

Summary
Repair of adult skeletal muscle depends on satellite cells, myogenic stem cells located between the basal lamina and the plasmalemma of the myofiber. Standardized protocols for the isolation and culture of satellite cells are key tools for understanding cell autonomous and extrinsic factors that regulate their performance. Knowledge gained from such studies can contribute important insights to developing strategies for the improvement of muscle repair following trauma and in muscle wasting disorders. This chapter provides an introduction to satellite cell biology and further describes the basic protocol used in our laboratory to isolate and culture satellite cells from adult skeletal muscle. The cell culture conditions detailed herein support proliferation and differentiation of satellite cell progeny and the development of reserve cells, which are thought to reflect the in vivo self-renewal ability of satellite cells. Additionally, this chapter describes our standard immunostaining protocol that allows the characterization of satellite cell progeny by the temporal expression of characteristic transcription factors and structural proteins associated with different stages of myogenic progression. While emphasis is given here to the isolation and characterization of satellite cells from mouse hindlimb muscles, the protocols are suitable for other muscle types (such as diaphragm and extraocular muscles) and for muscles from other species, including chicken and rat. Altogether, the basic protocols described are straightforward and facilitate the study of diverse aspects of skeletal muscle stem cells.
Keywords: Skeletal muscle, satellite cell, stem cell, myogenesis, Pronase, gelatin, Matrigel, Pax7, MyoD, myogenin
1. Introduction
This chapter aims to provide simple protocols for the isolation, culture and analysis of satellite cells from adult skeletal muscle. We first detail background information about satellite cells (Subheadings 1.1. through 1.3.) and the range of cell isolation approaches developed over the years by us and others to analyze satellite cells (Subheading 1.4.). We then introduce an overview of our basic satellite cell isolation and culture protocol (Subheading 2.) followed by practical details (starting with Subheading 3.). We provide what we consider the simplest protocol that can be performed in any basic tissue culture laboratory, and in Subheading 1.4. we briefly discuss alternative approaches to purifying satellite cells. The basic approach provided in this chapter is an excellent means for analysis of satellite cells in culture when extreme purity is not needed. With careful attention to minimize connective tissue contribution, our standard protocol can yield cultures that are 80– 95% pure based on staining for protein markers Pax7 and MyoD on culture day 4 (for additional details about these markers see Subheading 1.3.). Collectively, our simple protocol for satellite cell isolation and culture has allowed detailed analyses of tissue-dissociated satellite cells. Standardized protocols for the isolation and culture of satellite cells are essential tools to enhance our understanding of cell autonomous and extrinsic factors that regulate their performance.
1.1. The Satellite Cell Is Defined by Its Niche
The functional units responsible for skeletal muscle contraction are cylindrical, multinucleated muscle fibers (myofibers). These contractile structures are established during embryogenesis, when mononuclear cells known as myoblasts fuse into immature muscle fibers or myotubes. Myonuclei (the myofiber nuclei) are postmitotic and under normal conditions cannot re-enter a proliferative state to contribute additional nuclei. During postnatal life, myofiber growth, homeostasis and repair rely on a population of mononuclear myogenic cells known as satellite cells (1–3). Satellite cells were initially described fifty years ago by their anatomical location on the surface of muscle fibers, between the myofiber plasmalemma and the basal lamina (4, 5) (for a schematic and electron microscope image see Fig. 1). However, the ultimate experimental proof that satellite cells are indeed myogenic progenitors has only been obtained by showing that cells derived from isolated myofibers produce myogenic progeny, able to proliferate, differentiate and self-renew in vitro and in vivo (6–13).
Figure 1.

A schematic (A) and EM micrograph (B) of satellite cell location. The myofiber basement and plasma membranes have been routinely detected by immunostaining with antibodies against laminin and dystrophin, respectively. In panel A, myofiber nuclei depicted at the myofiber periphery represent the state of healthy adult myofibers; immature myofibers present in regenerating muscles display centralized myofiber nuclei (not shown). In panel B, black arrows depict the basal lamina, white arrows depict apposing satellite cell and myofiber membranes; note the sarcomeric organization within the myofiber. A color version of this figure appeared in ref. 3. Panel B was first published in ref. 15.
Satellite cells were initially described using electron microscopy (4, 5, 14, 15). More recent methods facilitate monitoring these cells by light microscopy based on expression of a range of specific markers that can be detected by immunostaining (16, 17). In particular, specific expression of the paired box transcription factor Pax7 and availability of an excellent antibody for immunodetection of this protein provides a uniform means to identify satellite cells in their native position in a range of species including mouse (10, 12, 13, 18, 19), rat (20), chicken (21, 22), and human (23, 24).
Additionally, genetically manipulated reporter mice permit direct detection of satellite cells based on specific expression of a fluorophore or β-galactosidase (β-gal) (13, 17, 19, 25, 26). We demonstrated that transgenic expression of GFP under the control of nestin regulatory elements (NES-GFP) allows detection of satellite cells in freshly isolated myofibers. NES-GFP mice also facilitate isolation of satellite cells using fluorescent-activated cell sorting (FACS) and subsequent studies of purified populations (13, 19). The Myf5nLacZ/+ mouse has also provided a means to identify satellite cells in intact muscle and isolated myofibers (2, 11, 19, 26, 27). In this mouse, one of the Myf5 alleles was modified to direct lacZ expression, resulting in β-gal expression in satellite cells as originally reported by Beauchamp and colleagues (26). We frequently use crosses of NES-GFP with Myf5nLacZ/+ mice, allowing the detection of satellite cells by means of direct fluorescence and X-gal staining (19).
Satellite cells are considered the major, if not only, source of myogenic progeny in adult muscle (2, 3). Other cell types isolated from skeletal muscle, such as mesoangioblasts, pericytes, and myoendothelial cells also seem to have some myogenic potency (28–30), but whether these cell types participate in normal muscle maintenance and repair remains unclear. The isolation of the latter cell types require special enrichment approaches and these cells do not appear to contribute to our myogenic preparations. The majority of cells in our standard preparations of freshly isolated myogenic progenitors display the satellite cell phenotype; i.e., preparations from Myf5nLacZ/+/NES-GFP mice are enriched with Pax7+/β-gal+/GFP+ cells (shown by cytospin and mRNA expression analyses of freshly isolated cells). Hence, we refer to our freshly isolated cells prepared by the basic approach detailed herein as preparations of satellite cells or myogenic progenitors. Once satellite cells are cultured and proliferate, the resulting cells are referred to as myogenic progeny.
1.2. Functional Satellite Cells Are Required Throughout Life
In the juvenile growth phase, when muscles enlarge, satellite cells are proliferative and add nuclei to growing myofibers (21, 31–34). In most adult muscles, satellite cells are typically quiescent until their activation is invoked by muscle injury (1, 35–37). Subtle injuries may lead to minimal proliferation of activated satellite cells whereas major trauma can recruit greater numbers of satellite cells and promote prolonged proliferation prior to differentiation. As small myofiber injuries can occur routinely during daily activity, a mechanism for repair is essential for muscle maintenance throughout life.
Activation of myogenic precursors is controlled by proximal signals from the muscle niche, microvasculature and from inflammatory cells (38–41). Systemic factors may also regulate satellite cell activation (42–44). Following their activation, satellite cells may contribute to repair of damaged myofibers and also generate new myofibers following cell division and fusion of myoblast progeny. Satellite cell behavior is under stringent regulatory control in order to balance various actively maintained states, including quiescence, entry into proliferation and continuity of the cell cycle, and terminal differentiation (45, 46). Furthermore, apart from their ability to fortify myofibers and contribute to muscle regeneration, satellite cells have the capacity to replenish a reserve pool and self-renew, qualifying them as tissue-specific stem cells (11, 47). It is not known, however, to what extent individual satellite cells differ with regard to their amplification and renewal potential (19, 47).
During early growth, muscle satellite cells may represent about 30% of the nuclei, whereas in the healthy adult satellite cells represent approximately 2–7% of nuclei within skeletal muscle (1, 21). The number of satellite cells per myofiber or per cross-sectional area may vary immensely between muscles. For example, the fast twitch extensor digitorum longus (EDL) contains fewer satellite cells compared to the slow twitch soleus (1, 12, 48). Additionally, myofiber ends may have a higher concentration of satellite cells than the rest of the myofiber (22). There are also reports of an age-associated decline in satellite cell number, where the presence and extent of decline may vary by muscle (12, 19, 20, 49)). Satellite cell performance may also decline in the aging environment, a possible contributory factor to age-associated muscle deterioration, also known as sarcopenia (20, 50). However, additional studies suggest that initial performance of skeletal muscle progenitors is delayed, but not necessarily impaired in old age and that factors beyond satellite cell activity alone may play a role in reducing muscle repair in old age (44, 51). Indeed, satellite cell activity can be rejuvenated upon exposure of old muscle to a juvenile environment by cross-transplantation or by parabiosis of young and old mice (42, 52). Muscle wasting associated with muscular dystrophy is also thought to lead to exhaustion of satellite cells due to the continuous demand for reparative myogenic cells (53–55). Overall, satellite cells are vital to skeletal muscle homeostasis and regeneration throughout life, and understanding the regulation of myogenic stem cells will likely provide valuable insights into muscle wasting in aging and disease.
1.3. Detection of Satellite Cell Progeny by Temporal Expression Patterns of Myogenic-Related Transcription Factors
At the molecular level, myogenesis of satellite cells is highly orchestrated to ensure that specific genes are regulated in a temporally organized manner according to genetic blueprints, cell cycle requirements, and environmental factors. The resulting pattern of gene expression yields terminally differentiated myoblasts, capable of adding myonuclei to existing myofibers in addition to fusing together to form new myofibers during muscle growth and repair (3, 45, 56, 57). To monitor various stages of satellite cell myogenesis in culture, we focus primarily on the expression patterns of Pax7 and the myogenic regulatory factors MyoD, myogenin and Myf5. As demonstrated in our published studies, the temporal expression patterns of these genes do not vary for mouse, rat, or chicken satellite cell progeny. For additional background information about the functional roles of Pax7 and the myogenic regulatory factors in myogenesis the reader should refer to additional publications (e.g., (58–60); for a comprehensive review see (3)).
Satellite cell progeny can be distinguished from their quiescent progenitors based on distinctive gene expression patterns (2, 3, 57). In particular, expressions of MyoD and myogenin have been used extensively in conjunction with Pax7 (8, 10, 12, 46) (Fig. 2). Proliferating progeny (myoblasts) continue to express Pax7, but distinctly from their quiescent progenitors, also express MyoD. A decline in Pax7 along with the induction of the muscle-specific transcription factor myogenin marks myoblasts that have entered the differentiation phase and initiated cell cycle withdrawal. Coinciding with or soon after the upregulation of myogenin, differentiating myoblasts initiate expression of various genes encoding structural proteins, such as sarcomeric myosin, and fuse into myotubes (12, 21, 39, 61). During myoblast differentiation, a subpopulation of mononucleated cells downregulate MyoD expression and exit the cell cycle, but maintain Pax7 expression. These cells define a reserve population that presumably reflects satellite cell self-renewal (10–12, 19, 46, 47, 57).
Figure 2.
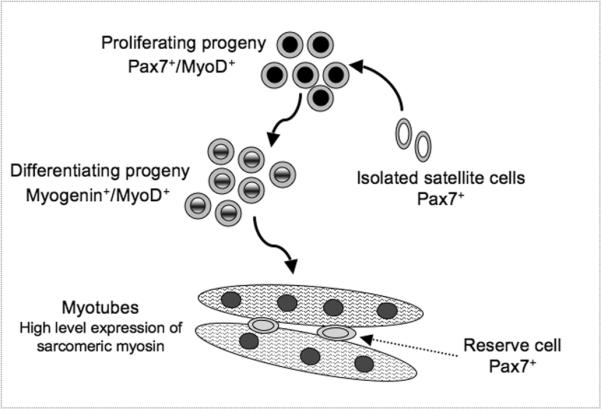
The molecular signature of satellite cell progeny in a primary cell culture: proliferation, differentiation and self-renewal. A color version appeared in ref. 3.
Both quiescent and proliferating satellite cells also express the myogenic regulatory factor Myf5 as determined by mRNA analysis (13, 19, 62). Myf5 promoter activity can also be observed through β-gal detection in satellite cells and their proliferating progeny in myogenic cultures from the aforementioned Myf5nlacZ/+ mice (19, 26). However, detection of the Myf5 protein has not been reported in quiescent satellite cells, though proliferating progeny do express Myf5 protein (46, 63). Thus, it is possible that while the Myf5 promoter is active in quiescent satellite cells, Myf5 protein is not produced until cells begin to proliferate. Ultimately, Myf5 expression declines when myoblasts enter differentiation, while MyoD expression persists well into the differentiation stage when satellite cells are maintained in our standard culture conditions (3, 12, 46).
1.4. Classic and Contemporary Approaches for Satellite Cell Isolation
Much of our understanding of satellite cell biology has arisen from cell culture studies. The information provided in this section focuses on primary cultures of bona fide satellite cells. Studies with myogenic cell lines (including rat L6 and L8, and mouse C2, C2C12 and MM14) have also permitted extensive biochemical and molecular analyses of aspects of myogenesis, though these models do not always fully adhere to the biology of satellite cells (64–68). A comprehensive description of myogenic cells lines from the American Tissue Culture Collection (ATCC) and other sources can be found in our recent review (3).
Two main cell culture approaches have been employed by us and other investigators in the study of bona fide satellite cells:
-
(i)
Cultures of isolated myofibers where the satellite cells remain in their native position underneath the myofiber basal lamina (8, 12, 69). This approach allows the study of satellite cells and their progeny in their in situ position and after they migrate out from the parent myofiber. We have described protocols for single myofiber isolation and culture as a means to study satellite cells at great details in other book chapters in this Methods in Molecular Biology series (70, 71).
-
(ii)
Primary myogenic cultures prepared from mononucleated cells dissociated from whole muscle. Protocols for obtaining primary myogenic cultures involve releasing satellite cells from their niche. Steps of mincing, enzymatic digestion and repetitive triturations of the muscle are required for breaking both the connective tissue network and the myofibers in order to release the satellite cells from the muscle bulk. Depending on the enzymatic procedure and the purpose for cell isolation, enrichment for satellite cells beyond the basic isolation protocol is often unnecessary. Indeed, the basic isolation protocol that is detailed next in this chapter, has been used by us in many cell culture studies of satellite cells (12, 21, 46, 72).
Alternatively, satellite cells can be enriched from whole muscle cell suspensions by various approaches that reduce the presence of fibroblastic cells, typically present to some degree in the preparation, and remove myofibril debris present in the initial cell suspension. Such approaches have included: (a) initial plating on uncoated tissue cultures dishes that results in separation of cells based on adhesion characteristics, where cells that remain in suspension after a short period are collected for culturing (i.e., differential plating) (73–75); (b) fractionation on Percoll density gradients (62, 76–78); c) cell sorting by forward and side scatter (79, 80).
In studies where further enrichment of satellite cells is warranted, cells can be isolated by FACS using antibodies that react with satellite cell surface antigens (47). First, cells are released from the muscle tissue using collagenase or collagenase-dispase, enzyme preparations that preserve cell surface antigens compared to Pronase or trypsin digestion methods. Studies from various laboratories (performed mainly with mouse tissue) have established that satellite cells can be isolated based on negative selection for CD45, CD31 and Sca1, and positive selection for CD34 and α7 integrin (25, 47, 81). Additional cell surface antigens, including CXCR4, β1 integrin, and syndecan-4 have also been used for isolation from adult muscle (82–84).
A range of fluorescence-based reporter systems in genetically manipulated mouse strains have also permitted reliable isolation of purified populations of satellite cells. For example, we have isolated satellite cells from different muscle groups of transgenic NES-GFP mice (13, 19), and Pax3- / Pax7-driven GFP reporter expression has also been used for isolation by FACS (25, 85), with the limitation that the Pax3 reporter is only expressed in satellite cells from selective muscles (25). Mice with a GFP reporter gene inserted into the Myf5 locus also permit isolation of myogenic cells by FACS (86–88); however, GFP expression is below detection level in many of the satellite cells, which reduces the usefulness of these Myf5GFP mice for satellite cell isolation by FACS.
Additionally, Cre-Lox mouse models are useful for isolating satellite cells and identifying their progeny. Fluorescent reporters can be permanently turned on in cells derived from myogenic progenitors upon expression of Cre-recombinase driven by promoters of myogenic genes such as Pax3, Myf5 and MyoD (89–92). When using such Cre-Lox mouse models to sort satellite cells, one should be careful to ensure that the reporter is not expressed in additional cell types during embryogenesis. For example, Myf5-Cre expression has been reported in nonmyogenic regions (93–95). It is also important to note that some head muscles (e.g., extraocular muscles) develop via Pax3-independent pathways and satellite cells in these muscles do not express the Pax3-Cre-driven reporter ((17, 96) and our unpublished studies).
2. About Our Basic Protocols for Satellite Cell Isolation and Analysis
In this chapter we describe the basic methodologies regularly used in our laboratory for the isolation, culture and characterization of myogenic progenitors from adult mouse skeletal muscle. As detailed in the previous section, we also use contemporary approaches for satellite cell isolation that are based on fluorescence reporter expression and/or based on expression of cell surface antigens. However, such approaches require the availability of special resources and reagents. Here, we describe a basic and straightforward method that we frequently use to isolate and characterize satellite cell performance in culture. This procedure can be performed in any tissue culture facility, using wildtype and mutant mouse muscles of various ages (12), and is suitable for satellite cell isolation from rat (62) and chicken (46) muscles. Fig. 3 shows representative micrographs of myogenic cultures emanating from satellite cells isolated using our basic procedure from adult mouse hindlimb muscles.
Figure 3.
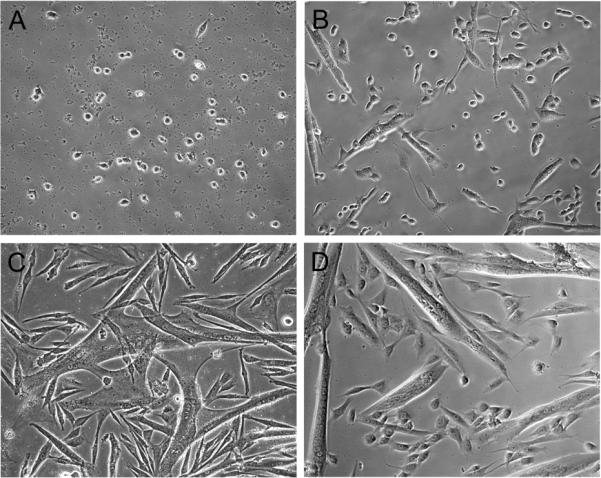
Phase micrographs depicting the morphology of mouse myogenic cultures seeded on gelatin-coated (A-C) and Matrigel-coated (D) dishes. Cells were isolated by Pronase digestion and cultures were maintained in rich growth medium according to protocols detailed in this chapter. Panels A, B, C, and D show the cultures on days 3, 5, 7, and 7, respectively. Round cells observed during early culture days (A and B) are proliferating myoblasts. Multinucleated myotubes can already be observed on day 5 (B) and enlarge on subsequent days (C and D). Residual debris resulting from tissue dissociation, which is present in early culture days and can be mistakenly considered a contamination (see step 30 in Subheading 4.1.), is noticeable at same focal level as the proliferating cells (A). The identity of myoblasts and myotubes can be further confirmed by their characteristic protein expression (see Fig. 2) using immunostaining with antibodies detailed in Table 1. Images were taken with a 20x objective.
Our standard protocol for immuncytochemical analysis of satellite cell cultures provides quantitative insight into the “myogenicity” of the cell preparation (i.e., the presence and frequency of myogenic cells) and progression of satellite cell progeny from proliferation to differentiation and production of reserve cells. Table 1 summarizes the source and characteristics of a set of monoclonal antibodies used in our laboratory for the analysis of myogenesis in primary cultures of mouse satellite cells, which are also applicable to rat satellite cells (8, 12, 13, 19, 20, 62). For analysis of chicken satellite cells, we rely on the same Pax7 and MF20 antibodies as in Table 1, but for the detection of myogenic regulatory factors we use rabbit polyclonal antibodies developed against the chicken proteins (21, 46, 97).
Table 1.
Mouse monoclonal antibodies frequently used in our studies for analyzing progeny of mouse satellite cells as they transit through proliferation, differentiation and renewal.
| Antibodya | Clone | Isotypeb | Sourcec | Refs. |
|---|---|---|---|---|
| Anti-Pax7 | Pax7 | IgG1 | DSHBd | (12, 13, 98) |
| Anti-MyoD | 5.8A | IgG1 | BD Biosciences | (12, 63, 99) |
| Anti-myogenin | F5D | IgG1 | DSHB | (12, 63, 100, 101) |
| Anti-sarcomeric myosin | MF20 | IgG2b | DSHB | (12, 102) |
The antibodies against Pax7 and sarcomeric myosin were prepared originally against chicken proteins (98, 102). The antibody against sarcomeric myosin recognizes an epitope shared by all isoforms of sarcomeric myosin heavy chain in skeletal and cardiac muscle in a wide range of species.
The isotype of each antibody is provided to help in designing double-immunostaining studies. We routinely perform such studies using the anti-sarcomeric myosin in combination with the antibodies against MyoD, myogenin and Pax7 (12). Isotype-specific secondary antibodies are available from a variety of commercial sources. We obtain such antibodies (Alexa Fluor conjugated) from Invitrogen.
The same monoclonal antibodies against Pax7, MyoD and myogenin are available from additional sources.
The Developmental Studies Hybridoma Bank (DSHB) is under the auspices of the National Institute of Child Health and Human Development and maintained by The University of Iowa, Department of Biology, Iowa City, IA 52242 <http://dshb.biology.uiowa.edu>
In the following subheadings, we discuss some important considerations that should be taken in mind when establishing satellite cell primary cultures.
2.1. Strain and Age of Animals
The protocols in this chapter focus on the isolation and culture of myogenic progenitors from adult (3–6 month-old) C57BL/6 mice. Aged mice and other mouse strains have also been used in our studies following the same procedures (12, 19). However, muscles from younger mice may contribute more cells due to age-associated decline in satellite cells in some muscles (12, 19). Also, the contribution of nonmyogenic cells in the preparation may increase with age or in different mouse strains, and consequently, some conditions may need modification (i.e. duration of enzymatic digestion, extent of tissue trituration, cell straining conditions to remove debris, centrifugation speed of harvested suspension, etc.) to minimize the proportion of undesired cell types.
2.2. Muscles
Herein we detail our standard procedure for the isolation of satellite cells from hindlimb muscles of adult mice. For this preparation, we typically pool the fast twitch muscles tibialis anterior (TA) and gastrocnemius from both hindlimbs, using one mouse per preparation. For additional details about TA and gastrocnemius anatomy and isolation procedures see Notes 1 and 2. This approach can also be used for isolating myogenic progenitors from limb, body and head muscles. However, the contribution of connective tissue and vasculature may vary between muscles, and the tissue isolation procedure should be modified accordingly to minimize cells derived from such structures. The purity of the resultant preparation of isolated satellite cells (and cultures emanating from this preparation) is directly dependent on the amount of effort spent meticulously cleaning the muscle of these additional structures.
2.3. Digestive Enzyme for Muscle Dissociation
Our procedure is based on cell dissociation from whole muscle using Pronase digestion (see item 7 in Subheading 3.4. and items 4 and 13 in Subheading 4.1.). Pronase (available from Calbiochem) consists of a mixture of proteases isolated from the extracellular fluid of Streptomyces griseus. Due to its particular protease content, which includes several types of endo- and exopeptidases, Pronase has a broad activity (103, 104).
Pronase digestion may not be optimal for prospective satellite cell enrichment by antigen-based cell sorting because of extended digestion of surface antigens. However, myogenic cell preparations isolated by Pronase digestions show a lower level of nonmyogenic cells compared to that observed when collagenase or collagenase/dispase enzyme solutions are used. It is possible that certain nonmyogenic populations do not survive well after Pronase digestion and this may lead to the increased purity of these cultures.
2.4. Cell Yield, Choice of Culture Dish and Cell Seeding Density
Cell yields can vary depending on the age of the animal. Muscles from neonatal and young mice (1-month old or less) yield considerably more myogenic progenitors than muscles from adult mice. As mentioned before, variations are also observed when working with different muscles. For the mouse strain (C57BL/6) and hindlimb muscles (TA and gastrocnemius) used for the protocol described herein, each preparation typically yields 2–5 × 105 cells.
We commonly use 24-well or 35-mm culture dishes. We generally use 35-mm dishes for training or when performing single comparisons. In such cases, we initiate the cultures at 5–10 × 104 cells per plate. For multiple replicates across multiple time points, we use 24-well plates where starting cell density can be proportionally matched with that of the 35-mm plates based on surface area. Alternatively, seeding densities can be further reduced and depending on experimental goals, may range from 5 × 104 to 1 × 103 for primary cultures. While not further detailed below in the protocol section, it is noteworthy that we also use in some of our studies 48-well trays where we seed 2–10 cells per well; in such studies we aim to achieve clonal growth for monitoring progeny of individual satellite cells.
2.5. Culture Medium
The standard growth medium used for our mouse satellite cell cultures consists of high glucose Dulbecco's Modified Eagle Medium (DMEM) supplemented with 20% fetal bovine serum, 10% horse serum and 1% chicken embryo extract (CEE). This serum-rich growth medium supports both proliferation and differentiation of myogenic cells (12). See Subheading 3.4. and Notes 3–6 for details about recommended cell culture reagents, our protocol for preselection of optimal sera lots and preparation of CEE, and final medium preparation.
Some variations can be found from laboratory to laboratory with regard to the basic culture media (e.g., Ham's F10 instead of DMEM, or a mixture of the two), serum type and concentration, and source of growth factors (e.g., purified growth factors, especially fibroblast growth factor, instead of CEE). Differences in culture conditions may explain some divergences in satellite cell behavior among different laboratories. For example, some published protocols rely on first using serum-rich growth medium that supports proliferation followed by a switch to serum-poor medium to support differentiation. There are also reported variations in medium composition when preparing cultures from other species. For example, for primary cultures of chicken satellite cells we typically use medium containing 10% horse serum and 5% chicken embryo extract (21, 46, 76, 97).
To study the effects of specific growth factors on myogenic cell performance, we typically maintain the cells for 3 days in our standard rich growth medium to allow for optimal cell adherence, then switch the cells into serum-low (e.g., DMEM containing 2% horse serum) or serum-deprived media. Prior to switching to serum-low medium, the cultures are rinsed extensively with DMEM to remove traces of the rich medium that otherwise adhere to the cell layer and reduce the observed effect of the additives being examined.
2.6. Plate Coating Matrices
Adhesion of myogenic progenitors to cell culture dishes can be significantly improved by coating the plastic substrate with a variety of extracellular matrix constituents or derivatives. In addition to cell adhesion, matrix components can influence the extent of myogenic cell proliferation, differentiation and renewal (12, 13, 105, 106). In our laboratory, the main matrices used for coating tissue culture plates for satellite cell cultures are Matrigel and gelatin.
Matrigel is a solubilized basement membrane preparation extracted from the Engelbreth-Holm-Swarm mouse sarcoma, a tumor rich in extracellular matrix proteins. Its major component is laminin, followed by collagen IV, entactin, and heparan sulfate proteoglycan (107). Matrigel is available from BD Biosciences and can be obtained in its standard format or in its growth factor reduced format. In our studies we have typically used the growth factor reduced format. Matrigel must be carefully handled on ice when aliquoting and coating tissue culture dishes with dilutions. For additional details on Matrigel source and handling, see item 8 in Subheading 3.4. and Notes 7 and 8 for additional details.
Gelatin is produced by partial hydrolysis of type I collagen extracted from connective tissues. It can be purchased in a tissue culture grade powder form and easily reconstituted in water to the desired concentration. For specific details about gelatin source and our preparation of gelatin solution, see item 8 in Subheading 3.4. and Notes 9 and 10.
Gelatin is readily available, inexpensive and easy to use, which makes it an ideal product for training new team members and for use in standard cultures. However, long-term high-density myogenic cultures may spontaneously detach from plates coated with gelatin. In addition, satellite cell progeny typically demonstrate a more limited proliferative period, earlier differentiation, smaller myotubes and more meager development of reserve cells when grown on this substrate compared to Matrigel-coated dishes. Matrigel also allows a more even cell distribution upon initial cell plating compared to that observed when cells are seeded on gelatin-coated dishes. Additionally, when plated on Matrigel-coated dishes, myogenic progenitors can reach high cell densities and form complex myotube networks, typically without detaching from the substrate. The latter features have prompted us to use Matrigel especially when seeding cells at low density or when aiming to obtain single cell clones. Disadvantages of Matrigel include higher cost and the requirement for more careful handling. Other commercially available matrices that we have tested in pilot experiments that may provide reasonable alternatives include: (a) GelTrex (a Matrigel-like product from Invitrogen) and (b) Attachment Factor (Invitrogen), a ready made gelatin-based product.
2.7. Fixation and Immunostaining
For immunostaining analyses using the antibodies listed in Table 1, we typically fix the cultures with a paraformaldehyde-sucrose solution that is prepared in our laboratory. For further details about fixation approach and fixative composition, see Subheading 4.2. and Notes 11 and 12. It should be noted that fixatives should be optimized for preservation of both the cells and the antigens being analyzed. We perform all immunostaining steps in a manner that maintains sterility; handling antibodies strictly in the tissue culture hood minimizes possible bacterial contamination and helps maintain antibody stocks for years.
3. Materials
3.1. General Comments
The quantities of glassware, media and reagents as well as the time intervals for enzymatic digestion described in this chapter are appropriate for the isolation of satellite cells from TA and gastrocnemius muscles of both hindlimbs of one adult (3–6 month-old) C57BL/6 mouse. We typically do not pool muscles from multiple mice into a single preparation as cell yields are not necessarily increased linearly when using more muscle bulk.
All procedures are performed using sterile materials, supplies and techniques. Before transferring solutions/media into the tissue culture hood, spray the glass/plastic containers with 70% ethanol and wipe dry.
3.2. General Equipment
The following facilities are required for the cultures described in this chapter:
Standard humidified tissue culture incubator (37°C, 5% CO2 in air).
Tissue-culture laminar flow hood.
Water bath (37°C).
Hair trimmer (optional, for shaving hair from the hindlimbs prior to muscle dissection).
Stereo dissecting microscope with transmitted light base (microscope is either placed inside a tissue culture hood or in an isolation box/clean area).
Surgical tools for harvesting the muscles. Two types of forceps with extra fine-tips are recommended in particular to clean the muscles: (a) straight 110-mm (41/4″), and (b) curved 115-mm (41/2″). We typically sterilize dissection tools with a glass bead sterilizer, which is useful for quick sterilizing of tools as needed.
Table-top centrifuge.
Inverted phase contrast microscope for monitoring cell culture.
Inverted fluorescence microscope for analysis of immunolabeled culture dishes.
Hemacytometer and cover glass. Cover glasses can be purchased separately if replacement is needed.
Pipette controller (motorized pipette filler), essential for triturating the tissue after enzymatic digestion.
3.3. Plastic and Glassware Supplies
Standard 9” Pasteur pipettes.
Wide-bore pipettes prepared from the standard 9” Pasteur pipettes. Using a file or a diamond knife cut the narrow end of these pipettes to prepare a set of them with a bore diameter of approximately 3 mm. Shake the pipette to remove any glass fragments, fire polish sharp ends and autoclave. These pipettes are used to transfer muscle fragments.
9” Pasteur pipettes with cotton plug.
1-mL serological glass pipettes.
10-mL serological glass pipettes.
Syringe filters, 0.22-μm PVDF low protein binding filters and 1- or 3-cc disposable plastic syringes. Bottle top filters, 0.22 μm.
Cell strainer, 40 μm nylon mesh.
Polypropylene conical centrifuge tubes, sterile, 15 and 50 mL.
Plastic Petri dishes, 100-mm.
Tissue culture dishes, 35-mm.
Twenty four-well multiwell tissue culture dishes.
3.4. Cell Isolation and Culture Reagents
DMEM (Dulbecco's Modified Eagle Medium; high glucose, with 4500 mg/L glucose, 4 mM L-glutamine, 110 mg/L sodium pyruvate), supplemented with 100 U/mL penicillin and 100 μg/mL streptomycin. The term DMEM used from here on in this chapter refers to DMEM with antibiotics.
Fetal bovine serum (FBS; standard, not heat inactivated; Invitrogen / Gibco; see Note 3). Original bottles are stored at −80oC for long term (years); once thawed and aliquoted, stored at −20°C.
Chicken embryo extract (CEE) (available commercially from several sources); or, as in our studies, prepared by the investigator (see Notes 4 and 5); stored at −80°C for long term (years) or at −20°C when aliquoted.
Horse serum (HS; standard, not heat inactivated; Hyclone; see Note 6). Original bottles are stored at −80°C for long term (years); once thawed and aliquoted, stored at −20°C.
Standard growth medium for satellite cell cultures is made up of DMEM, 20% fetal bovine serum, 10% HS and 1% CEE. Culture medium is stored at 4°C and used within 3 weeks from preparation.
DMEM containing 10% HS to resuspend cells after enzymatic digestion.
Pronase (Calbiochem, reconstituted in DMEM) used for muscle digestion as described in Subheading 4.1.4.
Matrigel (BD Biosciences) for coating tissue culture dishes (see Note 8 for instructions). Matrigel can be purchased in its standard format. We usually dispense Matrigel into aliquots of 0.1–0.2 ml and freeze them at −20°C. See Note 7 for handling details.
Gelatin (Type A, Sigma-Aldrich) can be used as an alternative for coating tissue culture dishes (see Note 10 for procedure). Prepare and store 5 mL aliquots of 2% gelatin solution as indicated in Note 9.
3.5. Reagents and Solutions for Fixing and Immunostaining
Unless otherwise stated, the following solutions are stored at 4°C and pre-warmed at room temperature before use.
Pre-fixation rinse solution: DMEM as in item 1 in Subheading 3.4.
Fixative: 4% paraformaldehyde in a sodium phosphate buffer containing 0.03M sucrose (for further hazardous material details and composition/preparation of the fixative solution see Notes 11 and 12). To maintain quality and effectiveness of fixative, only pre-warm the amount that is required for immediate use.
Post-fix rinse solution: Tris-buffered saline (TBS); 0.05 M Tris, 0.15 M NaCl, pH 7.4 (for preparing this solution, see Note 13).
Detergents: Triton X-100; Tween 20.
Detergent solutions: TBS containing 0.5% Triton X-100 (TBS-TRX100); TBS containing 0.05% Tween 20 (TBS-TW20).
Blocking reagent: Normal goat serum (standard serum, does not need to be a product that is sold specifically for immunostaining). Can be stored at −80°C for long term (years); once thawed and aliquoted, store at −20°C.
Blocking Solution: TBS containing 1% normal goat serum (TBS-NGS).
Mounting media: Vectashield (Vector Laboratories) and (i) sterile 25% glycerol solution in TBS for 24-well plates; or (ii) cover glass, 22 mm2, for 35-mm dishes.
4. Methods
4.1. Cell Isolation and Culture
Pre-warm 30 mL of DMEM to 37° C and then keep at room temperature throughout the procedure.
Coat the tissue culture dishes with gelatin or Matrigel following the instructions described in Notes 8 and 10, respectively.
Add 5 mL of DMEM to three 60-mm Petri dishes and place the dishes in the tissue culture incubator until muscle dissection begins.
Prepare 1 mL of 1 % Pronase solution; we prepare this solution fresh for each experiment, dissolving 0.01-gram Pronase in 1-mL DMEM. Use a 0.22-μm syringe filter attached to a 1- or 3-cc syringe to filter the Pronase solution into a 15-mL conical centrifuge tube. At the time of muscle digestion (see steps 13 and 14 in Subheading 4.1.), this solution will be diluted tenfold in the DMEM containing the muscle fragments to make 3 mL of a final digest in 0.1 % solution.
Euthanize one mouse according to institutional regulations, shave (optional) the hindlimbs and spray them (regardless if shaving or not) lightly with 70% ethanol.
Harvest the TA and gastrocnemius muscles from both hindlimbs and place them in a 60-mm Petri dish with DMEM (see Notes 1 and 2 for further description of these muscles and how to isolate them).
Rinse the muscles by gently swirling the plate and transfer them to the second 60-mm Petri dish.
Under the dissecting microscope, using the straight and curved fine point forceps (described in Subheading 3.2., item 6), carefully remove from each muscle the tendons, fat, vessels and bits of connective tissue as much as possible.
Transfer the cleaned muscles to the third 60-mm Petri dish with DMEM and cut into small fragments (about 3 mm3) but do not mince (if fragments are too small, the mechanical trituration that follows the enzymatic digestion step is less effective in releasing cells). Further inspect the muscle fragments to eliminate, as much as possible, any remaining connective tissue.
Using a sterilized wide-bore Pasteur pipette, transfer the suspension of muscle fragments to a 15-mL conical tube and allow the fragments to settle down. Alternatively, muscle fragments can be collected by low speed centrifugation (~ 200 × g) for 4 minutes.
Aspirate and discard supernatant. Add DMEM to the settled muscle fragments up to a final volume of 2 mL, including the muscle bulk. Shake the tube gently to loosen the pelleted tissue and transfer tube contents to a 35-mm dish using a wide-bore Pasteur pipette.
Use 700 μl of DMEM to rinse the 15-mL tube of any remaining muscle bits and add this volume to the 35-mm dish.
Add 300 μl of 1 % Pronase to the plate, generating a final volume of 3 mL (including muscle bits) and a final concentration of 0.1 % Pronase.
Place the 35-mm dish inside the tissue culture incubator for 60 min. Gently swirl the dish every 15–20 min during digestion (alternatively, one can use a low speed agitator placed inside the tissue culture incubator).
At the end of the digestion period, transfer the muscle fragments and Pronase solution to a 15 mL conical tube using a wide-bore Pasteur pipette.
Spin down the suspension by low-speed centrifugation at 400 × g for 5 min.
Aspirate the supernatant carefully, without disturbing the loose pellet of digested muscle pieces. Resuspend the muscle bulk in 5 mL of 10% HS in DMEM (pre-warmed at 37°C, then kept at room temperature until used). At this stage the still attached satellite cells can be released by mechanical trituration in a manner that avoids damaging the desired cells. We perform two cycles of muscle trituration (detailed below, steps18–22) so that cells released early in the process can be harvested and set aside, after which further trituration releases the remaining cells. It is critical that the enzymatic digestion does not fully dissociate the tissue, but only loosens the cells; without the mechanical trituration steps, cell yields are poor.
First muscle trituration: Vigorously triturate muscle fragments by passing them repetitively (about 20 times) through a 10-mL glass pipette until the tissue bits pass easily through the tip of the pipette. Shearing of the tissue with the mechanical trituration is critical to efficient cell release. Allow the suspension to settle in the 15 mL conical tube so that the remaining larger bits separate from the supernatant that contains the released cells.
Without disturbing the precipitated material, collect the supernatant and transfer it to a 15-mL conical tube.
Second muscle trituration: Add 5 mL of 10% HS into the 15-mL conical tube containing the remaining muscle pieces and repeat the muscle trituration process, now using a 9” cotton-plugged glass Pasteur pipette until all the muscle pieces easily passes through it.
Allow the suspension to settle as in step 19 and collect the supernatant in the same 15-mL conical tube as in step 20.
Place a 40-μm cell strainer onto a 50-mL conical tube.
Using a 10-mL glass pipette, transfer the pooled supernatants from the two triturations to the 40-μm cell strainer. Make sure the suspension passes through the strainer by carefully tapping the side. This step eliminates residual large debris from the cell suspension.
For maximal cell recovery, allow an additional 1-mL DMEM to drip through the cell strainer to recover the residual cells trapped by debris in the unit.
Centrifuge the strained cell suspension at ~1000 × g for 10 min (see Note 14) to recover the cells released during the trituration steps.
Carefully aspirate supernatant (which is discarded) and resuspend the cell pellet in 1 mL of standard growth medium (pre-warmed at 37° C and held at room temperature until needed) using a 1-mL glass pipette. We recommend removal of the supernatant manually with a Pasteur pipette, and not by mechanical aspiration to minimize the risk of aspirating the delicate cell pellet as well.
Using a micropipette, collect 10 μl of the cell suspension (ensure the suspension is mixed gently just before removing the aliquot for cell counting as cells settle very fast when held in the tube for processing) and transfer it to the edge of one of the hemacytometer chambers, previously cleaned with 70% ethanol, dried and covered with the cover glass (the cell suspension should run under the cover glass by capillarity). Count only the small round cells while avoiding red blood cells. For increased accuracy we recommend counting another 10-μl sample of the cell suspension in the second hemacytometer's chamber.
Plate cells in the Matrigel- or gelatin-coated culture dishes. When using 24-well trays, plate cells at a density of 1 – 2 × 104/well (standard density) or 1 – 2 × 103/well (low density). When using 35-mm dishes, plate 1 – 2 × 105 cells (standard density) or less, depending on the experimental goal.
Culture the cells undisturbed in the incubator for three days.
Rinse the cultures 1–2 times with 1 mL of pre-warmed DMEM before adding fresh medium at the first medium change. This helps to remove debris that is apparent in the primary cultures and can be easily mistaken for contamination to an inexperienced observer. Cultures should be rinsed very gently to minimize cell detachment. If warranted, the level of debris in the cell suspension can be further reduced before culturing the cells (see Note 15), but the debris also disappears with time as cultures get more dense.
Replace the culture medium with fresh medium every 3 days. Note, however, that medium may need to be changed more frequently at late time points depending on the density of the cells.
4.2. Cell Culture Fixation and Immunostaining
Warm DMEM and fixative solution to room temperature. DMEM can be first warmed in a water bath set at 37° C then held at room temperature until needed. The 4% paraformaldehyde fixative solution should be allowed time to equilibrate to room temperature prior to its use. For both items, warm only the required volume for the experiment. Extensive warming of the fixative solution multiple times results in deterioration of the paraformaldehyde.
Rinse cultures with DMEM three times. Following the final rinse add 250 μl of DMEM to each well in the 24-well plate or 500 μl to each 35-mm dish.
Add an equal volume of the 4% paraformaldehyde fixative solution to the culture medium in each well or dish (250 μl or 500 μl as above). Allow 10 min at room temperature for the fixation, then carefully remove the culture medium-paraformaldehyde fixative mixture and rinse each well three times with TBS.
-
Add 500 μl of TBS-TRX100 for 5 min at room temperature to permeabilize the cells. Alternatively, the permeabilization step can be omitted if considering using antibodies different from those listed in Table 1, as some antigens might be sensitive to this detergent. Also, cultures can be treated with TBS-TRX100 later (but then blocking solution detailed below needs to be reapplied prior to antibody staining).
Note that from this step on, unless otherwise stated, the volumes of each reagent are the same for either 24-well or 35-mm dishes.
Add 500 μl of blocking solution (TBS-NGS) to each well or dish to block nonspecific antibody binding.
Cultures are then kept at 4°C overnight or longer (see Note 16).
Allow plates to warm up to room temperature for at least 10 minutes before starting the antibody staining procedure.
Dilute the appropriate primary antibody in blocking solution. For antibodies listed in Table 1, we typically use antibody formulations as previously published (12, 19).
Rinse the cultures three times with TBS-TW20.
Aspirate the final TBS-TW20 rinse and add 150 μl of the primary antibody solution for 1 hour at room temperature, followed by an overnight incubation at 4°C in a humidified chamber. Primary (and secondary - see step 11) antibodies are applied at the center of the dish. When using 24-well plates, a light and continuous swirling on a flat surface is required to ensure optimal spreading of the antibody across the well; otherwise, antibody solution rapidly accumulates at the periphery (see Note 17). When working with 35-mm dishes, plates are manually swirled only upon applying the antibody then maintained without any disturbance during the labeling period, allowing the antibody solution to spread throughout the plate by capillarity.
Bring the plate to room temperature as in step 7 and dilute the appropriate secondary antibody in the blocking solution. For antibodies listed in Table 1, we typically use secondary antibodies diluted as previously published (12, 19).
Rinse cultures three times with TBS-TW20 warmed up at room temperature.
Aspirate the final TBS-TW20 rinse and add the diluted secondary antibody (same volume and swirling conditions as for the primary antibody, see step 10) for 1–2 h at room temperature.
Aspirate the secondary antibody and wash three times with TBS-TW20.
For nuclear visualization, add 100 μl of DAPI solution (4',6-diamidino-2-phenylindole, dihydrochloride; stock concentration 10 mg/mL, working concentration 1 μg/mL diluted in TBS-NGS prior to use) for 30 min at room temperature (see Note 18).
Rinse the cultures twice with TBS-TW20 followed by a final rinse with TBS.
Aspirate the TBS and mount cultures in Vectashield mounting medium. The mounting medium prevents the stained cultures from drying and retards fading of the immunofluorescent signal. Add 1 drop at the center of each well of the tray or each 35-mm culture dish. If working with a 35-mm culture dish, complete the mounting process by covering with a cover slip. We prefer not to use cover slips when working with 24-well trays. Instead, we add 300 μl of glycerol mounting solution (25% glycerol in TBS) following the initial drop of Vectashield to allow mounting medium coverage of individual wells in 24-multiwell trays.
5. Notes
- The information provided here is to assist in the identification and isolation of the tibialis anterior and gastrocnemius muscles. We recommend the following literature and links for anatomical descriptions and schematic images of mouse muscles, though they refer to rat and human muscles.
-
Tibialis anterior (TA): The TA is a superficial muscle of the anterior compartment of the lower hindlimb, located in a medial position (108, 109). It arises from the lateral condyle and the upper lateral surface of the tibia. Its tendon passes across the medial surface of the dorsum of the foot and inserts on the medial cuneiform bone and the first metatarsal. The TA muscle is responsible for the dorsiflexion and inversion of the foot.For a schematic image of this muscle see: http://www.bartleby.com/107/illus437.html
-
Gastrocnemius: The gastrocnemius muscle is the most superficial muscle in the posterior part of the lower hindlimb (108, 109). It consists of two heads that arise from the lateral and medial condyles of the femur. The distal end of the gastrocnemius muscle is the Achilles' or calcaneal tendon, which is attached to the posterior surface of the calcaneus. Located deep to the gastrocnemius and closely connected to it, is the soleus muscle. These two muscles are collectively called triceps surae and together, they are responsible for the plantarflexion of the foot.For a schematic image of the gastrocnemius location see: http://www.bartleby.com/107/illus438.html
-
- Harvesting hindlimb TA and gastrocnemius muscles:
- To begin with the TA extraction, secure the mouse in a supine position to the dissecting board by pinning down the hindlimb to be dissected and the diagonal forelimb.
- Use straight operating scissors to cut through the skin, opening a small incision above the knee.
- Holding the skin with fine forceps, insert rounded-tip scissors beneath the incision and carefully open the scissors to loosen the skin from the underlying muscles.
- Extend the incision to a point just in front of the digits.
- Loosen the skin as you go, being careful not to cut the underlying muscles or blood vessels.
- Cut and remove the skin from the knee to the paw.
- Identify the tendon at the insertion of the TA.
- Place one arm of the very fine point forceps underneath the tendon and carefully pull proximally, with the forceps under the TA muscle, to drag the fascia that covers the muscle. Then use the forceps to pull the fascia upward toward the knee and discard it.
- Use micro scissors to cut the tendon at the insertion of the TA, as far as possible from the muscle itself.
- Using very fine point forceps grasp the tendon and carefully pull it in order to lift the TA muscle gently away and upward.
- With the TA lifted, cut the proximal attachment against the knee with micro scissors and place the removed muscle in a 60-mm Petri dish with DMEM.
- Do the same (steps b–k) for the other hindlimb before proceeding to the next steps (gastrocnemius isolation).
- Turn the mouse over in a prone position, pin it appropriately and identify the gastrocnemius.
- Using dressing forceps, pull away the upper hindlimb muscles that cover the proximal portion of the gastrocnemius.
- Place very fine point forceps under the Achilles tendon and move it proximally underneath the gastrocnemius.
- Cut the Achilles tendon and lift the gastrocnemius as done for the TA.
- With the gastrocnemius lifted, cut its proximal side as close as possible to its origin. Remove the soleus muscle, which is intertwined with the main tendon of the gastrocnemius, and place the gastrocnemius in the same 60-mm Petri dish containing the isolated TA muscles. Repeat steps p and q for the other hindlimb.
Fetal bovine serum (FBS) should be pre-characterized by comparing sera from several suppliers. We select FBS based on the capacity of the serum to support proliferation and differentiation of mouse primary myoblasts cultured when seeding cultures at a wide range of cell concentrations. Only sera able to support good growth and differentiation at both high and low cell density, are employed in our studies. Primary myogenic cultures for these tests are prepared as described here. The vendor listed for this product in item 2, Subheading 3.4. is provided as an example for what we found to be optimal when the serum selection was performed.
We prepare chicken embryo extract (CEE) in our laboratory using 10-day old White Leghorn embryos (70, 110). The procedure is similar to a previously described method (111) but uses the entire embryo. We recommend this approach over purchasing CEE if the investigator can obtain embryonated chicken eggs, as the quality is higher and the cost lower than that of purchased CEE.
- Preparation of chicken embryo extract:
- Embryonated chicken eggs (8 dozen, White Leghorn; from Charles River or local sources with a good egg fertility index) are maintained in a standard egg incubator (incubation conditions: a dry temperature of 38°C, a wet temperature of 30°C and relative humidity of 56%).
- After 10 days, batches of 15–30 eggs are removed from the incubator and transferred into the tissue culture hood. All steps from here on are performed in a sterile manner.
- Place the eggs lengthwise in the rack and spray with 70% ethanol to sterilize. Wait for several minutes until the ethanol evaporates.
- Crack open one egg at a time into a 100-mm Petri dish.
- Remove the embryo from surrounding membranes by piercing it with fine forceps. Rinse the embryo by transferring it through three 100-mm Petri dishes containing DMEM supplemented with antibiotics (see item 1 in Subheading 3.4. for DMEM-antibiotics formulation). Swirl embryo a few times in each dish for a good rinse.
- Empty the egg remains from the initial 100-mm dish (described in step d) into a waste beaker and repeat steps d–f until the final rinse dish contains about 30 embryos.
- Transfer the embryos with fine forceps into a 60-mL disposable syringe, force through the opening with the syringe plunger, and collect the suspension into a 500-mL sterile glass bottle.
- The extract is diluted with approximately an equal volume of DMEM (supplemented with antibiotics) and gently agitated for 2 h at room temperature. To ensure good agitation, keep maximum volume to one-half bottle capacity and place the bottle at 45° angle during the agitation.
- The extract is frozen at −80°C for a minimum of 48 h. It is then thawed, dispensed into 50-mL conical tubes, and centrifuged at approximately 500 × g for 10 min to remove residual tissue.
- The supernatant is pooled, divided into 40-mL aliquots and kept frozen at −80°C for long-term storage. For short-term storage, we typically prepare aliquots of 2.5 mL that are kept frozen at −20°C.
- Prior to use, the CEE-thawed aliquot should again be centrifuged at about 800–1000 × g for 10 min to remove aggregates. The supernatant is then collected and added to the DMEM-based medium to prepare the rich growth medium for myogenic stem cell cultures. The growth medium is then passed through a sterile 0.22-μm filter to clear remaining particles and sterilize. All details of supplies for generating the medium are in Subheading 3.4. To ensure optimal cell growth conditions, we typically prepare only 250-mL medium each time, and use it within a few weeks.
Horse serum (HS) should be pre-characterized by comparing sera from various suppliers. We select HS based on its capacity to support proliferation and differentiation of primary chicken myoblasts cultured at standard and clonal densities (21, 46). The vendor product listed in item 4, Subheading 3.4. for HS source is provided as an example for what we found to be optimal when the serum selection was performed.
Matrigel (BD Biosciences) is shipped on dry ice and stored at −20°C until aliquoted. Matrigel should be thawed on ice; never use a warmer temperature, as it will prematurely gel. To ensure Matrigel stability, we follow the manufacturer's handling instructions, when thawing the product on ice (overnight in an ice bucket placed at 4°C). Once liquefied, Matrigel is aliquoted with pre-chilled 1-mL serological glass pipettes into tubes chilled on ice. Typically, we aliquot 0.1 and 0.2 ml each into 2-mL cryogenic vials sealed with O-rings. These aliquots are stored at −20°C.
- Coating Tissue Culture Dishes with Matrigel: All steps are done on ice, unless otherwise noted. Matrigel stock is first diluted to create a working mixture used to coat plates (see Note 7). We here describe the coating of 24-well plates only.
- Thaw the required amount of Matrigel by placing frozen aliquot(s) on ice for at least 30 min and as much as 1.5 h to allow the Matrigel stock to completely liquefy for subsequent dilution to the working solution. We observed some batch-to-batch variation in the time it takes to thaw the aliquots, therefore, for consistency, we typically allow Matrigel aliquots to thaw for 1.5 h.
- Pre-chill a 50-mL conical tube on ice and transfer the thawed Matrigel into the tube. Add ice-cold DMEM to dilute the Matrigel to a final concentration of 1 mg/mL. Gently mix the Matrigel and DMEM by several repetitive drawings through a 1-mL glass pipette. An optimal Matrigel stock is at ~10 mg/mL protein concentration, further diluted at 1:10 for the working Matrigel solution. Stock protein concentration can vary greatly from lot to lot and should be monitored. Allow the diluted Matrigel solution to cool on ice for 15 min.
- After 15 min, use a chilled 1 mL glass pipette to draw up the diluted Matrigel solution and coat the dishes with an appropriate volume (150 μl per well for a 24-well plate). In our experience, 2 mL of working Matrigel solution can be used to coat an entire 24-well plate; we typically coat 6–8 wells at a time as detailed next.
- Per each series of 6–8 wells, leave the culture plate/dish coated with the Matrigel working solution on ice for 7 min, then use the same pipette as before (held cooled in a tube on ice) to remove the Matrigel solution and place it back in the 50-mL conical tube that is kept on ice. This will leave a thin coat of Matrigel at the bottom of the wells/dishes.
- Once all of Matrigel solution has been placed back in the tube, use the same pipette to coat the next set of wells in the tray. Always be sure to leave the aliquot of diluted Matrigel in each well for 7 min.
- Having coated all the desired wells per 1 tray, tilt the dishes and use a 20-μl pipette tip to carefully remove residual Matrigel and place it back in the 50-ml conical tube kept on ice.
- Incubate the Matrigel-coated dishes in the tissue culture incubator for at least 1 h.
- About 10 min before plating the isolated cells, take the Matrigel-coated dishes out of the incubator to the tissue culture hood and open the lid. This will allow evaporation of water that otherwise will condense on the underside of the lid when moving the dish from the warm incubator to room temperature. If allowed to form, the condensation will drip into the well, disturbing the Matrigel coating.
- The working Matrigel solution can be used to coat additional dishes after completing one tray coating. Matrigel that has been used to coat too many dishes, however, is less effective in supporting cell adhesion. We typically limit reuse of diluted Matrigel to three rounds of coating and work with a larger volume of diluted Matrigel if coating more than 1 tray. Also, we only use Matrigel that has been diluted the day of the fiber isolation to maintain consistency.
- Preparation of 2% gelatin solution:
- Weigh 2 g of gelatin powder and transfer it to a 250-mL glass bottle containing 100 mL of deionized water.
- Autoclave (only at this stage will gelatin powder completely dissolve).
- Allow the solution to cool to room temperature.
- Aliquot 5 ml into 15 ml conical tubes and store at 4°C. Gelatin solution will solidify upon refrigeration. Aliquots stored at 4°C are good for years.
- Coating tissue culture dishes with gelatin:
- Place 2% gelatin aliquot in a 37°C water bath until completely liquefied; then keep in the tissue culture hood until used.
- Distribute 150–200 μl of gelatin solution into each well of the 24-well plate, or 300–500 μl into 35-mm culture dishes.
- Swirl gently the 24-well plate or the 35-mm dish to allow even coating of the plating surface. Inspect plates to ensure even spreading of the gelatin solution as some regions may remain uncoated initially.
- Allow the gelatin-coated dish to sit at room temperature for at least 1 hour.
- Using a Pasteur pipette remove the entire volume of gelatin solution from the wells. That will leave a thin coat of gelatin at the bottom of the wells/dishes. Gelatin solution can be reused several times (at least 10 times) without affecting cell adhesion and growth.
- Let the gelatin-coated dishes sit in the tissue culture hood for at least 30 min before plating the isolated cells.
Paraformaldehyde is a white powder with a formaldehyde-like odor. It is a rapid fixative and a potential carcinogen. When handling paraformaldehyde, wear gloves, a mask, and goggles. It is important to refer to the MSDS instructions and institutional regulations for further information regarding storage, handling and first-aid.
- Preparation of 100 mL of 4% paraformaldehyde with 0.03M sucrose, in a fume hood:
- Mix 4 g of paraformaldehyde powder and 80 mL of deionized water in a glass beaker; cover with parafilm.
- Warm the solution to 60°C with continuous stirring to dissolve the powder.
- Allow the solution to cool to room temperature.
- Add about 1–4 drops of 1N NaOH, until the opaque color of the solution clears.
- Add 10 mL 1M sodium phosphate.
- Adjust the pH to 7.2–7.4 using color pH strips.
- Add 1.026 g of sucrose.
- Bring volume to 100 mL.
- Filter through a 0.22-μm disposable filter unit into a bottle.
- Store at 4°C in an aluminum foil-wrapped bottle for no more than 1 month.
-
Preparation of Tris buffered saline (TBS):
To make one liter of 10X TB:- Weigh 60.5 g of Tris-Base into a beaker.
- Add 700 mL deionized water to the beaker.
- Place the beaker on top of a magnetic stirrer.
- When the powder has dissolved, adjust the pH to 7.4.
- Add deionized water to bring the volume up to 1 liter, mix well, autoclave or sterile through filter, and store at 4°C.
To make one liter of TBS:- Weigh 8.766g NaCl in a beaker
- Add 100 mL of 10X TB to the beaker and mix vigorously.
- When the powder has dissolved, add deionized water to bring the volume up to 1 liter; mix well, sterile through filter and store at 4°C.
The optimal speed to centrifuge the cell suspension should be further “fine tuned” by the investigator. High centrifugation speed results in preparations with better cell yields, but also with higher debris content. Debris may represent an obstacle not only for further analysis and/or treatments, such as flow cytometry and cell sorting, but also for the survival and adhesion of isolated myogenic stem cells.
In order to minimize debris resulting from muscle digestion, cell suspensions of freshly isolated satellite cells can be further be purified by Percoll density centrifugation. While this approach aims mostly to remove debris (76), a modification of this procedure that includes a multi-step Percoll gradient can also fractionate cell subpopulations (62).
For some antibodies the cultures may be blocked for just 2–4 h at room temperature if overnight blocking is not desired.
For even and continuous distribution of the antibodies (both primary and secondary), it is recommended to place 24-well plates on a gyrating platform rotator. This is important since, without agitation, the antibody solution tends to rapidly accumulate at the well periphery, leading to uneven staining across the culture.
DAPI is potentially harmful. Avoid prolonged or repeated exposure; we typically dissolve the entire powder in its original container and generate a concentrated stock solution. A ready-made DAPI reagent is available from Molecular Probes. It is important to refer to the MSDS instructions and institutional regulations for further information regarding storage, handling and first aid.
Acknowledgements
We thank Lindsey Muir for reviewing this manuscript and providing valuable comments. We are also grateful to the granting agencies that funded this study. Our current research is supported by grants to Z.Y.R. from the National Institutes of Health (AG021566; AG035377; AR057794) and the Muscular Dystrophy Association (135908). M.E.D is supported by the Genetic Approaches to Aging Training Program (T32 AG000057). The development of the protocols described here could not be possible without early support to Z.Y.R from the American Heart Association, the USDA Cooperative State Research, Education and Extension Service, and the National Institutes of Health (AG013798).
References
- 1.Hawke TJ, Garry DJ. Myogenic satellite cells: physiology to molecular biology. J Appl Physiol. 2001;91:534–551. doi: 10.1152/jappl.2001.91.2.534. [DOI] [PubMed] [Google Scholar]
- 2.Zammit PS, Partridge TA, Yablonka-Reuveni Z. The skeletal muscle satellite cell: the stem cell that came in from the cold. J Histochem Cytochem. 2006;54:1177–1191. doi: 10.1369/jhc.6R6995.2006. [DOI] [PubMed] [Google Scholar]
- 3.Yablonka-Reuveni Z, Day K. Skeletal muscle stem cells in the spotlight: the satellite cell. In: Cohen I, Gaudette G, editors. Regenerating the Heart: Stem Cells and the Cardiovascular System. Springer, Human Press; 2011. pp. 173–200. Chapter 11. [Google Scholar]
- 4.Mauro A. Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol. 1961;9:493–495. doi: 10.1083/jcb.9.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Katz B. The terminations of the afferent nerve fibre in the muscle spindle of the frog. Philos Trans Royal Soc Lond. 1961;243,:221–240. [Google Scholar]
- 6.Bischoff R. Regeneration of single skeletal muscle fibers in vitro. Anat Rec. 1975;182:215–235. doi: 10.1002/ar.1091820207. [DOI] [PubMed] [Google Scholar]
- 7.Konigsberg UR, Lipton BH, Konigsberg IR. The regenerative response of single mature muscle fibers isolated in vitro. Dev Biol. 1975;45:260–275. doi: 10.1016/0012-1606(75)90065-2. [DOI] [PubMed] [Google Scholar]
- 8.Yablonka-Reuveni Z, Rivera AJ. Temporal expression of regulatory and structural muscle proteins during myogenesis of satellite cells on isolated adult rat fibers. Dev Biol. 1994;164:588–603. doi: 10.1006/dbio.1994.1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosenblatt JD, Lunt AI, Parry DJ, Partridge TA. Culturing satellite cells from living single muscle fiber explants. In Vitro Cell Dev Biol Anim. 1995;31:773–779. doi: 10.1007/BF02634119. [DOI] [PubMed] [Google Scholar]
- 10.Zammit PS, Golding JP, Nagata Y, Hudon V, Partridge TA, Beauchamp JR. Muscle satellite cells adopt divergent fates: a mechanism for self-renewal? J Cell Biol. 2004;166:347–357. doi: 10.1083/jcb.200312007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Collins CA, Olsen I, Zammit PS, Heslop L, Petrie A, Partridge TA, Morgan JE. Stem cell function, self-renewal, and behavioral heterogeneity of cells from the adult muscle satellite cell niche. Cell. 2005;122:289–301. doi: 10.1016/j.cell.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 12.Shefer G, Van de Mark DP, Richardson JB, Yablonka-Reuveni Z. Satellite-cell pool size does matter: defining the myogenic potency of aging skeletal muscle. Dev Biol. 2006;294:50–66. doi: 10.1016/j.ydbio.2006.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Day K, Shefer G, Richardson JB, Enikolopov G, Yablonka-Reuveni Z. Nestin-GFP reporter expression defines the quiescent state of skeletal muscle satellite cells. Dev Biol. 2007;304:246–259. doi: 10.1016/j.ydbio.2006.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muir AR, Kanji AH, Allbrook D. The structure of the satellite cells in skeletal muscle. J Anat. 1965;99:435–444. [PMC free article] [PubMed] [Google Scholar]
- 15.Yablonka-Reuveni Z. Development and postnatal regulation of adult myoblasts. Microsc Res Tech. 1995;30:366–380. doi: 10.1002/jemt.1070300504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boldrin L, Muntoni F, Morgan JE. Are Human and Mouse Satellite Cells Really the Same? J Histochem Cytochem. 2010;58:941–955. doi: 10.1369/jhc.2010.956201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Biressi S, Rando TA. Heterogeneity in the muscle satellite cell population. Semin Cell Dev Biol. 2010;21:845–854. doi: 10.1016/j.semcdb.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seale P, Sabourin LA, Girgis-Gabardo A, Mansouri A, Gruss P, Rudnicki MA. Pax7 is required for the specification of myogenic satellite cells. Cell. 2000;102:777–786. doi: 10.1016/s0092-8674(00)00066-0. [DOI] [PubMed] [Google Scholar]
- 19.Day K, Shefer G, Shearer A, Yablonka-Reuveni Z. The depletion of skeletal muscle satellite cells with age is concomitant with reduced capacity of single progenitors to produce reserve progeny. Dev Biol. 2010;340:330–343. doi: 10.1016/j.ydbio.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shefer G, Rauner G, Yablonka-Reuveni Z, Benayahu D. Reduced satellite cell numbers and myogenic capacity in aging can be alleviated by endurance exercise. PLoS One. 2010;5:e13307. doi: 10.1371/journal.pone.0013307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Halevy O, Piestun Y, Allouh MZ, Rosser BW, Rinkevich Y, Reshef R, Rozenboim I, Wleklinski-Lee M, Yablonka-Reuveni Z. Pattern of Pax7 expression during myogenesis in the posthatch chicken establishes a model for satellite cell differentiation and renewal. Dev Dyn. 2004;231:489–502. doi: 10.1002/dvdy.20151. [DOI] [PubMed] [Google Scholar]
- 22.Allouh MZ, Yablonka-Reuveni Z, Rosser BW. Pax7 reveals a greater frequency and concentration of satellite cells at the ends of growing skeletal muscle fibers. J Histochem Cytochem. 2008;56:77–87. doi: 10.1369/jhc.7A7301.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lindstrom M, Thornell LE. New multiple labelling method for improved satellite cell identification in human muscle: application to a cohort of power-lifters and sedentary men. Histochem Cell Biol. 2009;132:141–157. doi: 10.1007/s00418-009-0606-0. [DOI] [PubMed] [Google Scholar]
- 24.Lindstrom M, Pedrosa-Domellof F, Thornell LE. Satellite cell heterogeneity with respect to expression of MyoD, myogenin, Dlk1 and c-Met in human skeletal muscle: application to a cohort of power lifters and sedentary men. Histochem Cell Biol. 2010;134:371–385. doi: 10.1007/s00418-010-0743-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Montarras D, Morgan J, Collins C, Relaix F, Zaffran S, Cumano A, Partridge T, Buckingham M. Direct isolation of satellite cells for skeletal muscle regeneration. Science. 2005;309:2064–2067. doi: 10.1126/science.1114758. [DOI] [PubMed] [Google Scholar]
- 26.Beauchamp JR, Heslop L, Yu DS, Tajbakhsh S, Kelly RG, Wernig A, Buckingham ME, Partridge TA, Zammit PS. Expression of CD34 and Myf5 defines the majority of quiescent adult skeletal muscle satellite cells. J Cell Biol. 2000;151:1221–1234. doi: 10.1083/jcb.151.6.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ono Y, Boldrin L, Knopp P, Morgan JE, Zammit PS. Muscle satellite cells are a functionally heterogeneous population in both somite-derived and branchiomeric muscles. Dev Biol. 2010;337:29–41. doi: 10.1016/j.ydbio.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dellavalle A, Sampaolesi M, Tonlorenzi R, Tagliafico E, Sacchetti B, Perani L, Innocenzi A, Galvez BG, Messina G, Morosetti R, Li S, Belicchi M, Peretti G, Chamberlain JS, Wright WE, Torrente Y, Ferrari S, Bianco P, Cossu G. Pericytes of human skeletal muscle are myogenic precursors distinct from satellite cells. Nat Cell Biol. 2007;9:255–267. doi: 10.1038/ncb1542. [DOI] [PubMed] [Google Scholar]
- 29.Zheng B, Cao B, Crisan M, Sun B, Li G, Logar A, Yap S, Pollett JB, Drowley L, Cassino T, Gharaibeh B, Deasy BM, Huard J, Peault B. Prospective identification of myogenic endothelial cells in human skeletal muscle. Nat Biotechnol. 2007;25:1025–1034. doi: 10.1038/nbt1334. [DOI] [PubMed] [Google Scholar]
- 30.Tedesco FS, Dellavalle A, Diaz-Manera J, Messina G, Cossu G. Repairing skeletal muscle: regenerative potential of skeletal muscle stem cells. J Clin Invest. 2010;120:11–19. doi: 10.1172/JCI40373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moss FP, Leblond CP. Satellite cells as the source of nuclei in muscles of growing rats. Anat Rec. 1971;170:421–435. doi: 10.1002/ar.1091700405. [DOI] [PubMed] [Google Scholar]
- 32.Campion DR. The muscle satellite cell: a review. Int Rev Cytol. 1984;87:225–251. doi: 10.1016/s0074-7696(08)62444-4. [DOI] [PubMed] [Google Scholar]
- 33.Schultz E. Satellite cell proliferative compartments in growing skeletal muscles. Dev Biol. 1996;175:84–94. doi: 10.1006/dbio.1996.0097. [DOI] [PubMed] [Google Scholar]
- 34.White RB, Bierinx AS, Gnocchi VF, Zammit PS. Dynamics of muscle fibre growth during postnatal mouse development. BMC Dev Biol. 2010;10:21. doi: 10.1186/1471-213X-10-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schultz E, Gibson MC, Champion T. Satellite cells are mitotically quiescent in mature mouse muscle: an EM and radioautographic study. J Exp Zool. 1978;206:451–456. doi: 10.1002/jez.1402060314. [DOI] [PubMed] [Google Scholar]
- 36.Snow MH. An autoradiographic study of satellite cell differentiation into regenerating myotubes following transplantation of muscles in young rats. Cell Tissue Res. 1978;186:535–540. doi: 10.1007/BF00224941. [DOI] [PubMed] [Google Scholar]
- 37.Grounds MD, Yablonka-Reuveni Z. Molecular and cell biology of skeletal muscle regeneration. Mol Cell Biol Hum Dis Ser. 1993;3:210–256. doi: 10.1007/978-94-011-1528-5_9. [DOI] [PubMed] [Google Scholar]
- 38.Bischoff R. Analysis of muscle regeneration using single myofibers in culture. Med Sci Sports Exerc. 1989;21:S164–172. [PubMed] [Google Scholar]
- 39.Yablonka-Reuveni Z, Seger R, Rivera AJ. Fibroblast growth factor promotes recruitment of skeletal muscle satellite cells in young and old rats. J Histochem Cytochem. 1999;47:23–42. doi: 10.1177/002215549904700104. [DOI] [PubMed] [Google Scholar]
- 40.Anderson JE. The satellite cell as a companion in skeletal muscle plasticity: currency, conveyance, clue, connector and colander. J Exp Biol. 2006;209:2276–2292. doi: 10.1242/jeb.02088. [DOI] [PubMed] [Google Scholar]
- 41.Gopinath SD, Rando TA. Stem cell review series: aging of the skeletal muscle stem cell niche. Aging Cell. 2008;7:590–598. doi: 10.1111/j.1474-9726.2008.00399.x. [DOI] [PubMed] [Google Scholar]
- 42.Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, Rando TA. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature. 2005;433:760–764. doi: 10.1038/nature03260. [DOI] [PubMed] [Google Scholar]
- 43.Carlson ME, Conboy MJ, Hsu M, Barchas L, Jeong J, Agrawal A, Mikels AJ, Agrawal S, Schaffer DV, Conboy IM. Relative roles of TGF-beta1 and Wnt in the systemic regulation and aging of satellite cell responses. Aging Cell. 2009;8:676–689. doi: 10.1111/j.1474-9726.2009.00517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shavlakadze T, McGeachie J, Grounds MD. Delayed but excellent myogenic stem cell response of regenerating geriatric skeletal muscles in mice. Biogerontology. 2010;11:363–366. doi: 10.1007/s10522-009-9260-0. [DOI] [PubMed] [Google Scholar]
- 45.Shefer G, Yablonka-Reuveni Z. Ins and outs of satellite cell myogenesis: the role of the ruling growth factors. In: Schiaffino S, Partridge T, editors. Skeletal Muscle Repair and Regeneration. Springer, Dordrecht; Netherlands: 2008. pp. 107–144. [Google Scholar]
- 46.Day K, Paterson B, Yablonka-Reuveni Z. A distinct profile of myogenic regulatory factor detection within Pax7+ cells at S phase supports a unique role of Myf5 during posthatch chicken myogenesis. Dev Dyn. 2009;238:1001–1009. doi: 10.1002/dvdy.21903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sacco A, Doyonnas R, Kraft P, Vitorovic S, Blau HM. Self-renewal and expansion of single transplanted muscle stem cells. Nature. 2008;456:502–506. doi: 10.1038/nature07384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zammit PS, Heslop L, Hudon V, Rosenblatt JD, Tajbakhsh S, Buckingham ME, Beauchamp JR, Partridge TA. Kinetics of myoblast proliferation show that resident satellite cells are competent to fully regenerate skeletal muscle fibers. Exp Cell Res. 2002;281:39–49. doi: 10.1006/excr.2002.5653. [DOI] [PubMed] [Google Scholar]
- 49.Collins CA, Zammit PS, Ruiz AP, Morgan JE, Partridge TA. A population of myogenic stem cells that survives skeletal muscle aging. Stem Cells. 2007;25:885–894. doi: 10.1634/stemcells.2006-0372. [DOI] [PubMed] [Google Scholar]
- 50.Thompson LV. Age-related muscle dysfunction. Exp Gerontol. 2009;44:106–111. doi: 10.1016/j.exger.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grounds MD. Age-associated changes in the response of skeletal muscle cells to exercise and regeneration. Ann N Y Acad Sci. 1998;854:78–91. doi: 10.1111/j.1749-6632.1998.tb09894.x. [DOI] [PubMed] [Google Scholar]
- 52.Carlson BM, Faulkner JA. Muscle transplantation between young and old rats: age of host determines recovery. Am J Physiol. 1989;256:C1262–1266. doi: 10.1152/ajpcell.1989.256.6.C1262. [DOI] [PubMed] [Google Scholar]
- 53.Aguennouz M, Vita GL, Messina S, Cama A, Lanzano N, Ciranni A, Rodolico C, Di Giorgio RM, Vita G. Telomere shortening is associated to TRF1 and PARP1 overexpression in Duchenne muscular dystrophy. Neurobiol Aging. 2010 Feb 4; doi: 10.1016/j.neurobiolaging.2010.01.008. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 54.Blau HM, Webster C, Pavlath GK. Defective myoblasts identified in Duchenne muscular dystrophy. Proc Natl Acad Sci U S A. 1983;80:4856–4860. doi: 10.1073/pnas.80.15.4856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Webster C, Blau HM. Accelerated age-related decline in replicative life-span of Duchenne muscular dystrophy myoblasts: implications for cell and gene therapy. Somat Cell Mol Genet. 1990;16:557–565. doi: 10.1007/BF01233096. [DOI] [PubMed] [Google Scholar]
- 56.Charge SB, Rudnicki MA. Cellular and molecular regulation of muscle regeneration. Physiol Rev. 2004;84:209–238. doi: 10.1152/physrev.00019.2003. [DOI] [PubMed] [Google Scholar]
- 57.Yablonka-Reuveni Z, Day K, Vine A, Shefer G. Defining the transcriptional signature of skeletal muscle stem cells. J Anim Sci. 2008;86:E207–216. doi: 10.2527/jas.2007-0473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Olguin HC, Yang Z, Tapscott SJ, Olwin BB. Reciprocal inhibition between Pax7 and muscle regulatory factors modulates myogenic cell fate determination. J Cell Biol. 2007;177:769–779. doi: 10.1083/jcb.200608122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Collins CA, Gnocchi VF, White RB, Boldrin L, Perez-Ruiz A, Relaix F, Morgan JE, Zammit PS. Integrated functions of Pax3 and Pax7 in the regulation of proliferation, cell size and myogenic differentiation. PLoS One. 2009;4:e4475. doi: 10.1371/journal.pone.0004475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lepper C, Conway SJ, Fan CM. Adult satellite cells and embryonic muscle progenitors have distinct genetic requirements. Nature. 2009;460:627–631. doi: 10.1038/nature08209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Andres V, Walsh K. Myogenin expression, cell cycle withdrawal, and phenotypic differentiation are temporally separable events that precede cell fusion upon myogenesis. J Cell Biol. 1996;132:657–666. doi: 10.1083/jcb.132.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kastner S, Elias MC, Rivera AJ, Yablonka-Reuveni Z. Gene expression patterns of the fibroblast growth factors and their receptors during myogenesis of rat satellite cells. J Histochem Cytochem. 2000;48:1079–1096. doi: 10.1177/002215540004800805. [DOI] [PubMed] [Google Scholar]
- 63.Yablonka-Reuveni Z, Rudnicki MA, Rivera AJ, Primig M, Anderson JE, Natanson P. The transition from proliferation to differentiation is delayed in satellite cells from mice lacking MyoD. Dev Biol. 1999;210:440–455. doi: 10.1006/dbio.1999.9284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Clegg CH, Linkhart TA, Olwin BB, Hauschka SD. Growth factor control of skeletal muscle differentiation: commitment to terminal differentiation occurs in G1 phase and is repressed by fibroblast growth factor. J Cell Biol. 1987;105:949–956. doi: 10.1083/jcb.105.2.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yablonka-Reuveni Z, Balestreri TM, Bowen-Pope DF. Regulation of proliferation and differentiation of myoblasts derived from adult mouse skeletal muscle by specific isoforms of PDGF. J Cell Biol. 1990;111:1623–1629. doi: 10.1083/jcb.111.4.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yablonka-Reuveni Z, Rivera AJ. Influence of PDGF-BB on proliferation and transition through the MyoD-myogenin-MEF2A expression program during myogenesis in mouse C2 myoblasts. Growth Factors. 1997;15:1–27. doi: 10.3109/08977199709002109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Graves DC, Yablonka-Reuveni Z. Vascular smooth muscle cells spontaneously adopt a skeletal muscle phenotype: a unique Myf5(−)/MyoD(+) myogenic program. J Histochem Cytochem. 2000;48:1173–1193. doi: 10.1177/002215540004800902. [DOI] [PubMed] [Google Scholar]
- 68.Kwiatkowski BA, Kirillova I, Richard RE, Israeli D, Yablonka-Reuveni Z. FGFR4 and its novel splice form in myogenic cells: Interplay of glycosylation and tyrosine phosphorylation. J Cell Physiol. 2008;215:803–817. doi: 10.1002/jcp.21365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yablonka-Reuveni Z, Anderson JE. Satellite cells from dystrophic (mdx) mice display accelerated differentiation in primary cultures and in isolated myofibers. Dev Dyn. 2006;235:203–212. doi: 10.1002/dvdy.20602. [DOI] [PubMed] [Google Scholar]
- 70.Shefer G, Yablonka-Reuveni Z. Isolation and culture of skeletal muscle myofibers as a means to analyze satellite cells. Methods Mol Biol. 2005;290:281–304. doi: 10.1385/1-59259-838-2:281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Keire P, Shearer A, Shefer G, Yablonka-Reuveni Z. Isolation and culture of skeletal muscle myofibers as a means to analyze satellite cells. Methods Mol Biol ('Basic Cell Culture Protocols', 4th edition) 2012 doi: 10.1007/978-1-62703-128-8_28. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yablonka-Reuveni Z. Isolation and culture of myogenic stem cells. In: Lanza R, Blau D, Melton D, Moore M, Thomas ED, Verfaillie C, Weissman I, West M, editors. Handbook of Stem Cells - Vol 2: Adult and Fetal Stem Cells. Elsevier: Academic Press; San Diego: 2004. pp. 571–580. [Google Scholar]
- 73.Richler C, Yaffe D. The in vitro cultivation and differentiation capacities of myogenic cell lines. Dev Biol. 1970;23:1–22. doi: 10.1016/s0012-1606(70)80004-5. [DOI] [PubMed] [Google Scholar]
- 74.Rando TA, Blau HM. Primary mouse myoblast purification, characterization, and transplantation for cell-mediated gene therapy. J Cell Biol. 1994;125:1275–1287. doi: 10.1083/jcb.125.6.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Qu-Petersen Z, Deasy B, Jankowski R, Ikezawa M, Cummins J, Pruchnic R, Mytinger J, Cao B, Gates C, Wernig A, Huard J. Identification of a novel population of muscle stem cells in mice: potential for muscle regeneration. J Cell Biol. 2002;157:851–864. doi: 10.1083/jcb.200108150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yablonka-Reuveni Z, Quinn LS, Nameroff M. Isolation and clonal analysis of satellite cells from chicken pectoralis muscle. Dev Biol. 1987;119:252–259. doi: 10.1016/0012-1606(87)90226-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yablonka-Reuveni Z, Nameroff M. Skeletal muscle cell populations. Separation and partial characterization of fibroblast-like cells from embryonic tissue using density centrifugation. Histochemistry. 1987;87:27–38. doi: 10.1007/BF00518721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Morgan JE. Myogenicity in vitro and in vivo of mouse muscle cells separated on discontinuous Percoll gradients. J Neurol Sci. 1988;85:197–207. doi: 10.1016/0022-510x(88)90156-6. [DOI] [PubMed] [Google Scholar]
- 79.Yablonka-Reuveni Z. Discrimination of myogenic and nonmyogenic cells from embryonic skeletal muscle by 90 degrees light scattering. Cytometry. 1988;9:121–125. doi: 10.1002/cyto.990090204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yablonka-Reuveni Z. Application of density centrifugation and flow cytometry for the isolation and characterization of myogenic and fibroblast-like cells from skeletal muscle. In: Kedes LH, editor. Cellular and Molecular Biology of Muscle Development. John Wiley & Sons Inc; New York: 1989. pp. 869–879. [Google Scholar]
- 81.Ieronimakis N, Balasundaram G, Rainey S, Srirangam K, Yablonka-Reuveni Z, Reyes M. Absence of CD34 on murine skeletal muscle satellite cells marks a reversible state of activation during acute injury. PLoS One. 2010;5:e10920. doi: 10.1371/journal.pone.0010920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cerletti M, Jurga S, Witczak CA, Hirshman MF, Shadrach JL, Goodyear LJ, Wagers AJ. Highly efficient, functional engraftment of skeletal muscle stem cells in dystrophic muscles. Cell. 2008;134:37–47. doi: 10.1016/j.cell.2008.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tanaka KK, Hall JK, Troy AA, Cornelison DD, Majka SM, Olwin BB. Syndecan-4-expressing muscle progenitor cells in the SP engraft as satellite cells during muscle regeneration. Cell Stem Cell. 2009;4:217–225. doi: 10.1016/j.stem.2009.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Uezumi A, Fukada S, Yamamoto N, Takeda S, Tsuchida K. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat Cell Biol. 2010;12:143–152. doi: 10.1038/ncb2014. [DOI] [PubMed] [Google Scholar]
- 85.Bosnakovski D, Xu Z, Li W, Thet S, Cleaver O, Perlingeiro RC, Kyba M. Prospective isolation of skeletal muscle stem cells with a Pax7 reporter. Stem Cells. 2008;26:3194–3204. doi: 10.1634/stemcells.2007-1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Biressi S, Tagliafico E, Lamorte G, Monteverde S, Tenedini E, Roncaglia E, Ferrari S, Ferrari S, Cusella-De Angelis MG, Tajbakhsh S, Cossu G. Intrinsic phenotypic diversity of embryonic and fetal myoblasts is revealed by genome-wide gene expression analysis on purified cells. Dev Biol. 2007;304:633–651. doi: 10.1016/j.ydbio.2007.01.016. [DOI] [PubMed] [Google Scholar]
- 87.Christov C, Chretien F, Abou-Khalil R, Bassez G, Vallet G, Authier FJ, Bassaglia Y, Shinin V, Tajbakhsh S, Chazaud B, Gherardi RK. Muscle satellite cells and endothelial cells: close neighbors and privileged partners. Mol Biol Cell. 2007;18:1397–1409. doi: 10.1091/mbc.E06-08-0693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gayraud-Morel B, Chretien F, Flamant P, Gomes D, Zammit PS, Tajbakhsh S. A role for the myogenic determination gene Myf5 in adult regenerative myogenesis. Dev Biol. 2007;312:13–28. doi: 10.1016/j.ydbio.2007.08.059. [DOI] [PubMed] [Google Scholar]
- 89.Joe AW, Yi L, Natarajan A, Le Grand F, So L, Wang J, Rudnicki MA, Rossi FM. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat Cell Biol. 2010;12:153–163. doi: 10.1038/ncb2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kanisicak O, Mendez JJ, Yamamoto S, Yamamoto M, Goldhamer DJ. Progenitors of skeletal muscle satellite cells express the muscle determination gene, MyoD. Dev Biol. 2009;332:131–141. doi: 10.1016/j.ydbio.2009.05.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kuang S, Kuroda K, Le Grand F, Rudnicki MA. Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell. 2007;129:999–1010. doi: 10.1016/j.cell.2007.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schienda J, Engleka KA, Jun S, Hansen MS, Epstein JA, Tabin CJ, Kunkel LM, Kardon G. Somitic origin of limb muscle satellite and side population cells. Proc Natl Acad Sci U S A. 2006;103:945–950. doi: 10.1073/pnas.0510164103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jinno H, Morozova O, Jones KL, Biernaskie JA, Paris M, Hosokawa R, Rudnicki MA, Chai Y, Rossi F, Marra MA, Miller FD. Convergent Genesis of an Adult Neural Crest-Like Dermal Stem Cell From Distinct Developmental Origins. Stem Cells. 2010 Sep 16; doi: 10.1002/stem.525. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Seale P, Bjork B, Yang W, Kajimura S, Chin S, Kuang S, Scime A, Devarakonda S, Conroe HM, Erdjument-Bromage H, Tempst P, Rudnicki MA, Beier DR, Spiegelman BM. PRDM16 controls a brown fat/skeletal muscle switch. Nature. 2008;454:961–967. doi: 10.1038/nature07182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gensch N, Borchardt T, Schneider A, Riethmacher D, Braun T. Different autonomous myogenic cell populations revealed by ablation of Myf5-expressing cells during mouse embryogenesis. Development. 2008;135:1597–1604. doi: 10.1242/dev.019331. [DOI] [PubMed] [Google Scholar]
- 96.Harel I, Nathan E, Tirosh-Finkel L, Zigdon H, Guimaraes-Camboa N, Evans SM, Tzahor E. Distinct origins and genetic programs of head muscle satellite cells. Dev Cell. 2009;16:822–832. doi: 10.1016/j.devcel.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yablonka-Reuveni Z, Paterson BM. MyoD and myogenin expression patterns in cultures of fetal and adult chicken myoblasts. J Histochem Cytochem. 2001;49:455–462. doi: 10.1177/002215540104900405. [DOI] [PubMed] [Google Scholar]
- 98.Kawakami A, Kimura-Kawakami M, Nomura T, Fujisawa H. Distributions of PAX6 and PAX7 proteins suggest their involvement in both early and late phases of chick brain development. Mech Dev. 1997;66:119–130. doi: 10.1016/s0925-4773(97)00097-x. [DOI] [PubMed] [Google Scholar]
- 99.Dias P, Parham DM, Shapiro DN, Tapscott SJ, Houghton PJ. Monoclonal antibodies to the myogenic regulatory protein MyoD1: epitope mapping and diagnostic utility. Cancer Res. 1992;52:6431–6439. [PubMed] [Google Scholar]
- 100.Wright WE, Binder M, Funk W. Cyclic amplification and selection of targets (CASTing) for the myogenin consensus binding site. Mol Cell Biol. 1991;11:4104–4110. doi: 10.1128/mcb.11.8.4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wright WE, Dac-Korytko I, Farmer K. Monoclonal antimyogenin antibodies define epitopes outside the bHLH domain where binding interferes with protein-protein and protein-DNA interactions. Dev Genet. 1996;19:131–138. doi: 10.1002/(SICI)1520-6408(1996)19:2<131::AID-DVG4>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 102.Bader D, Masaki T, Fischman DA. Immunochemical analysis of myosin heavy chain during avian myogenesis in vivo and in vitro. J Cell Biol. 1982;95:763–770. doi: 10.1083/jcb.95.3.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Narahashi Y, Yanagita M. Studies on proteolytic enzymes (pronase) of Streptomyces griseus K-1. I. Nature and properties of the proteolytic enzyme system. J Biochem. 1967;62:633–641. doi: 10.1093/oxfordjournals.jbchem.a128718. [DOI] [PubMed] [Google Scholar]
- 104.Narahashi Y. Pronase. Methods Enzymol. 1970;19:651–664. [Google Scholar]
- 105.Hartley RS, Yablonka-Reuveni Z. Long-term maintenance of primary myogenic cultures on a reconstituted basement membrane. In Vitro Cell Dev Biol. 1990;26:955–961. doi: 10.1007/BF02624469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gilbert PM, Havenstrite KL, Magnusson KE, Sacco A, Leonardi NA, Kraft P, Nguyen NK, Thrun S, Lutolf MP, Blau HM. Substrate elasticity regulates skeletal muscle stem cell self-renewal in culture. Science. 2010;329:1078–1081. doi: 10.1126/science.1191035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kleinman HK, McGarvey ML, Liotta LA, Robey PG, Tryggvason K, Martin GR. Isolation and characterization of type IV procollagen, laminin, and heparan sulfate proteoglycan from the EHS sarcoma. Biochemistry. 1982;21:6188–6193. doi: 10.1021/bi00267a025. [DOI] [PubMed] [Google Scholar]
- 108.Greene EC. Anatomy of the rat. Hafner Publishing Company; New York, NY: 1963. [Google Scholar]
- 109.Hebel R, Stromberg MW. Anatomy of the laboratory rat. Williams & Wilkins; Baltimore: 1976. [Google Scholar]
- 110.Yablonka-Reuveni Z. Myogenesis in the chicken: the onset of differentiation of adult myoblasts is influenced by tissue factors. Basic Appl. Myology (BAM) 1995;5:33–42. [PMC free article] [PubMed] [Google Scholar]
- 111.O'Neill MC, Stockdale FE. A kinetic analysis of myogenesis in vitro. J Cell Biol. 1972;52:52–65. doi: 10.1083/jcb.52.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]