

The inhibition of bacterial cell division offers a new approach to controlling resistant bacterial infections.[1, 2] FtsZ is a protein of central importance to cell division that is often described as the prokaryotic homolog of tubulin because of the structural similarity of these two proteins, in spite of low sequence homology.[3–5] Both proteins undergo GTP-driven oligomerization and the active sites of both proteins are similar. Tubulin forms microtubules in the bipolar spindle assembly, which mediates chromosome separation in eukaryotes. FtsZ oligomerizes at midcell during bacterial cell division to form the Z-ring, which constricts to induce septation. Inhibition of FtsZ has been validated as a potential new therapeutic strategy for fighting resistant infections, including MRSA.[6–8] In stark contrast to tubulin, few FtsZ-targeting natural products are known and no information regarding the molecular basis of their inhibition of FtsZ has been documented.[9–12] The development of efficient syntheses for FtsZ-targeting natural products will enable elucidation of their mechanisms of inhibition and enable further development of this target. This communication describes the synthesis of viriditoxin, one the most potent FtsZ-targeting natural products and the first 6,6'-binapthopyranone to be synthesized.[13]
Viriditoxin was discovered as an FtsZ inhibitor by researchers at Merck in a high-throughput biochemical screen.[14] The 6,6'-binaphthopyranone structure, previously isolated from Aspergillus viridinutans, (Scheme 1) was originally misassigned as having 8-8' linkage like that of vioxanthin (6a, Scheme 1).[15–17] 6,6'-Binaphthopyran-2-ones are uncommon natural products and so far only four others have been reported.[18–20] A small number of the biosynthetically related binaphthopyran-4-ones, such as cephalochromin (6b, Scheme 1), have also been reported.[21–25] Although the absolute stereochemistry at C3 of semiviriditoxin was recently confirmed by synthesis,[26] the axial configuration of viriditoxin was not known unequivocally at the outset of our studies
Scheme 1.

A) Structures of viriditoxin, semiviriditoxin and semi-vioxanthin; retrosynthesis of viriditoxin. B) Structures of vioxanthin (6a), an example of an 8,8'-binaphthopyran-2-one and cephalochromin (6b), an example 6,6'-binaphthopyran-4-one
The key bond construction in viriditoxin is the stereoselective formation of the 6-6' biaryl linkage. Strategies for achieving atropselectivity in the construction of complex molecules have been recently reviewed.[27, 28] Subsequent recent examples for asymmetric biaryl construction often employ auxillary- and catalyst-controlled oxidative homo-coupling[29–31] and cross-coupling reactions.[32–35] In two complementary approaches, atropselectivity can also be realized by the asymmetric de novo assembly of aromatic rings in cycloadditions[36–41] and by dynamic kinetic resolution polycyclic lactones through aminolysis[42] or reduction.[43] or reduction.[43] Most of these strategies rely on either the influence of chiral catalysts or proximal stereogenic centers in the substrate or covalently-linked chiral auxiliary. We set out to explore the influence of remote stereogenic centers, i.e. those located more than three bonds from the reacting aromatic carbon center, in the catalytic oxidative dimerization of naphthols. Two previous examples of similarly remote influence in the oxidative dimerization of arylcuprate intermediates have been reported in studies leading to the synthesis of phleichrome and related perylenequnione natural products.[44, 45]
Several oxidation catalysts have been employed in the dimerization of naphthols and the vanadium-catalyzed process reported by Uang was best suited to the substitution pattern of4.[46] Two diastereomeric transition states (A and B, Scheme 2) could lead to the two possible isomers of biaryl intermediate 4. Although the mechanism of this reaction is unclear, several studies suggest that the active catalyst might be bi-metallic,[47] possibly a μ-oxo dimer,[48] albeit of undefined oxidation state and ligation. Under this assumption, the naphthopyranone rings could be expected to react in an anti-parallel fashion in which the stereogenic centers of the pyranone ring would be forced into proximity. This ring system favors the equatorial conformation (shown), which forces an interaction between either the axial hydrogen atoms (A) or the equatorial alkyl substituents (B). These two transition structures lead to the (M)/(Ra) or (P)/(Sa), isomers of binaphthopyranone 7, the latter of which leads to the proposed configuration of viriditoxin.
Scheme 2.

Remote asymmetric induction in the dimerization of 4.
A suitable tricyclic precursor for viriditoxin was prepared from orsellinic acid derivative 9 and pyranone 12, each of which was available in two steps from known compounds (Scheme 3).[49–52] A Michael addition-Dieckman condensation sequence was employed to yield naphthopyranone 13 after subsequent oxidation and methylation. Although this route is ostensibly less efficient than the Staunton-Weinreb condensation of the corresponding β-alkoxy pyranone,[17, 53, 54] we observed significantly higher yields in the two-step process[55, 56] The ethoxymethyl (EOM) ether was readily cleaved by propylene glycol providing the dimerization precursor 14.[57] When 14 was treated with 20 mol% of VO(acac)2, a rapid reaction ensued, producing a single regioisomer of desired product 15 with respectable diastereoselection favoring proposed transition state B (Scheme 2). This is one of the few cases of biaryl bond formation in which appreciable levels of stereocontrol are induced by a distal stereogenic center of the substrate without concomitant ring-formation.[44, 45] The stereochemistry of 15 was established though X-ray crystallography (Figure 1).
Scheme 3.

Stereoselective assembly of core of viriditoxin.
LDA=lithium diisopropylamide; DMPU = N,N'-Dimethylpropyleneurea; DDQ = 2,3-dichloro-5,6-dicyanobenzoquinone; DMS = dimethylsulfide
Figure 1.

X-ray crystal structure of 15.
In order to enhance the atropselectivity of the biaryl bond-formation, we explored the possibility of double diastereo-differentiation[58] induced by a chiral vanadium catalyst. Gong has previously demonstrated that BINOL-derived bimetallic vanadium catalysts exhibit appreciable levels of enantioselectivity in the oxidative dimerization of naphthols.[48] We prepared four catalysts derived from R- and S-BINOL and D- and L-valine (Scheme 4).
Scheme 4.

Chiral vanadium catalysts and variously substituted naphthopyranones for evaluating substrate scope.
Although substrate 14 is central to the goal of preparing viriditoxin, we wanted to explore the scope of diastereoselectivity enabled by this new remotely induced axial chirality. Pyranones 17–19, prepared in three steps from the requisite chiral epoxides, were converted to naphthopyranones 20–22 in analogy to 14 (Scheme 4).
The chiral bimetallic vanadium catalysts exhibited superior diastereoselectivity and reactivity (eq 1, Table 1). Naphthopyranone 14 was treated with all four isomers of Gong-type catalyst 16 and showed pair-wise matched and mismatched selectivity. Specifically, (Sa, R)-16 produced the desired isomer of 15 with selectivity that was enhanced to 89:11, (Table 1, entry 4) whereas the catalyst differing only in the amino acid configuration reversed the selectivity to 12:88 (Table 1, entry 5). The isomeric catalysts derived from R-BINOL were mis-matched and showed the same sense of induction controlled by the amino acid with lesser degrees of induction. A similar trend was observed for the other substrates for which the selectivity of VO(acac)2 was modest and the choice of Gong- type catalysts could produce either isomer as the major product. In addition, the bimetallic catalysts generally provided higher yields than VO(acac)2.
Table 1.
Double diastereo-differentiating oxidative couplings with chiral bimetallic vanadium catalysts.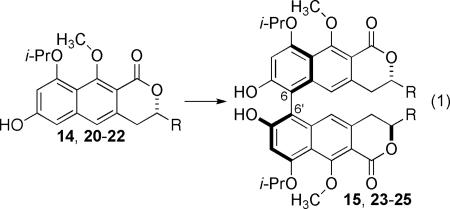
| entry | substrate (R) | product | catalyst | dr (yield) |
|---|---|---|---|---|
| 1 | 14 (CH2CH2OTIPS) | 15 | VO(acac)2 | 76:24 (67%) |
| 2 | 14 | 15 | (Ra,S)-18 | 19:81 |
| 3 | 14 | 15 | (Ra,R)-18 | 82:18 |
| 4 | 14 | 15 | (Sa,S)-18 | 12:88 |
| 5 | 14 | 15 | (Sa,R)-18 | 89:11 (87%) |
| 6 | 20 (CH3) | 23 | VO(acac)2 | 60:40 (90%) |
| 7 | 20 | 23 | (Sa,R)-18 | 90:10 (85%) |
| 8 | 20 | 23 | (Sa,S)-18 | 18:82 (57%) |
| 9 | 21 (CH2OTIPS) | 24 | VO(acac)2 | 61:39 (68%) |
| 10 | 21 | 24 | (Sa,R)-18 | 96:04 (84%) |
| 11 | 21 | 24 | (Sa,S)-18 | 30:70 (76%) |
| 12 | 22 (c-C6H11) | 25 | VO(acac)2 | 56:44 (78%) |
| 13 | 22 | 25 | (Sa,R)-18 | 83:17 (67%) |
| 14 | 22 | 25 | (Sa,S)-18 | 11:89 (74%) |
[a] Diastereomer rations determined from 1H NMR spectra of crude reaction mixtures. [b] Axial configuration determined by X-ray crystallography (15) and chemical shift comparison (23–25), see supporting information. [c] All reactions performed on a 10 mg scale. A larger scale run (70 mg) for entry 7 proceeded in identical isolated yield.
TIPS = triisoproylsilyl
The synthesis of viriditoxin was completed in five steps (eq 2). The 7 and 7' hydroxyl groups of 15 were methylated with dimethyl sulfate followed by removal of the TIPS protecting groups. The resultant diol was oxidized to the corresponding diacid and converted to the requisite diester in 57% overall yield. The 9,9'-isopropyl and 10,10'-methyl ethers were cleaved readily with BCl3 to yield (−)-viriditoxin. The synthetic material exhibited NMR spectra (1H and 13C) that compared favourably with reported values.[59] Synthetic viriditoxin was thus produced in a longest linear sequence of 12 steps from the alkene precursor of 10.[60]
In conclusion, we have described the first synthesis of a 6,6'-binaphthopyranone natural product and, in so doing, established the relative configuration of viriditoxin. This synthesis represents a general route to related natural products[18, 20] and it will enable the l products exploration of the interactions that result in inhibition of the bacterial cell division protein FtsZ.
Supplementary Material
Figure 2.

Acknowledgments
This research was supported by start-up funds from the University of California, Davis, the Petroleum Research Fund (administered by the ACS), and the NIH/NIAID (R56AI80931-01 & R01AI080931-01). This work was initiated at the Broad Institute of Harvard and MIT, where it was supported by the NIH/NIAID (R03 AI062905-01) and the Scientific Planning and Allocation of Resources Committee (SPARC). MGL thanks the Fundación Ramon Areces for a postdoctoral fellowship. The authors thank Prof. Jon Clardy (Harvard Medical School) for helpful discussions, Prof. Dean Tantillo for DFT calculations and Dr. Sheo Singh (Merck Research Laboratories) for providing copies of NMR spectra for viriditoxin.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org.
References
- [1].Lock RL, Harry EJ. Nat. Rev. Drug Discovery. 2008;7:324. doi: 10.1038/nrd2510. [DOI] [PubMed] [Google Scholar]
- [2].Callaway E. Nature (London, U. K.) 2008;451:124. doi: 10.1038/451124a. [DOI] [PubMed] [Google Scholar]
- [3].Romberg L, Levin PA. Ann. Rev. Microbiol. 2003;57:125. doi: 10.1146/annurev.micro.57.012903.074300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Dajkovic A, Lutkenhaus J. J. Mol. Microbiol. Biotechnol. 2006;11:140. doi: 10.1159/000094050. [DOI] [PubMed] [Google Scholar]
- [5].Margolin W. Nat. Rev. Mol. Cell Biol. 2005;6:862. doi: 10.1038/nrm1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Haydon DJ, Stokes NR, Ure R, Galbraith G, Bennett JM, Brown DR, Baker PJ, Barynin VV, Rice DW, Sedelnikova SE, Heal JR, Sheridan JM, Aiwale ST, Chauhan PK, Srivastava A, Taneja A, Collins I, Errington J, Czaplewski LG. Science (Washington, DC, U. S.) 2008;321:1673. doi: 10.1126/science.1159961. [DOI] [PubMed] [Google Scholar]
- [7].Czaplewski LG, Collins I, Boyd EA, Brown D, East SP, Gardiner M, Fletcher R, Haydon DJ, Henstock V, Ingram P, Jones C, Noula C, Kennison L, Rockley C, Rose V, Thomaides-Brears HB, Ure R, Whittaker M, Stokes NR. Bioorg. Med. Chem. Lett. 2009;19:524. doi: 10.1016/j.bmcl.2008.11.021. [DOI] [PubMed] [Google Scholar]
- [8].Haydon DJ, Bennett JM, Brown D, Collins I, Galbraith G, Lancett P, Macdonald R, Stokes NR, Chauhan PK, Sutariya JK, Nayal N, Srivastava A, Beanland J, Hall R, Henstock V, Noula C, Rockley C, Czaplewski L. J. Med. Chem. 2010;53:3927. doi: 10.1021/jm9016366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Urgaonkar S, La Pierre HS, Meir I, Lund H, RayChaudhuri D, Shaw JT. Org. Lett. 2005;7:5609. doi: 10.1021/ol052269z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Jaiswal R, Beuria TK, Mohan R, Mahajan SK, Panda D. Biochemistry. 2007;46:4211. doi: 10.1021/bi602573e. [DOI] [PubMed] [Google Scholar]
- [11].Domadia PN, Bhunia A, Sivaraman J, Swarup S, Dasgupta D. Biochemistry. 2008;47:3225. doi: 10.1021/bi7018546. [DOI] [PubMed] [Google Scholar]
- [12].Kanoh K, Adachi K, Matsuda S, Shizuri Y, Yasumoto K, Kusumi T, Okumura K, Kirikae T. J. Antibiot. 2008;61:192. doi: 10.1038/ja.2008.29. [DOI] [PubMed] [Google Scholar]
- [13].Vollmer W. Appl. Microbiol. Biotechnol. 2006;73:37. doi: 10.1007/s00253-006-0586-0. [DOI] [PubMed] [Google Scholar]
- [14].Wang J, Galgoci A, Kodali S, Herath KB, Jayasuriya H, Dorso K, Vicente F, Gonzalez A, Cully D, Bramhill D, Singh S. J. Biol. Chem. 2003;278:44424. doi: 10.1074/jbc.M307625200. [DOI] [PubMed] [Google Scholar]
- [15].Weisleder D, Lillehoj EB. Tetrahedron Lett. 1971;4705.
- [16].Suzuki K, Nozawa K, Nakajima S, Kawai K. Chem. Pharm. Bull. 1990;38:3180. doi: 10.1248/cpb.38.73. [DOI] [PubMed] [Google Scholar]
- [17].Bode SE, Drochner D, Mueller M. Angew. Chem., Int. Ed. 2007;46:5916. doi: 10.1002/anie.200701014. [DOI] [PubMed] [Google Scholar]
- [18].Suzuki K, Nozawa K, Nakajima S, Udagawa S, Kawai K. Chem. Pharm. Bull. 1992;40:1116. doi: 10.1248/cpb.40.1116. [DOI] [PubMed] [Google Scholar]
- [19].Arone A, Assante G, Montorsi M, Nasini G. Phytochemistry. 1995;38:595. [Google Scholar]
- [20].Elix JA, Wardlaw JH. Aust. J. Chem. 2004;57:681. [Google Scholar]
- [21].Shibata S, Ogihara Y. Chem. Pharm. Bull. 1963;11:1576. doi: 10.1248/cpb.11.1576. [DOI] [PubMed] [Google Scholar]
- [22].Shibata S, Ohta A, Ogihara Y. Chem. Pharm. Bull. 1963;11:1174. doi: 10.1248/cpb.11.1174. [DOI] [PubMed] [Google Scholar]
- [23].Matsumoto M, Minato H, Kondo E, Mitsugi T, Katagiri K. J. Antibiot. 1975;28:602. doi: 10.7164/antibiotics.28.602. [DOI] [PubMed] [Google Scholar]
- [24].Koyama K, Natori S, Iitaka Y. Chem. Pharm. Bull. 1987;35:4049. doi: 10.1248/cpb.35.578. [DOI] [PubMed] [Google Scholar]
- [25].Kock I, Draeger S, Schulz B, Elsaesser B, Kurtan T, Kenez A, Antus S, Pescitelli G, Salvadori P, Speakman J-B, Rheinheimer J, Krohn K. Eur. J. Org. Chem. 2009;1427 [Google Scholar]
- [26].Tan NPH, Donner CD. Tetrahedron. 2009;65:4007. [Google Scholar]
- [27].Bringmann G, Mortimer AJP, Keller PA, Gresser MJ, Garner J, Breuning M. Angew. Chem., Int. Ed. 2005;44:5384. doi: 10.1002/anie.200462661. [DOI] [PubMed] [Google Scholar]
- [28].Kozlowski MC, Morgan BJ, Linton EC. Chem. Soc. Rev. 2009;38:3193. doi: 10.1039/b821092f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Chen W-W, Zhao Q, Xu M-H, Lin G-Q. Org. Lett. 2010;12:1072. doi: 10.1021/ol1000632. [DOI] [PubMed] [Google Scholar]
- [30].Chen Q-A, Dong X, Chen M-W, Wang D-S, Zhou Y-G, Li Y-X. Org. Lett. 2010;12:1928. doi: 10.1021/ol100536e. [DOI] [PubMed] [Google Scholar]
- [31].Morgan BJ, Mulrooney CA, O'Brien EM, Kozlowski MC. J. Org. Chem. 2010;75:30. doi: 10.1021/jo901384h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Baudoin O. Eur. J. Org. Chem. 2005;4223 [Google Scholar]
- [33].Joncour A, Decor A, Liu J-M, Dau M-ETH, Baudoin O. Chem. Eur. J. 2007;13:5450. doi: 10.1002/chem.200601764. [DOI] [PubMed] [Google Scholar]
- [34].Colobert F, Valdivia V, Choppin S, Leroux FR, Fernandez I, Alvarez E, Khiar N. Org. Lett. 2009;11:5130. doi: 10.1021/ol9020755. [DOI] [PubMed] [Google Scholar]
- [35].Shen X, Jones GO, Watson DA, Bhayana B, Buchwald SL. J. Am. Chem. Soc. 2010;132:11278. doi: 10.1021/ja104297g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Nishida G, Suzuki N, Noguchi K, Tanaka K. Org. Lett. 2006;8:3489. doi: 10.1021/ol0611550. [DOI] [PubMed] [Google Scholar]
- [37].Nishida G, Noguchi K, Hirano M, Tanaka K. Angew. Chem., Int. Ed. 2007;46:3951. doi: 10.1002/anie.200700064. [DOI] [PubMed] [Google Scholar]
- [38].Liu Y, Lu K, Dai M, Wang K, Wu W, Chen J, Quan J, Yang Z. Org. Lett. 2007;9:805. doi: 10.1021/ol063013b. [DOI] [PubMed] [Google Scholar]
- [39].Shibata T, Yoshida S, Arai Y, Otsuka M, Endo K. Tetrahedron. 2007;64:821. [Google Scholar]
- [40].Tanaka K. Chem.--Asian J. 2009;4:508. doi: 10.1002/asia.200800378. [DOI] [PubMed] [Google Scholar]
- [41].Hapke M, Kral K, Fischer C, Spannenberg A, Gutnov A, Redkin D, Heller B. J. Org. Chem. 75:3993. doi: 10.1021/jo100122d. [DOI] [PubMed] [Google Scholar]
- [42].Bringmann G, Scharl H, Maksimenka K, Radacki K, Braunschweig H, Wich P, Schmuck C. Eur. J. Org. Chem. 2006;4349 [Google Scholar]
- [43].Ashizawa T, Tanaka S, Yamada T. Org. Lett. 2008;10:2521. doi: 10.1021/ol800802c. [DOI] [PubMed] [Google Scholar]
- [44].Broka CA. Tetrahedron Lett. 1991;32:859. [Google Scholar]
- [45].Coleman RS, Grant EB. J. Am. Chem. Soc. 1995;117:10889. [Google Scholar]
- [46].Hwang D-R, Chen C-P, Uang B-J. Chem. Commun. 1999;1207 [Google Scholar]
- [47].Tohma H, Morioka H, Takizawa S, Arisawa M, Kita Y. Tetrahedron. 2001;57:345. [Google Scholar]
- [48].Guo Q-X, Wu Z-J, Luo Z-B, Liu Q-Z, Ye J-L, Luo S-W, Cun L-F, Gong L-Z. J. Am. Chem. Soc. 2007;129:13927. doi: 10.1021/ja074322f. [DOI] [PubMed] [Google Scholar]
- [49].Barrett AGM, Morris TM, Barton DHR. J. Chem. Soc., Perkin Trans. 1980;1:2272. [Google Scholar]
- [50].Tangdenpaisal K, Sualek S, Ruchirawat S, Ploypradith P. Tetrahedron. 2009;65:4316. [Google Scholar]
- [51].Keck GE, Krishnamurthy D. Org. Synth. 1998;75:12. [Google Scholar]
- [52].Keck GE, Li X-Y, Knutson CE. Org. Lett. 1999;1:411. doi: 10.1021/ol990632u. [DOI] [PubMed] [Google Scholar]
- [53].Evans GE, Leeper FJ, Murphy JA, Staunton J. J. Chem. Soc., Chem. Commun. 1979;205 [Google Scholar]
- [54].Dodd JH, Weinreb SM. Tetrahedron Lett. 1979;3593 [Google Scholar]
- [55].Tatsuta K, Yamazaki T, Yoshimoto T. J. Antibiot. 1998;51:383. doi: 10.7164/antibiotics.51.383. [DOI] [PubMed] [Google Scholar]
- [56].White JD, Demnitz FWJ, Xu Q, Martin WHC. Org. Lett. 2008;10:2833. doi: 10.1021/ol8009732. [DOI] [PubMed] [Google Scholar]
- [57].Miyake H, Tsumura T, Sasaki M. Tetrahedron Lett. 2004;45:7213. [Google Scholar]
- [58].Huang S, Petersen TB, Lipshutz BH. J. Am. Chem. Soc. 2010;132:14021. doi: 10.1021/ja1065202. [DOI] [PubMed] [Google Scholar]
- [59].See supporting information for a copy of the 1HNMR spectrum supplied by Dr. Singh (Merck). Our observed optical rotation for viriditoxin [−118° and −110°, two different batches] is significantly lower than the original reported value of −202° (see reference 15) for which no experimental details are given. The high yield observed in the coupling step, i.e. the lack of heterodimer formation and a chiral HPLC trace of 28 (see supporting information) are both consistent with high optical purity.
- [60].Nicolaou KC, Lim YH, Piper JL, Papageorgiou CD. J. Am. Chem. Soc. 2007;129:4001. doi: 10.1021/ja0685708. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.