh C, Jin Y, Kramer JM. The basement membrane components nidogen and type XVIII collagen regulate organization of neuromuscular junctions in Caenorhabditis elegans. J Neurosci. 2003;23:3577–3587. doi: 10.1523/JNEUROSCI.23-09-03577.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackley BD, Harrington RJ, Hudson ML, Williams L, Kenyon CJ, Chisholm AD, Jin Y. The two isoforms of the Caenorhabditis elegans leukocyte-common antigen related receptor tyrosine phosphatase PTP-3 function independently in axon guidance and synapse formation. J Neurosci. 2005;25:7517–7528. doi: 10.1523/JNEUROSCI.2010-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao H, Berlanga ML, Xue M, Hapip SM, Daniels RW, Mendenhall JM, Alcantara AA, Zhang B. The atypical cadherin flamingo regulates synaptogenesis and helps prevent axonal and synaptic degeneration in Drosophila. Mol Cell Neurosci. 2007;34:662–678. doi: 10.1016/j.mcn.2007.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolte S, Cordelières FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown HM, Van Epps HA, Goncharov A, Grant BD, Jin Y. The JIP3 scaffold protein UNC-16 regulates RAB-5 dependent membrane trafficking at C. elegans synapses. Dev Neurobiol. 2009;69:174–190. doi: 10.1002/dneu.20690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casal J, Lawrence PA, Struhl G. Two separate molecular systems, Dachsous/Fat and Starry night/Frizzled, act independently to confer planar cell polarity. Development. 2006;133:4561–4572. doi: 10.1242/dev.02641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PL, Clandinin TR. The cadherin Flamingo mediates level-dependent interactions that guide photoreceptor target choice in Drosophila. Neuron. 2008;58:26–33. doi: 10.1016/j.neuron.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WS, Antic D, Matis M, Logan CY, Povelones M, Anderson GA, Nusse R, Axelrod JD. Asymmetric homotypic interactions of the atypical cadherin flamingo mediate intercellular polarity signaling. Cell. 2008;133:1093–1105. doi: 10.1016/j.cell.2008.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cólon-Ramos DA, Margeta MA, Shen K. Glia promote local synaptogenesis through UNC-6 (netrin) signaling in C. elegans. Science. 2007;318:103–106. doi: 10.1126/science.1143762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtin JA, Quint E, Tsipouri V, Arkell RM, Cattanach B, Copp AJ, Henderson DJ, Spurr N, Stanier P, Fisher EM, Nolan PM, Steel KP, Brown SD, Gray IC, Murdoch JN. Mutation of Celsr1 disrupts planar polarity of inner ear hair cells and causes severe neural tube defects in the mouse. Curr Biol. 2003;13:1129–1133. doi: 10.1016/s0960-9822(03)00374-9. [DOI] [PubMed] [Google Scholar]
- Dougherty RP. Extensions of DAMAS and benefits and limitations of deconvolution in beamforming. 2005 AIAA Paper 2005–2961, May, 2005. [Google Scholar]
- Formstone CJ, Mason I. Expression of the Celsr/flamingo homologue, c-fmi1, in the early avian embryo indicates a conserved role in neural tube closure and additional roles in asymmetry and somitogenesis. Dev Dyn. 2005;232:408–413. doi: 10.1002/dvdy.20228. [DOI] [PubMed] [Google Scholar]
- Gally C, Bessereau JL. GABA is dispensable for the formation of junctional GABA receptor clusters in Caenorhabditis elegans. J Neurosci. 2003;23:2591–2599. doi: 10.1523/JNEUROSCI.23-07-02591.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao FB, Kohwi M, Brenman JE, Jan LY, Jan YN. Control of dendritic field formation in Drosophila: the roles of flamingo and competition between homologous neurons. Neuron. 2000;28:91–101. doi: 10.1016/s0896-6273(00)00088-x. [DOI] [PubMed] [Google Scholar]
- Grill B, Bienvenut WV, Brown HM, Ackley BD, Quadroni M, Jin Y. C. elegans RPM-1 regulates axon termination and synaptogenesis through the Rab GEF GLO-4 and the Rab GTPase GLO-1. Neuron. 2007;55:587–601. doi: 10.1016/j.neuron.2007.07.009. [DOI] [PubMed] [Google Scholar]
- Hall DH, Russell RL. The posterior nervous system of the nematode Caenorhabditis elegans: serial reconstruction of identified neurons and complete pattern of synaptic interactions. J Neurosci. 1991;11:1–22. doi: 10.1523/JNEUROSCI.11-01-00001.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallam SJ, Goncharov A, McEwen J, Baran R, Jin Y. SYD-1, a presynaptic protein with PDZ, C2 and rhoGAP-like domains, specifies axon identity in C. elegans. Nat Neurosci. 2002;5:1137–1146. doi: 10.1038/nn959. [DOI] [PubMed] [Google Scholar]
- Huang X, Cheng HJ, Tessier-Lavigne M, Jin Y. MAX-1, a novel PH/MyTH4/FERM domain cytoplasmic protein implicated in netrin-mediated axon repulsion. Neuron. 2002;34:563–576. doi: 10.1016/s0896-6273(02)00672-4. [DOI] [PubMed] [Google Scholar]
- Huang X, Huang P, Robinson MK, Stern MJ, Jin Y. UNC-71, a disintegrin and metalloprotease (ADAM) protein, regulates motor axon guidance and sex myoblast migration in C. elegans. Development. 2003;130:3147–3161. doi: 10.1242/dev.00518. [DOI] [PubMed] [Google Scholar]
- Inaki M, Yoshikawa S, Thomas JB, Aburatani H, Nose A. Wnt4 is a local repulsive cue that determines synaptic target specificity. Curr Biol. 2007;17:1574–1579. doi: 10.1016/j.cub.2007.08.013. [DOI] [PubMed] [Google Scholar]
- Jin Y. Synaptogenesis, (WormBook, ed.) The C. elegans Research Community, WormBook. 2005. doi: 10.1895/wormbook.1.44.1, http://www.workbook.org.
- Kennedy S, Wang D, Ruvkun G. A conserved siRNA-degrading RNase negatively regulates RNA interference in C. elegans. Nature. 2004;427:645–649. doi: 10.1038/nature02302. [DOI] [PubMed] [Google Scholar]
- Killeen MT. The dual role of the ligand UNC-6/Netrin in both axon guidance and synaptogenesis in C. elegans. Cell Adh Migr. 2009;3:268–271. doi: 10.4161/cam.3.3.8398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klassen MP, Shen K. Wnt signaling positions neuromuscular connectivity by inhibiting synapse formation in C. elegans. Cell. 2007;130:704–716. doi: 10.1016/j.cell.2007.06.046. [DOI] [PubMed] [Google Scholar]
- Koushika SP, Richmond JE, Hadwiger G, Weimer RM, Jorgensen EM, Nonet ML. A post-docking role for active zone protein Rim. Nat Neurosci. 2001;4:997–1005. doi: 10.1038/nn732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RC, Clandinin TR, Lee CH, Chen PL, Meinertzhagen IA, Zipursky SL. The protocadherin Flamingo is required for axon target selection in the Drosophila visual system. Nat Neurosci. 2003;6:557–563. doi: 10.1038/nn1063. [DOI] [PubMed] [Google Scholar]
- Lewis A, Wilson N, Stearns G, Johnson N, Nelson R, Brockerhoff SE. Celsr3 is required for normal development of GABA circuits in the inner retina. PLoS Genet. 2011;7:e1002239. doi: 10.1371/journal.pgen.1002239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Usui T, Uemura T, Jan L, Jan YN. Flamingo controls the planar polarity of sensory bristles and asymmetric division of sensory organ precursors in Drosophila. Curr Biol. 1999;9:1247–1250. doi: 10.1016/s0960-9822(99)80505-3. [DOI] [PubMed] [Google Scholar]
- McCaffrey LM, Macara IG. Widely conserved signaling pathways in the establishment of cell polarity. Cold Spring Harb Perspect Biol. 2009;1:a001370. doi: 10.1101/cshperspect.a001370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonet ML, Grundahl K, Meyer BJ, Rand JB. Synaptic function is impaired but not eliminated in C. elegans mutants lacking synaptotagmin. Cell. 1993;73:1291–1305. doi: 10.1016/0092-8674(93)90357-v. [DOI] [PubMed] [Google Scholar]
- Pettitt J. The cadherin superfamily, (WormBook, ed.) The C. elegans Research Community, WormBook. 2005. doi: 10.1895/wormbook.1.50.1, http://www.wormbook.org.
- Poon VY, Klassen MP, Shen K. UNC-6/netrin and its receptor UNC-5 locally exclude presynaptic components from dendrites. Nature. 2008;455:669–673. doi: 10.1038/nature07291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakasam AK, Maruthamuthu V, Leckband DE. Similarities between heterophilic and homophilic cadherin adhesion. Proc Natl Acad Sci U S A. 2006;103:15434–15439. doi: 10.1073/pnas.0606701103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond J. Synaptic function, (WormBook, ed.) The C. elegans Research Community, WormBook. 2005. doi. 10.1895/wormbook.1.69.1, http://www.workbook.org.
- Schmitz C, Wacker I, Hutter H. The Fat-like cadherin CDH-4 controls axon fasciculation, cell migration and hypodermis and pharynx development in Caenorhabditis elegans. Dev Biol. 2008;316:249–259. doi: 10.1016/j.ydbio.2008.01.024. [DOI] [PubMed] [Google Scholar]
- Senti KA, Usui T, Boucke K, Greber U, Uemura T, Dickson BJ. Flamingo regulates R8 axon-axon and axon-target interactions in the Drosophila visual system. Curr Biol. 2003;13:828–832. doi: 10.1016/s0960-9822(03)00291-4. [DOI] [PubMed] [Google Scholar]
- Shen K, Fetter RD, Bargmann CI. Synaptic specificity is generated by the synaptic guidepost protein SYG-2 and its receptor, SYG-1. Cell. 2004;116:869–881. doi: 10.1016/s0092-8674(04)00251-x. [DOI] [PubMed] [Google Scholar]
- Shima Y, Kengaku M, Hirano T, Takeichi M, Uemura T. Regulation of dendritic maintenance and growth by a mammalian 7-pass transmembrane cadherin. Dev Cell. 2004;7:205–216. doi: 10.1016/j.devcel.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Shima Y, Kawaguchi SY, Kosaka K, Nakayama M, Hoshino M, Nabeshima Y, Hirano T, Uemura T. Opposing roles in neurite growth control by two seven-pass transmembrane cadherins. Nat Neurosci. 2007;10:963–969. doi: 10.1038/nn1933. [DOI] [PubMed] [Google Scholar]
- Steimel A, Wong L, Najarro EH, Ackley BD, Garriga G, Hutter H. The Flamingo ortholog FMI-1 controls pioneer-dependent navigation of follower axons in C. elegans. Development. 2010;137:3663–3673. doi: 10.1242/dev.054320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC. Alpha-latrotoxin and its receptors: neurexins and CIRL/latrophilins. Annu Rev Neurosci. 2001;24:933–962. doi: 10.1146/annurev.neuro.24.1.933. [DOI] [PubMed] [Google Scholar]
- Tissir F, Bar I, Jossin Y, De Backer O, Goffinet AM. Protocadherin Celsr3 is crucial in axonal tract development. Nat Neurosci. 2005;8:451–457. doi: 10.1038/nn1428. [DOI] [PubMed] [Google Scholar]
- Trujillo G, Nakata K, Yan D, Maruyama IN, Jin Y. A ubiquitin E2 variant protein acts in axon termination and synaptogenesis in Caenorhabditis elegans. Genetics. 2010;186:135–145. doi: 10.1534/genetics.110.117341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usui T, Shima Y, Shimada Y, Hirano S, Burgess RW, Schwarz TL, Takeichi M, Uemura T. Flamingo, a seven-pass transmembrane cadherin, regulates planar cell polarity under the control of Frizzled. Cell. 1999;98:585–595. doi: 10.1016/s0092-8674(00)80046-x. [DOI] [PubMed] [Google Scholar]
- White JG, Southgate E, Thomson JN, Brenner S. The structure of the ventral nerve cord of Caenorhabditis elegans. Philos Trans R Soc Lond B Biol Sci. 1976;275:327–348. doi: 10.1098/rstb.1976.0086. [DOI] [PubMed] [Google Scholar]
- White JG, Southgate E, Thomson JN, Brenner S. The structure of the nervous system of the nematode Caenorhabditis elegans. Philos Trans R Soc Lond B Biol Sci. 1986;314:1–340. doi: 10.1098/rstb.1986.0056. [DOI] [PubMed] [Google Scholar]
- Wu Z, Ghosh-Roy A, Yanik MF, Zhang JZ, Jin Y, Chisholm AD. Caenorhabditis elegans neuronal regeneration is influenced by life stage, ephrin signaling, and synaptic branching. Proc Natl Acad Sci U S A. 2007;104:15132–15137. doi: 10.1073/pnas.0707001104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita H, Goto A, Kadowaki T, Kitagawa Y. Mammalian and Drosophila cells adhere to the laminin alpha4 LG4 domain through syndecans, but not glypicans. Biochem J. 2004;382:933–943. doi: 10.1042/BJ20040558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D, Wu Z, Chisholm AD, Jin Y. The DLK-1 kinase promotes mRNA stability and local translation in C. elegans synapses and axon regeneration. Cell. 2009;138:1005–1018. doi: 10.1016/j.cell.2009.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh E, Kawano T, Weimer RM, Bessereau JL, Zhen M. Identification of genes involved in synaptogenesis using a fluorescent active zone marker in Caenorhabditis elegans. J Neurosci. 2005;25:3833–3841. doi: 10.1523/JNEUROSCI.4978-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Macara IG. The polarity protein PAR-3 and TIAM1 cooperate in dendritic spine morphogenesis. Nat Cell Biol. 2006;8:227–237. doi: 10.1038/ncb1368. [DOI] [PubMed] [Google Scholar]
- Zhen M, Jin Y. The liprin protein SYD-2 regulates the differentiation of presynaptic termini in C. elegans. Nature. 1999;401:371–375. doi: 10.1038/43886. [DOI] [PubMed] [Google Scholar]