

Abstract
Objective
Endothelial cell activation results in altered cell-cell interactions with adjacent endothelial cells and with infiltrating leukocytes. Eph receptors and their ephrin ligands regulate cell-cell interactions during tissue remodeling, and multiple proinflammatory mediators induce endothelial EphA receptor and ephrinA ligand expression. Therefore, we sought to elucidate the role of EphA receptors and ephrinA ligands in endothelial cell activation and atherosclerosis.
Methods and Results
qRT-PCR screening for EphA/ephrinA expression in atherosclerosis-prone macrovascular endothelium identified EphA2, EphA4, and ephrinA1 as the dominant isoforms. Endothelial activation with oxidized LDL (oxLDL) and proinflammatory cytokines induced EphA2 and ephrinA1 expression and sustained EphA2 activation, whereas EphA4 expression was unaffected. Atherosclerotic plaques from mice and humans show enhanced EphA2 and ephrinA1 expression colocalizing in the endothelial cell layer. EphA2 activation with recombinant Fc-ephrinA1 induces proinflammatory gene expression (ex. VCAM-1, E-Selectin) and stimulates monocyte adhesion, while inhibiting EphA2 (siRNA, pharmacological inhibitors) abrogated both ephrinA1-induced and oxLDL-induced VCAM-1 expression.
Conclusions
The current data suggest that enhanced EphA2 signaling during endothelial cell activation perpetuates proinflammatory gene expression. Coupled with EphA2 expression in mouse and human atherosclerotic plaques, these data implicate EphA2 as a novel proinflammatory mediator and potential regulator of atherosclerotic plaque development.
Keywords: Eph receptor tyrosine kinase, Atherogenesis, Endothelium, Inflammation, Gene expression
The endothelium regulates multiple aspects of atherosclerotic plaque formation and progression. Early during atherogenesis, proinflammatory factors such as cytokines (TNFα, IL-1β) and oxidized LDL (oxLDL) stimulate endothelial cell activation, resulting in enhanced endothelial permeability, increased cell adhesion molecules expression driving leukocyte recruitment, and dysfunction of the endothelium’s anti-thrombotic properties1. Proinflammatory cell adhesion molecules such as vascular cell adhesion molecule (VCAM-1), intercellular adhesion molecule (ICAM-1), and E-selectin mediate monocyte extravasation into the atherosclerotic plaque2 where they differentiate into macrophages and phagocytose accumulated oxLDL3. These activated, lipid-laden macrophages secrete cytokines and mitogenic factors into the vessel wall perpetuating local inflammation and plaque progression1. Clinical manifestations of atherosclerosis most often involve acute thrombosis due to plaque rupture or endothelial denudation (superficial plaque erosion)4. A smooth muscle cell and extracellular matrix-rich fibrotic cap resists plaque rupture, whereas leukocyte-derived proteases degrade the fibrotic cap and destabilize the plaque4. Therefore, endothelial cell activation contributes to plaque initiation, progression, and rupture by promoting local inflammation.
The Eph family of receptor tyrosine kinases, the largest in the mammalian genome, form intercellular interactions with their ephrin ligands that affect cytoskeletal remodeling, cell motility, and cell adhesion/repulsion depending upon cell type and Eph/ephrin expression gradients5. Mammals express 15 Eph receptors divided into A-class (EphA1-8 & 10) and B-class (EphB1-6) receptors based on sequence homology and binding preferences for their ephrin ligands (ephrinA1-5, ephrinB1-3)6. In vascular biology, Ephs and ephrins classically regulate segregation of endothelial cells into veins (EphB4) and arteries (ephrinB2) during development7. Additionally, both B-class and A-class Ephs and ephrins mediate angiogenesis in the adult8. Recent evidence suggests B-class ephrins show altered expression in the atherosclerotic plaque and may mediate monocyte targeting to the plaque9, 10. However, a causal role for Eph/ephrin signaling in endothelial activation has not been shown.
While no studies have linked A-class Ephs/ephrins to atherosclerosis, multiple lines of evidence implicate EphA receptors and ephrinA ligands in inflammatory responses. Systemic LPS administration results in increased EphA2 expression in the liver, lungs, and brains of rats11, and M. tuberculosis infection stimulates both ephrinA1 and EphA2 expression in the mouse lung12. The ephrinA1 gene was originally identified as a TNFα-inducible immediate-early response gene in human umbilical vein endothelial cells (HUVEC), and TNFα-induced vascular tube formation requires EphA2-ephrinA1 interactions13. EphA receptors and ephrinA ligands on the endothelial cell surface function as counter-receptors that stimulate either lymphocyte adhesion or repulsion in a highly cell type specific manner14, 15. Interestingly, the EphA2 gene resides on a region of chromosome 1 linked to premature myocardial infarction in humans (1p36)16 and on a region of mouse chromosome 4 linked to enhanced susceptibility to atherosclerosis (Athsq1 locus)17. Therefore, we sought to characterize the role of EphA/ephrinA interactions in the endothelial cell inflammatory response and their association with atherosclerotic plaque formation.
Methods
EphA/ephrinA expression in human aortic endothelial cells (HAEC), human coronary artery endothelial cells (HCAEC), or HUVECs was determined by qRT-PCR and Western blotting. Endothelial cell activation was induced by treatment with cytokines (TNFα, IL-1β), highly oxidized LDL (CuSO4 oxidized; relative electrophoretic mobility between 2 and 318, 19), or recombinant, pre-conjugated Fc-ephrinA1. Changes in endothelial gene expression after treatment with Fc-ephrinA1 were determined using the StellARray Endothelial Cell Biology qRT-PCR array plate (Lonza). Animal protocols were approved by the LSU Health Sciences Center-Shreveport IACUC committee, and all animals were cared for according to the National Institute of Health guidelines for the care and use of laboratory animals. All experiments using human tissue were deemed non human research by the local IRB due to exclusive use of postmortem samples. Changes in gene expression in vessels harvested from male ApoE null mice fed standard chow or a high fat Western type diet (Teklad 88137) for either 2 or 6 months was detected by classic immunohistochemistry (DAB), immunofluorescence microscopy, and qRT-PCR analysis. Atherosclerotic plaques from human vessels were analyzed by immunohistochemistry. EphA2 activation was assessed by EphA2 immunoprecipitation and Western blotting with a phosphotyrosine-specific antibody (4G10), and EphA2 inhibition was accomplished using 3 different anti-EphA2 siRNA oligos (Sigma, Dharmacon) and a chemical compound (2,5-dimethylpyrrol benzoic acid; Chembridge) that blocks ligand binding to both EphA2 and EphA4.
Results
EphA2 expression is responsive to atherogenic mediators
Eph/ephrin signaling differs greatly between cell types, and much of the previous work studying EphA signaling in endothelial cells utilized microvascular endothelium and HUVECs. Therefore, we first compared EphA/ephrinA expression profiles in macrovascular endothelial cells (HAECs, HCAECs) to that of HUVECs by qRT-PCR (Fig. 1A). Of the nine mammalian EphA receptors, only EphA2 and EphA4 receptors showed significant expression. Overall, mRNA derived from HAECs, HCAECs, and HUVECs (isolated from three different donors) revealed no difference in EphA receptor expression pattern between macrovascular endothelium and HUVECs.
Figure 1.
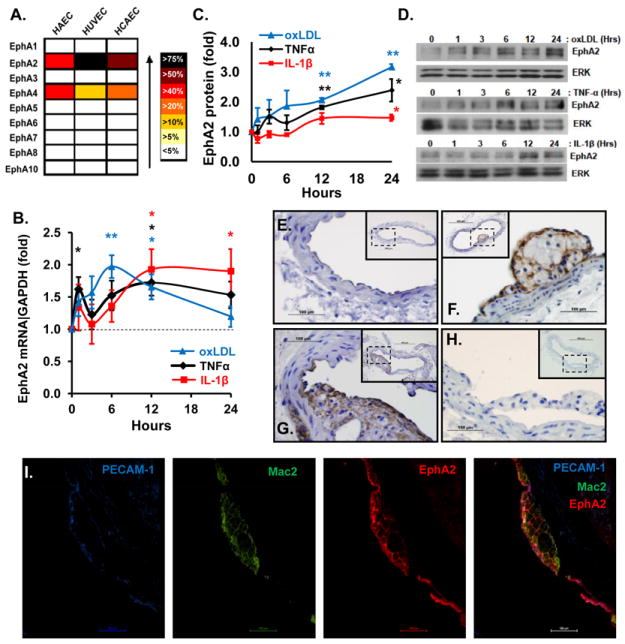
A, Heat map of endothelial cell EphA receptor gene expression by qRT-PCR analysis normalized to 18S (n = 3 donors per cell type) and expressed as a percent of total EphA receptor expression. Validity of the qRT-PCR primers (Table SI) was verified by melt curve analysis and by sequencing of the PCR products. B, qRT-PCR analysis of EphA2 expression in HAECs treated with 10 ng/ml TNFα (black), 5 ng/ml IL-1β (red), or 50-100 μg/ml oxLDL (blue) for 0-24 hrs and lysed simultaneously. Results are normalized to GAPDH and to untreated controls. (n = 3-7). C/D, EphA2 protein analysis of HAECs treated as in B. (n = 4-5); * p< 0.05, ** p< 0.01. E-H, ApoE null mice were fed standard chow (E) or a high fat Western diet for 2 months (F) or 6 months (G). The carotid sinus was stained for EphA2 (brown) or with no primary antibody (H). n = 4 mice for each time point. I, Immunofluorescent co-staining of an early atherosclerotic plaque with PECAM-1 (endothelium, blue), Mac2 (macrophages, green), and EphA2 (red). Representative 40X images are shown.
Local inflammatory stimuli drive endothelial cell activation and atherogenesis1 and promote EphA2 expression in other systems20, 21. Therefore, we tested whether the proinflammatory atherogenic stimuli TNFα (10 ng/ml), IL-1β (5 ng/ml) and oxidized LDL (50-100 μg/ml) affect endothelial EphA2 expression. The cytokines TNFα and IL-1β stimulated a slow induction of EphA2 mRNA maximal at 1.7 and 1.9-fold at 12 hours post-treatment (Fig. 1B) followed by EphA2 protein expression maximal at 2.4- and 1.5-fold between 12 and 24 hours post-treatment (Fig. 1C/D). In contrast, oxLDL stimulated a transient induction of EphA2 mRNA levels maximal at 2-fold between 6-12 hours post-treatment but returning to near baseline levels by 24 hours (Fig. 1B). However, oxLDL stimulated a sustained increase in EphA2 protein expression to greater than 3-fold at 24 hours post-treatment (Fig. 1C/D). These proinflammatory stimuli significantly decreased EphA4 mRNA expression (Fig. SI-A) but did not affect EphA4 protein levels (Fig. SI-B), suggesting that proinflammatory stimuli specifically enhance EphA2 expression but not EphA4.
Since the EphA2 gene resides in a susceptibility locus for premature myocardial infarction in humans (1p36)16 and atherosclerosis in mice (Athsq1)17, we next tested whether EphA2 expression correlates with plaque formation in atherosclerosis-prone ApoE null mice. Eight to ten week-old male ApoE null mice were fed standard chow or a high fat (21%), Western diet for 8 or 24 weeks to stimulate plaque development. EphA2 expression in the atherosclerotic carotid sinus, innominate artery, aortic arch, and abdominal aorta near the renal artery and femoral artery branch points was determined by immunohistochemistry using an antibody previously verified as EphA2-specific in EphA2 knockout mice22. EphA2 displayed minimal expression in atherosclerosis prone vessels of ApoE null mice fed standard chow (Fig. 1E) but showed enhanced expression in vessels isolated from mice fed the high fat Western diet (Fig 1F/G). We observed prominent EphA2 staining in the endothelium overlying both developing fatty streaks (Fig. 1F) and more advanced atheromas (Fig. 1G); however EphA2 remained undetectable in unaffected regions of the same vessels and in non-atherosclerotic vessels. Control tissue incubated with secondary antibody alone showed no staining (Fig. 1H). Immunofluorescent co-staining for endothelial cells (PECAM-1), macrophages (Mac2), and Epha2 demonstrated pronounced EphA2 staining in the endothelial cell layer and moderate EphA2 staining in the cell-cell junctions between adjacent foam cells (Fig. 1I). While representative plaques from the carotid sinus are shown, similar EphA2 staining patterns in atherosclerotic plaques from other regions (Fig. SII) implicate EphA2 as a common atherosclerotic plaque component.
To determine if human atherosclerotic plaques also show enhanced EphA2 expression, we obtained atherosclerosis-prone (carotid sinus, right coronary artery, left anterior descending artery) and resistant (common carotid) arteries excised post-mortem during autopsies. H&E-stained sections were examined by a pathologist and scored for plaque grade based on the Stary Classification method23. Stary scoring of 45 vessels from 25 patients identified a range of plaques from grade 1 to grade 5 with 5 to 14 plaques identified per grade (Table SII and SIII). Nearby sections were stained for EphA2 expression as well as markers of endothelial cells (vWF) and macrophages (CD68). Consistent with murine tissues, the endothelium of non-atherosclerotic vessels lacked EphA2 expression, whereas the endothelium overlying atherosclerotic plaques showed prominent EphA2 expression (Fig. 2) beginning in early fatty streaks (grade 1/2 plaques) and sustained in intermediate (grade 3 and 4) and advanced atheromas (grade 5). Neointimal EphA2 staining corresponded with regions of macrophage recruitment as assessed by CD68 stains, and marked EphA2 expression was observed in CD68 and ApoB-positive foam cells within human fatty streaks (Fig. SIII). Taken together, these data suggest that endothelial EphA2 expression occurs concomitant with atherosclerosis-associated endothelial cell activation in both mouse and human atherosclerotic plaques.
Figure 2.
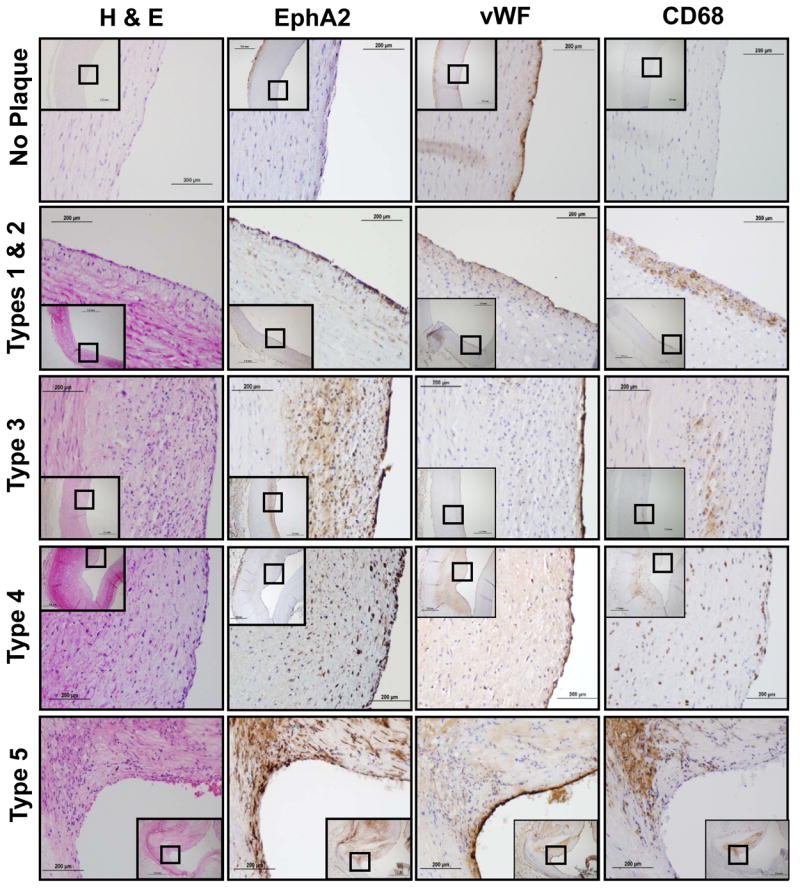
Serial sections of human coronary and carotid arteries were scored via the Stary Classification method and stained by H&E or for EphA2, wWF (endothelium), or CD68 (macrophages) by immunohistochemistry (brown DAB stain). Images are representative of 5-14 vessels per plaque grade.
EphrinA expression in models of atherosclerosis
Previous reports demonstrate that TNFα stimulates ephrinA1 expression and EphA2 activation in HUVECs13, suggesting that proinflammatory stimuli likely promote EphA2 ligand expression as well. To test this, we next examined ephrinA ligand expression in endothelial cells by qRT-PCR. EphrinA1 expression significantly exceeded ephrinA4 or ephrinA5 expression, whereas ephrinA2 and ephrinA3 exhibited very low to no expression in all three endothelial cell types (Fig. 3A). Like EphA2, cytokine treatment stimulated a rapid and sustained increase in ephrinA1 mRNA (Fig. 3B) whereas ephrinA4 (Fig. 3C) and ephrinA5 (Fig. 3D) mRNA was unaffected. The upregulation in ephrinA1 mRNA coincided with a sustained increase in ephrinA1 protein expression following TNFα treatment (1.5-fold from 6h to 24h) but only a transient induction by IL-1β (1.5 to 2-fold from 1h to 6h; Fig. 3E). While oxLDL did not affect ephrinA4 or ephrinA5 mRNA expression (Fig. 3C/D), oxLDL significantly reduced ephrinA1 mRNA expression by 24 hours (Fig. 3B) but induced a sustained increase in ephrinA1 protein expression up to 3-fold between 6 and 24 hours post-treatment (Fig. 3E), suggesting that oxLDL promotes ephrinA1 expression independent of mRNA transcription.
Figure 3.
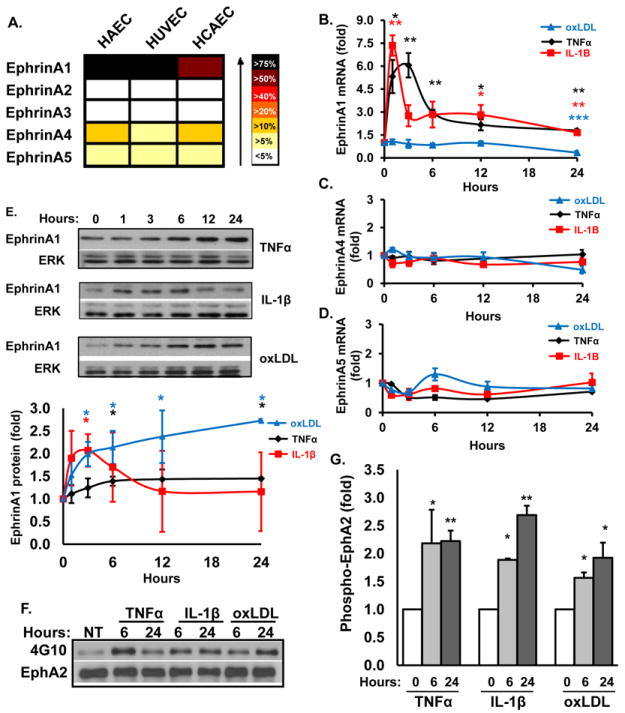
A, EphrinA ligand gene expression in endothelial cells analyzed as in Fig 1A. B, mRNA expression of ephrinA1 (B), ephrinA4 (C), and ephrinA5 (D) was determined as in Fig. 1B. (n = 3-7). E, EphrinA1 protein analysis of HAECs treated as in Fig. 1B. (n = 3-4). Representative immunoblots are shown. F/G, EphA2 phosphorylation (activation) in HAECs treated for 6 or 24 hrs with TNFα, IL-1β, or oxLDL was determined by EphA2 immunoprecipitation and immunoblotting with an anti-pTyr antibody (4G10). Results were normalized to total immunoprecipitated EphA2 and to untreated controls (NT); n = 4-5, * p< 0.05, ** p< 0.01.
Since these atherogenic mediators stimulate ephrinA1 ligand expression, we assessed subsequent EphA2 activation and tyrosine phosphorylation by immunoblotting EphA2 immunoprecipitates with a phosphotyrosine-specific antibody (4G10). Consistent with the time course for ligand expression, TNFα and oxLDL both induced a 1.5 to 2.0-fold increase in EphA2 phosphorylation by 6hrs that was sustained for at least 24 hrs post-treatment (Fig. 3F/G). Surprisingly, IL-1β also induced a sustained increase in EphA2 phosphorylation despite only transient inducing ephrinA1 expression. Addition of media alone over this same time course did not affect EphA2 receptor phosphorylation (Fig. SIV-A). While EphA2 transactivation has been described for other receptors24, EphA2 transactivation occurs early following receptor activation as opposed to the delayed response to proinflammatory stimuli. As such, none of these proinflammatory stimuli activated EphA2 early following treatment (5-15 minutes) (Fig. SIV-B) but instead induced a significant reduction in EphA2 phosphorylation, suggesting that trans-activation of EphA2 is not likely to be a dominant mechanism of EphA2 activation. Taken together, these data demonstrate that multiple atherogenic mediators induce expression of both the EphA2 receptor and its ephrinA1 ligand resulting in EphA2 activation.
Since EphrinA1 expression in atherosclerotic plaques in vivo may induce endothelial EphA2 activation, we measured EphA2 and ephrinA1 staining in healthy and atherosclerotic vessels harvested from ApoE null mice. In contrast to EphA2, expression of ephrinA1 was most prominent in the endothelial cell layer overlying the atherosclerotic plaque with minimal staining in the leukocyte infiltrates (Fig. 4A). To verify colocalization of EphA2 and ephrinA1 in the plaque, individual vessels were costained with antibodies for EphA2 and ephrinA1, and visualized by epifluorescence microscopy. Consistent with conventional immunohistochemistry, EphA2 staining was apparent in both the endothelium and neointima whereas ephrinA1 staining co-localized with EphA2 in the endothelial layer (Fig. 4B). EphrinA1 and EphA2 similarly showed enhanced mRNA expression in plaque-prone regions (aortic arch) compared to healthy regions (thoracic aorta) in ApoE null mice, verifying their concurrent upregulation during atherosclerotic plaque formation (Fig. 4C)
Figure 4.
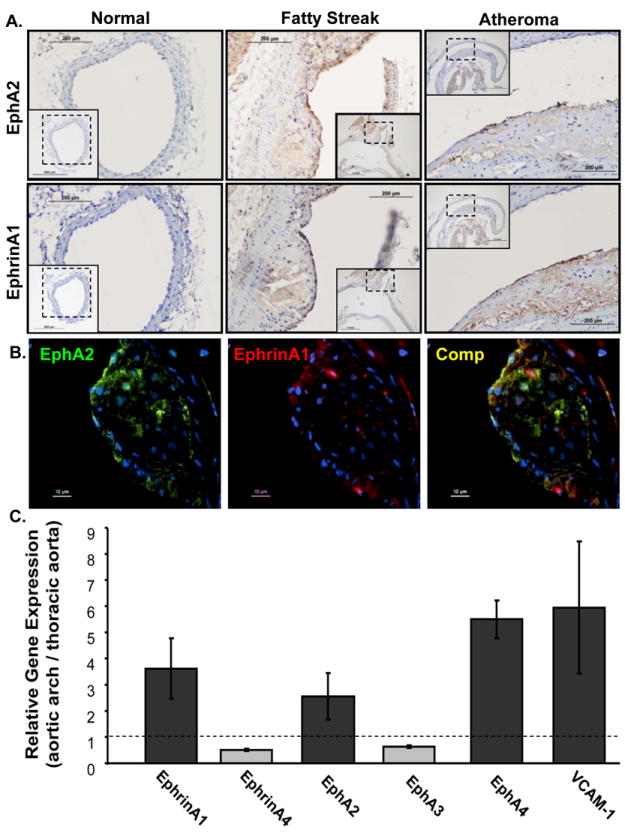
A, The common carotid (normal) and aortic arch (fatty streak, atheroma) of ApoE null mice fed a Western diet were stained for EphA2 (upper) or ephrinA1 (middle) in serial sections. B, Co-localization of EphA2 (green) and ephrinA1 (red) in the carotid sinus of Western diet-fed ApoE null mice was determined by epiflourescence microscopy. C, Gene expression in the aortic arch vs. the thoracic aorta by qRT-PCR analysis (normalized to PGK1 and PPIA; n = 3).
EphrinA1-induced human aortic endothelial cell gene expression
To determine how EphA2 signaling affects endothelial cell gene expression, HAECs were stimulated with recombinant ephrinA1 for 3 hours and changes in mRNA expression were analyzed using a commercial qRT-PCR array for genes involved in endothelial cell physiology (Endothelial Cell Biology array; Lonza StellARray®). Changes in gene expression were analyzed by Global Pattern Recognition software (Lonza GPR 2.0) identifying 8 genes significantly regulated in response to ephrinA1 (Table 1, p < 0.05 highlighted in yellow). As expected, ephrinA1 enhanced the expression of pro-angiogenic, pro-survival, and proliferation-associated genes (VEGFR225, 26, COX-227, TYMP28), but decreased expression of anti-angiogenic (COL18A129) and lymphangiogenic genes (VEGFR330). However, ephrinA1 also induced a marked increase in classic atherosclerosis-associated genes1, 31 involved in inflammatory cell targeting (VCAM-1, 26-fold; E-Selectin, 10-fold) and thrombosis (tissue factor, 9-fold). Interestingly, ephrinA1 also reduced the expression of known atheroprotective genes including collagen XVIII (2.6 fold decrease32) and the calcification inhibitor osteoprotegerin33 (4-fold decrease), although the latter only reached statistical significance within an 85% confidence interval. Taken together, these data suggest that EphA2 signaling promotes endothelial proinflammatory and prothrombotic gene expression.
Table 1.
EphrinA1-induced endothelial cell gene expression.
| Common Name | Symbol | P-value | Fold |
|---|---|---|---|
| Vascular cellular adhesion molecule-1 | VCAM-1 | 0.0015 | 26.17 |
| Tissue Factor | F3 | 0.0026 | 9.05 |
| E-Selectin | SELE | 0.0100 | 10.37 |
| Cyclooxygenase | PTGS2/COX-2 | 0.0107 | 13.77 |
| Thymidine Phosphorylase | TYMP | 0.0262 | 3.79 |
| Vascular endothelial growth factor receptor-2 | KDR/VEGFR-2 | 0.0273 | 3.60 |
| Vascular endothelial growth factor receptor-3 | FLT4/VEGFR-3 | 0.0329 | -2.68 |
| Collagen XVIII alpha-1 | COL18A1 | 0.0458 | -2.59 |
| Protein kinase B | AKT1/PKB | 0.0736 | -3.33 |
| Endothelial differentiation- related factor-1 | EDF-1 | 0.0845 | -2.84 |
| Survivin | BIRC5 | 0.1159 | -2.23 |
| Osteoprotegerin | TNFR11B | 0.1180 | -4.36 |
| Endothelial cell-specific molecule-1 | ESM1 | 0.1187 | 2.76 |
| v-raf-leukemia viral oncogene-1 | RAF1 | 0.1270 | -1.93 |
| B-cell CLL/lymphoma-2 | BCL2 | 0.1513 | 2.60 |
HAECs were treated with Fc-ephrinA1 for 3 hours. mRNA was isolated and analyzed by qRT-PCR in a 96-well Endothelial Cell Biology array (Lonza StellARray) and analyzed with Global Pattern Recognition 2.0 software. n = 5; Values highlighted in yellow are significant at p < 0.05.
To confirm Fc-ephrinA1-induced gene expression, we analyzed ephrinA1-induced VCAM-1 expression at the protein level in various endothelial cell types. As expected, Fc-ephrinA1 induced VCAM-1 expression in HAECs and HCAECs (Fig. 5A) whereas Fc treatment did not (Fig. SV-A). However, ephrinA1 failed to induce VCAM-1 expression in HUVECs (Fig. 5A), potentially explaining why ephrinA1’s proinflammatory effect has not been described to date. Fc-ephrinA1 similarly failed to induce VCAM-1, E-selectin, and tissue factor mRNA expression in HUVECs (Fig. SV-B), suggesting that HUVECs lack a critical signaling component linking EphA2 activation to proinflammatory gene expression. To verify that ephrinA1 possesses proinflammatory properties, we performed static monocytes adhesion assays on endothelial cells treated for 5 hours with Fc-ephrinA1. As expected, Fc-ephrinA1 treatment resulted in a ~50% increase in the binding of human monocytic THP-1 cells (Fig. SV-C).
Figure 5.
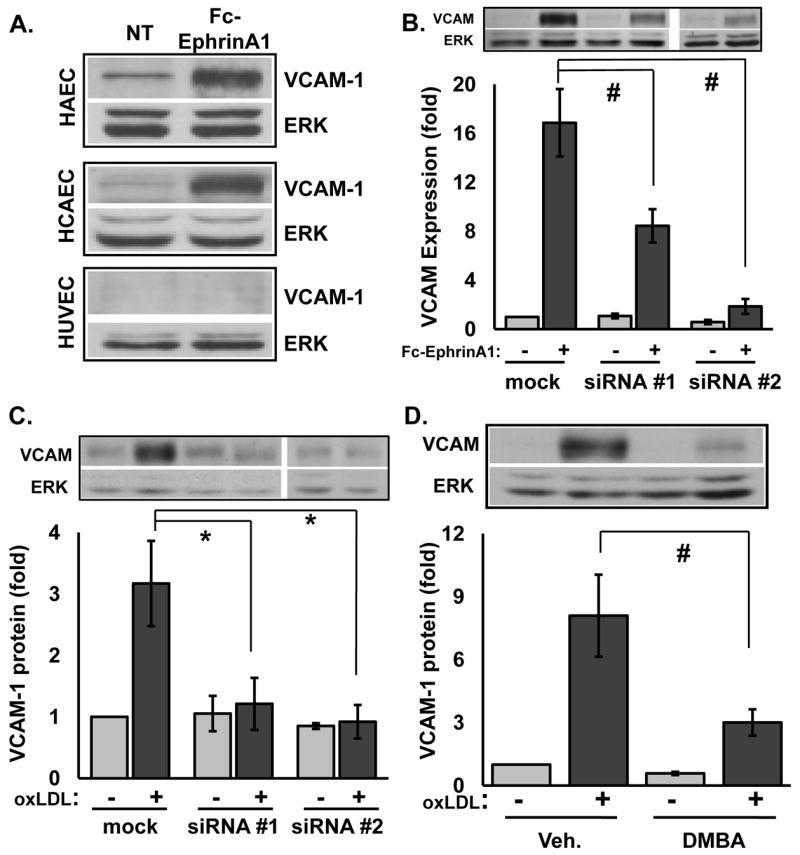
A, VCAM-1 expression in endothelial cells stimulated for 5 hours with 2 μg/ml Fc-ephrinA1 by Western blot analysis (n = 3 per cell type). B-C, HAE cells transiently transfected with two different anti-EphA2 siRNAs (>85% and >90% knockdown) were treated as in A or with 50-100 μg/ml oxLDL (n=3). D, HAECs pre-incubated for 15 minutes with the EphA2/EphA4 inhibitor DMBA (400 μmol/L) were treated with 50-100 μg/ml oxLDL. * p <0.05, # p<0.01.
Eph receptor and ephrin ligand interactions are notoriously promiscuous within the A- and B-subfamilies34. Since macrovascular endothelial cells also express EphA4 (Fig. 1), we next tested which EphA receptor (EphA2 or EphA4) mediates ephrinA1-induced VCAM-1 expression. Selectively reducing EphA2 expression by transient transfection with either of two separate EphA2 siRNA constructs significantly blunted ephrinA1-induced VCAM-1 expression (Fig. 5B) with the siRNA efficacy correlating with their degree of EphA2 knockdown (85% and >90% knockdown; Fig. SVI-A). While siRNA #1 resulted in a greater EphA2 knockdown (85%) than VCAM-1 inhibition (~50%), this effect may result from a low threshold of EphA2 receptors required to mediate the ephrinA1-induced proinflammatory response. Neither EphA2-targeted siRNA affected EphA4 expression (Fig. SVI-A), and EphA4-targeted siRNA oligos did not significantly affect VCAM-1 expression (Fig. SVI-B), suggesting that EphA2 is the primary EphA receptor mediating ephrinA1-induced VCAM-1 expression in HAECs.
TNFα, IL-1β, and oxLDL all stimulate EphA2 activation, suggesting EphA2 may facilitate VCAM-1 expression in response to these stimuli. OxLDL-induced VCAM-1 expression was reduced to near-baseline levels after EphA2 knockdown (Fig. 5C), while a chemical competitor for EphA2/EphA4 ligand binding (DMBA; 2,5-dimethylpyrrol benzoic acid) 35 similarly blocked oxLDL-induced VCAM expression (Fig. 5D). In contrast, EphA2 depletion did not affect cytokine-induced VCAM-1 expression (Fig. SVII). Consistent with an important role for EphA2 in oxLDL-induced VCAM-1 expression, EphA2 co-localized with ApoB (present on LDL) and VCAM-1 in the endothelial cell layer of human atherosclerotic plaques (Fig. SVIII). Taken together, these data suggest that enhanced EphA2 activation mediates oxLDL-induced but not cytokine-induced VCAM-1 expression.
Discussion
The current work suggests a novel role for endothelial Eph/ephrin signaling in atherosclerotic plaque formation. Atherosclerotic plaques from humans and hypercholesterolemic mice show robust EphA2 expression in the endothelium and in leukocytes, whereas EphA2 expression is undetectable in non-atherosclerotic vessels. Multiple atherogenic mediators induce endothelial EphA2 expression and enhance EphA2 activation concomitant with expression of its ligand ephrinA1. EphrinA1 co-localizes with EphA2 in the endothelial cell layer of mouse plaques suggesting that EphA2 expressed within the atherosclerotic plaques likely becomes activated. EphA2 activation with recombinant ephrinA1 stimulates proinflammatory and prothrombotic gene expression as well as enhanced monocyte adhesion, and oxLDL-induced but not cytokine-induced VCAM-1 expression requires EphA2 activation. Taken together, these data suggest that EphA2 activation perpetuates proinflammatory gene expression and atherosclerosis-associated endothelial cell activation.
Following the discovery that EphB4/ephrinB2 demarcated arterial and venous endothelium7, only a paucity of studies have characterized EphA/ephrinA signaling in the vasculature. Our current data suggest a novel role for EphA2 as a secondary, chronic effector of inflammatory stimuli as EphA2 shows both sustained expression and activation by atherogenic mediators, as well as the ability to promote inflammatory gene expression. Interestingly, EphA2 is required for oxLDL-induced but not TNFα- or IL-1β-induced VCAM-1 expression despite similar levels of EphA2 activation by all three treatments. While the cause of this differential requirement is not immediately obvious, these stimuli differ markedly in the mechanism, amplitude, and duration of VCAM-1 expression36, 37. The early, robust proinflammatory response to cytokines stimulates negative feedback pathways, such as IκB family member and A20 expression, that limits NF-κB activation and proinflammatory gene expression38. Therefore, the self-limiting nature of inflammation may minimize the proinflammatory role of EphA2 activation at the later stages following cytokine treatment but less so following oxLDL treatment. However, from these data we cannot exclude a role for cytokine-induced EphA2 activation in the regulation of endothelial cell phenotype or in the differential expression of proinflammatory genes other than VCAM-1.
While TNFα, IL-1β, and oxLDL induce both EphA2 transcription and protein expression, the expression of ephrinA ligands appears to involve more complex post-transcriptional regulatory mechanisms. IL-1β stimulates a sustained increase in ephrinA1 mRNA with levels remaining over 2-fold baseline by 24 hours; however, protein expression is transient and returns to baseline by 12 hours post-treatment. More strikingly, oxLDL stimulates the greatest induction of ephrinA1 protein expression but does not induce ephrinA1 mRNA transcription. Rather, oxLDL treatment reduces ephrinA1 mRNA after 24 hours. This disconnect between mRNA levels and protein expression could occur through multiple mechanisms, but perhaps the most intriguing potential mechanism involves regulation by endothelial microRNAs (miRNA). MiRNAs can repress protein translation without affecting mRNA levels39, suggesting that oxLDL could promote ephrinA1 protein but not mRNA expression by depleting a miRNA construct targeting ephrinA1. Work by Alisi et al. found that ephrinA1 protein levels, but not mRNA levels, were negatively correlated with the miR-200b-429 cluster40, and this miRNA cluster is down-regulated in endothelial cells during hypoxia consistent with ephrinA1’s role in angiogenesis41. However, more work will be required to determine whether miRNAs play a role in oxLDL-induced ephrinA1 expression.
Much of the previous work on endothelial cell EphA2 signaling utilized HUVECs, and the absence of ephrinA1-induced inflammation in HUVECs likely explains why our proposed proinflammatory role has gone unnoticed. However, it is unclear why HUVECs do not display ephrinA1-induced inflammatory gene expression, since EphA2 is the dominant EphA receptor in these cells and becomes equally activated by ephrinA1. Interestingly, a previous study by Amberger et al. demonstrated that oxLDL stimulates VCAM-1 expression in arterial endothelial cells but not in venous endothelial cells42, consistent with the role for EphA2 in mediating both ephrinA1- and oxLDL-induced proinflammatory responses in HAECs and HCAECs but not HUVECs. Since EphA2 receptor expression is similar (Fig. 1A), the lack of a proinflammatory effect in the HUVECs most likely results from the reduced expression of a critical downstream mediator involved in coupling EphA2 activation with proinflammatory gene expression. Future work will utilize this model system to delineate which signaling pathways are required for EphA2’s proinflammatory response.
Monocyte homing to the atherosclerotic plaque critically regulates plaque initiation and is thought to enhance plaque vulnerability through monocyte-dependent degradation of the fibrotic cap43. EphA2 and ephrinA1 may affect multiple steps during monocyte homing including endothelial proinflammatory gene expression, monocyte activation, and transendothelial migration. Our data shows that EphA2/ephrinA1 interactions stimulate the expression of cell adhesion molecules (E-selectin, VCAM-1) that promote monocyte binding2, and ephrinA1-activated endothelial cells exhibited enhanced monocyte adhesion. Additionally, EphA2 and ephrinA1 on the luminal endothelial cell surface might interact with counter-receptors on rolling monocytes to regulate monocyte attachment. A similar scenario was recently shown for the classic arterial ephrin, ephrinB2; luminal ephrinB2 on the endothelium interacts with EphB2 on monocytes enhancing monocyte adhesion9. T cell ephrinA ligation by endothelial EphA receptors stimulates T cell adhesion to endothelial cell surface ligands, whereas endothelial ephrinA ligation of T cell EphA receptors inhibits adhesion15. In contrast to T cells, activation of dendritic cell-expressed EphA2 with ephrinA3 stimulates their adhesion to fibronectin14, suggesting that the ability of ephrinA1 and EphA2 to serve as leukocyte counter-receptors may be cell type specific. Furthermore, co-expression of ephrinA1 and EphA2 on atherosclerotic endothelium suggests that the relative balance of ephrinA1 to EphA2 may be an important determinant affecting the availability of these proteins to serve as counter-receptors.
In addition to inflammation, ephrinA1 and EphA2 classically mediate vessel remodeling responses during angiogenesis44. In ischemic tissue, angiogenesis plays protective and regenerative roles in clearance and remodeling, and a recent study demonstrated that enhanced ephrinA1 expression on cardiomyocytes led to activation and recruitment of cardiac stem cells, resulting in increased vessel density and reduced tissue scarring45. However, angiogenesis perpetuates inflammation during chronic inflammatory responses by providing additional routes for leukocyte targeting46. In the growing atherosclerotic plaque core, hypoxic cells stimulate intraplaque angiogenesis, and angiogenic inhibitors are sufficient to reduce plaque progression32. Furthermore, the presence of intraplaque vessels correlates with poor clinical prognosis and enhanced likelihood of plaque instability and hemorrhage47. Therefore, limiting EphA2 activity or expression may prevent atherosclerotic plaque formation by reducing both inflammation and intraplaque angiogenesis.
Taken together, our data demonstrate a novel proinflammatory role for EphA2 in macrovascular endothelial cells and implicate EphA2 as a potential proinflammatory mediator of atherosclerotic plaque formation.
Supplementary Material
Acknowledgments
Special thanks go to Sushil Jain for the donation of THP-1 monocytes and to Christopher Kevil for advice on monocyte cell culture and adhesion assays.
Sources of Funding
This work was funded by an LSU Health Sciences Center-Shreveport/Biomedical Research Foundation Grant-in-Aid to A.W.O., and by a VA Merit Award, and NIH RO1 CA95004 and CA114301 to J.C.
Footnotes
Disclosures
None
Works Cited
- 1.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 2.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 3.Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 4.Farb A, Burke AP, Tang AL, Liang TY, Mannan P, Smialek J, Virmani R. Coronary plaque erosion without rupture into a lipid core. A frequent cause of coronary thrombosis in sudden coronary death. Circulation. 1996;93:1354–1363. doi: 10.1161/01.cir.93.7.1354. [DOI] [PubMed] [Google Scholar]
- 5.Pasquale EB. Eph-ephrin bidirectional signaling in physiology and disease. Cell. 2008;133:38–52. doi: 10.1016/j.cell.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 6.Lackmann M, Boyd AW. Eph, a protein family coming of age: more confusion, insight, or complexity? Sci Signal. 2008;1:re2. doi: 10.1126/stke.115re2. [DOI] [PubMed] [Google Scholar]
- 7.Wang HU, Chen ZF, Anderson DJ. Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell. 1998;93:741–753. doi: 10.1016/s0092-8674(00)81436-1. [DOI] [PubMed] [Google Scholar]
- 8.Brantley-Sieders DM, Chen J. Eph receptor tyrosine kinases in angiogenesis: from development to disease. Angiogenesis. 2004;7:17–28. doi: 10.1023/B:AGEN.0000037340.33788.87. [DOI] [PubMed] [Google Scholar]
- 9.Braun J, Hoffmann SC, Feldner A, Ludwig T, Henning R, Hecker M, Korff T. Endothelial cell ephrinB2-dependent activation of monocytes in arteriosclerosis. Arteriosclerosis, thrombosis, and vascular biology. 2011;31:297–305. doi: 10.1161/ATVBAHA.110.217646. [DOI] [PubMed] [Google Scholar]
- 10.Sakamoto A, Ishibashi-Ueda H, Sugamoto Y, Higashikata T, Miyamoto S, Kawashiri MA, Yagi K, Konno T, Hayashi K, Fujino N, Ino H, Takeda Y, Yamagishi M. Expression and function of ephrin-B1 and its cognate receptor EphB2 in human atherosclerosis: from an aspect of chemotaxis. Clin Sci (Lond) 2008;114:643–650. doi: 10.1042/CS20070339. [DOI] [PubMed] [Google Scholar]
- 11.Ivanov AI, Romanovsky AA. Putative dual role of ephrin-Eph receptor interactions in inflammation. IUBMB Life. 2006;58:389–394. doi: 10.1080/15216540600756004. [DOI] [PubMed] [Google Scholar]
- 12.Khounlotham M, Subbian S, Smith R, 3rd, Cirillo SL, Cirillo JD. Mycobacterium tuberculosis interferes with the response to infection by inducing the host EphA2 receptor. J Infect Dis. 2009;199:1797–1806. doi: 10.1086/599096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pandey A, Shao H, Marks RM, Polverini PJ, Dixit VM. Role of B61, the ligand for the Eck receptor tyrosine kinase, in TNF-alpha-induced angiogenesis. Science. 1995;268:567–569. doi: 10.1126/science.7536959. [DOI] [PubMed] [Google Scholar]
- 14.de Saint-Vis B, Bouchet C, Gautier G, Valladeau J, Caux C, Garrone P. Human dendritic cells express neuronal Eph receptor tyrosine kinases: role of EphA2 in regulating adhesion to fibronectin. Blood. 2003;102:4431–4440. doi: 10.1182/blood-2003-02-0500. [DOI] [PubMed] [Google Scholar]
- 15.Sharfe N, Nikolic M, Cimpeon L, Van De Kratts A, Freywald A, Roifman CM. EphA and ephrin-A proteins regulate integrin-mediated T lymphocyte interactions. Mol Immunol. 2008;45:1208–1220. doi: 10.1016/j.molimm.2007.09.019. [DOI] [PubMed] [Google Scholar]
- 16.Wang Q, Rao S, Shen GQ, Li L, Moliterno DJ, Newby LK, Rogers WJ, Cannata R, Zirzow E, Elston RC, Topol EJ. Premature myocardial infarction novel susceptibility locus on chromosome 1P34-36 identified by genomewide linkage analysis. Am J Hum Genet. 2004;74:262–271. doi: 10.1086/381560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Welch CL, Bretschger S, Latib N, Bezouevski M, Guo Y, Pleskac N, Liang CP, Barlow C, Dansky H, Breslow JL, Tall AR. Localization of atherosclerosis susceptibility loci to chromosomes 4 and 6 using the Ldlr knockout mouse model. Proc Natl Acad Sci U S A. 2001;98:7946–7951. doi: 10.1073/pnas.141239098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Noble RP. Electrophoretic separation of plasma lipoproteins in agarose gel. J Lipid Res. 1968;9:693–700. [PubMed] [Google Scholar]
- 19.Yoshida H, Quehenberger O, Kondratenko N, Green S, Steinberg D. Minimally oxidized low-density lipoprotein increases expression of scavenger receptor A, CD36, and macrosialin in resident mouse peritoneal macrophages. Arterioscler Thromb Vasc Biol. 1998;18:794–802. doi: 10.1161/01.atv.18.5.794. [DOI] [PubMed] [Google Scholar]
- 20.Okazaki T, Ni A, Baluk P, Ayeni OA, Kearley J, Coyle AJ, Humbles A, McDonald DM. Capillary defects and exaggerated inflammatory response in the airways of EphA2-deficient mice. Am J Pathol. 2009;174:2388–2399. doi: 10.2353/ajpath.2009.080949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Larson J, Schomberg S, Schroeder W, Carpenter TC. Endothelial EphA receptor stimulation increases lung vascular permeability. Am J Physiol Lung Cell Mol Physiol. 2008;295:L431–439. doi: 10.1152/ajplung.90256.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fang WB, Ireton RC, Zhuang G, Takahashi T, Reynolds A, Chen J. Overexpression of EPHA2 receptor destabilizes adherens junctions via a RhoA-dependent mechanism. J Cell Sci. 2008;121:358–368. doi: 10.1242/jcs.017145. [DOI] [PubMed] [Google Scholar]
- 23.Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W, Jr, Rosenfeld ME, Schwartz CJ, Wagner WD, Wissler RW. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Arteriosclerosis, thrombosis, and vascular biology. 1995;15:1512–1531. doi: 10.1161/01.atv.15.9.1512. [DOI] [PubMed] [Google Scholar]
- 24.Chan B, Sukhatme VP. Receptor tyrosine kinase EphA2 mediates thrombin-induced upregulation of ICAM-1 in endothelial cells in vitro. Thromb Res. 2009;123:745–752. doi: 10.1016/j.thromres.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246:1306–1309. doi: 10.1126/science.2479986. [DOI] [PubMed] [Google Scholar]
- 26.Terman BI, Carrion ME, Kovacs E, Rasmussen BA, Eddy RL, Shows TB. Identification of a new endothelial cell growth factor receptor tyrosine kinase. Oncogene. 1991;6:1677–1683. [PubMed] [Google Scholar]
- 27.Greenhough A, Wallam CA, Hicks DJ, Moorghen M, Williams AC, Paraskeva C. The proapoptotic BH3-only protein Bim is downregulated in a subset of colorectal cancers and is repressed by antiapoptotic COX-2/PGE(2) signalling in colorectal adenoma cells. Oncogene. 29:3398–3410. doi: 10.1038/onc.2010.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jeung HC, Che XF, Haraguchi M, Zhao HY, Furukawa T, Gotanda T, Zheng CL, Tsuneyoshi K, Sumizawa T, Roh JK, Akiyama S. Protection against DNA damage-induced apoptosis by the angiogenic factor thymidine phosphorylase. FEBS Lett. 2006;580:1294–1302. doi: 10.1016/j.febslet.2006.01.047. [DOI] [PubMed] [Google Scholar]
- 29.O’Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, Flynn E, Birkhead JR, Olsen BR, Folkman J. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88:277–285. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- 30.Alitalo K, Tammela T, Petrova TV. Lymphangiogenesis in development and human disease. Nature. 2005;438:946–953. doi: 10.1038/nature04480. [DOI] [PubMed] [Google Scholar]
- 31.Collins T, Cybulsky MI. NF-kappaB: pivotal mediator or innocent bystander in atherogenesis? J Clin Invest. 2001;107:255–264. doi: 10.1172/JCI10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moulton KS, Heller E, Konerding MA, Flynn E, Palinski W, Folkman J. Angiogenesis inhibitors endostatin or TNP-470 reduce intimal neovascularization and plaque growth in apolipoprotein E-deficient mice. Circulation. 1999;99:1726–1732. doi: 10.1161/01.cir.99.13.1726. [DOI] [PubMed] [Google Scholar]
- 33.Emery JG, McDonnell P, Burke MB, Deen KC, Lyn S, Silverman C, Dul E, Appelbaum ER, Eichman C, DiPrinzio R, Dodds RA, James IE, Rosenberg M, Lee JC, Young PR. Osteoprotegerin is a receptor for the cytotoxic ligand TRAIL. J Biol Chem. 1998;273:14363–14367. doi: 10.1074/jbc.273.23.14363. [DOI] [PubMed] [Google Scholar]
- 34.Pasquale EB. Eph receptor signalling casts a wide net on cell behaviour. Nat Rev Mol Cell Biol. 2005;6:462–475. doi: 10.1038/nrm1662. [DOI] [PubMed] [Google Scholar]
- 35.Noberini R, Koolpe M, Peddibhotla S, Dahl R, Su Y, Cosford ND, Roth GP, Pasquale EB. Small molecules can selectively inhibit ephrin binding to the EphA4 and EphA2 receptors. J Biol Chem. 2008;283:29461–29472. doi: 10.1074/jbc.M804103200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mitra S, Goyal T, Mehta JL. Oxidized LDL, LOX-1 and Atherosclerosis. Cardiovasc Drugs Ther. 2011;25:419–429. doi: 10.1007/s10557-011-6341-5. [DOI] [PubMed] [Google Scholar]
- 37.Tedgui A, Mallat Z. Cytokines in atherosclerosis: pathogenic and regulatory pathways. Physiol Rev. 2006;86:515–581. doi: 10.1152/physrev.00024.2005. [DOI] [PubMed] [Google Scholar]
- 38.Werner SL, Kearns JD, Zadorozhnaya V, Lynch C, O’Dea E, Boldin MP, Ma A, Baltimore D, Hoffmann A. Encoding NF-kappaB temporal control in response to TNF: distinct roles for the negative regulators IkappaBalpha and A20. Genes Dev. 2008;22:2093–2101. doi: 10.1101/gad.1680708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chekulaeva M, Filipowicz W. Mechanisms of miRNA-mediated post-transcriptional regulation in animal cells. Curr Opin Cell Biol. 2009;21:452–460. doi: 10.1016/j.ceb.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 40.Alisi A, Da Sacco L, Bruscalupi G, Piemonte F, Panera N, De Vito R, Leoni S, Bottazzo GF, Masotti A, Nobili V. Mirnome analysis reveals novel molecular determinants in the pathogenesis of diet-induced nonalcoholic fatty liver disease. Lab Invest. 2011;91:283–293. doi: 10.1038/labinvest.2010.166. [DOI] [PubMed] [Google Scholar]
- 41.Chan YC, Khanna S, Roy S, Sen CK. miR-200b targets Ets-1 and is down-regulated by hypoxia to induce angiogenic response of endothelial cells. J Biol Chem. 2011;286:2047–2056. doi: 10.1074/jbc.M110.158790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Amberger A, Maczek C, Jurgens G, Michaelis D, Schett G, Trieb K, Eberl T, Jindal S, Xu Q, Wick G. Co-expression of ICAM-1, VCAM-1, ELAM-1 and Hsp60 in human arterial and venous endothelial cells in response to cytokines and oxidized low-density lipoproteins. Cell Stress Chaperones. 1997;2:94–103. doi: 10.1379/1466-1268(1997)002<0094:ceoive>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Falk E, Shah PK, Fuster V. Coronary plaque disruption. Circulation. 1995;92:657–671. doi: 10.1161/01.cir.92.3.657. [DOI] [PubMed] [Google Scholar]
- 44.Cheng N, Brantley DM, Liu H, Lin Q, Enriquez M, Gale N, Yancopoulos G, Cerretti DP, Daniel TO, Chen J. Blockade of EphA receptor tyrosine kinase activation inhibits vascular endothelial cell growth factor-induced angiogenesis. Mol Cancer Res. 2002;1:2–11. [PubMed] [Google Scholar]
- 45.Goichberg P, Bai Y, D’Amario D, Ferreira-Martins J, Fiorini C, Zheng H, Signore S, del Monte F, Ottolenghi S, D’Alessandro DA, Michler RE, Hosoda T, Anversa P, Kajstura J, Rota M, Leri A. The ephrin A1-EphA2 system promotes cardiac stem cell migration after infarction. Circ Res. 2011;108:1071–1083. doi: 10.1161/CIRCRESAHA.110.239459. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 46.Moulton KS, Vakili K, Zurakowski D, Soliman M, Butterfield C, Sylvin E, Lo KM, Gillies S, Javaherian K, Folkman J. Inhibition of plaque neovascularization reduces macrophage accumulation and progression of advanced atherosclerosis. Proc Natl Acad Sci U S A. 2003;100:4736–4741. doi: 10.1073/pnas.0730843100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Virmani R, Kolodgie FD, Burke AP, Finn AV, Gold HK, Tulenko TN, Wrenn SP, Narula J. Atherosclerotic plaque progression and vulnerability to rupture: angiogenesis as a source of intraplaque hemorrhage. Arterioscler Thromb Vasc Biol. 2005;25:2054–2061. doi: 10.1161/01.ATV.0000178991.71605.18. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.