

Abstract
Transthyretin (TTR) tetramer dissociation and misfolding facilitate assembly into amyloid fibrils that putatively cause senile systemic amyloidosis and familial amyloid polyneuropathy. We have previously discovered more than 50 small molecules that bind to and stabilize tetrameric TTR, inhibiting amyloid fibril formation in vitro. A method is presented here to evaluate the binding selectivity of these inhibitors to TTR in human plasma, a complex biological fluid composed of more than 60 proteins and numerous small molecules. Our immunoprecipitation approach isolates TTR and bound small molecules from a biological fluid such as plasma, and quantifies the amount of small molecules bound to the protein by HPLC analysis. This approach demonstrates that only a small subset of the inhibitors that saturate the TTR binding sites in vitro do so in plasma. These selective inhibitors can now be tested in animal models of TTR amyloid disease to probe the validity of the amyloid hypothesis. This method could be easily extended to evaluate small molecule binding selectivity to any protein in a given biological fluid without the necessity of determining or guessing which other protein components may be competitors. This is a central issue to understanding the distribution, metabolism, activity, and toxicity of potential drugs.
Amyloid diseases are characterized by the conversion of soluble proteins or peptides into insoluble β-sheet-rich amyloid fibrils. There are currently 17 different human proteins known to form amyloid fibrils in vivo (1–4). These fibrils, or their oligomeric precursors, are thought to cause pathology either through disruption of normal cellular function or by direct toxicity (5–8). X-ray fibril diffraction and electron microscopy reconstruction of amyloid fibrils reveal filaments that have a lamellar cross β-sheet structure wrapped around one another (9–13). Folded proteins form amyloid fibrils through partial unfolding triggered by a change of local environment, a mutation in the protein, or both (8, 14–20).
Transthyretin (TTR) is a tetrameric protein composed of identical 127-aa subunits that fold into a β-sandwich tertiary structure. It is found in both the plasma (3.6 μM) and cerebrospinal fluid (CSF) (0.36 μM) of humans. The TTR tetramer has two negatively cooperative C2 symmetric thyroxine (T4)-binding sites (21–23). In the CSF, it binds and transports the thyroid hormone T4 and the retinol-binding protein (RBP), which in turn transports vitamin A. In the plasma, only 10–15% of TTR has T4 bound to it, as thyroid-binding globulin has an order of magnitude higher affinity for T4 than does TTR (24). TTR fibril formation is linked to two amyloid diseases in which the fibrils are composed of full-length protein. Deposition of wild-type TTR is associated with cardiac dysfunction in the disease senile systemic amyloidosis (SSA) (25, 26). More than 70 different single-site mutants have been linked to early-onset amyloid deposition in diseases with a spectrum of clinical manifestations, collectively referred to as familial amyloid polyneuropathy (FAP) (27–35).
We have discovered compounds, through both screening and structure-based design, that dramatically inhibit TTR amyloid fibril formation in vitro (36–42). To stabilize the TTR tetramer and thus prevent amyloid fibril formation in SSA and FAP, these small molecules must be able to selectively bind to TTR in human blood plasma over all other plasma proteins. Possible competitors include thyroid-binding globulin (TBG), which has an order of magnitude higher affinity for TTR's natural ligand, T4; and albumin, which binds numerous hydrophobic small molecules and is present at a concentration two orders of magnitude higher than TTR, as well as the other plasma proteins. Historically, one was forced to choose two or three of the most likely protein competitors and evaluate their relative affinities for the small molecule in comparison to the protein of interest. The advantage of the approach outlined within this article is that the binding selectivity of TTR amyloid inhibitors in human plasma is determined without having to make assumptions as to which proteins may competitively bind the TTR ligand. Compounds that bind to TTR selectively in plasma are the best candidates for further evaluation in animal models and, ultimately, in human clinical trials.
Materials and Methods
TTR Polyclonal Antibody Production.
Rabbits were injected with a 1:1 mixture of complete Freund's adjuvant and 1 mg/ml recombinant human TTR with an additional methionine at the N terminus. After 5 weeks, the rabbits were given boosters of 1:1 incomplete Freund's adjuvant/TTR (1 mg/ml) every 2 weeks for 2 months. Subsequently, the boosters were given once a month. Fifty milliliters of serum was drawn from each rabbit 30 days after each booster injection, and the blood serum was isolated.
TTR Antibody Purification and Conjugation to Sepharose.
Antibodies were isolated from rabbit serum by passage over a recombinant staphylococcal protein A column (Amersham Pharmacia Biotech). The column was washed with 5 column volumes of 50 mM sodium phosphate (pH 7.2), and the antibodies were eluted with 5 column volumes of 100 mM sodium citrate (pH 3.0). The elution fractions were returned to neutral pH with the addition of 1 ml of 1 M Tris⋅HCl (pH 9.0) to each 5-ml fraction. The fractions were pooled and exchanged into 100 mM sodium bicarbonate, pH 8.2. This solution was concentrated, and the polyclonal TTR antibodies were coupled to cyanogen bromide-activated Sepharose (Amersham Pharmacia Biotech) according to the manufacturer's protocol (43), yielding 10 mg of antibody per ml of gel. The gel was stored as a 1:1 slurry in TSA (10 mM Tris⋅HCl, pH 8.0/140 mM NaCl/0.025% NaN3). In addition, quenched Sepharose was prepared by coupling 200 mM Tris⋅HCl, pH 8.0, to the gel instead of the antibody.
Western Blot Analysis of TTR Antibodies.
Recombinant human TTR and 10-fold diluted human blood plasma were loaded onto a 12% polyacrylamide SDS gel and subjected to electrophoresis at 125 V. The proteins were electrotransferred to a nitrocellulose membrane at 100 V by using a Western Transfer Apparatus (Bio-Rad). The nitrocellulose was blocked with 5% dried milk (Carnation) in TBST (20 mM Tris⋅HCl, pH 7.5/500 mM NaCl/0.05% Tween-20) for 18 h and washed twice with TBST for 10 min. The membrane was incubated with a 1:1000 dilution of the antibody solution for 1 h at 25°C. The nitrocellulose was washed with TBST (three times, 10 min each), followed by an incubation with goat-anti-rabbit IgG antibody conjugated to alkaline phosphatase (Sigma) (1:1000 dilution in TBST) for 1 h at 25°C. The membrane was subsequently washed twice with TBST and once with TBS (20 mM Tris⋅HCl, pH 7.5/500 mM NaCl). Finally, the nitrocellulose membrane was incubated with 10 ml of BCIP/NBT (Sigma Fast 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium) solution until bands appeared, at which time the reaction was stopped with the addition of 5 ml of a 1 mM EDTA solution.
Human Plasma Preparation.
Whole blood was drawn from healthy volunteers at the Scripps General Clinical Research Center's Normal Blood Drawing Program and transferred to 50-ml conical tubes. The tubes were centrifuged at 3,000 rpm (1,730 × g) in a Sorvall RT7 benchtop centrifuge equipped with a swinging-bucket rotor for 10 min at 25°C. The plasma supernatant was removed and centrifuged again at 3,000 rpm for 10 min to remove the remaining cells. Sodium azide was added to give a 0.05% solution. The plasma was stored at 4°C until use.
ELISA Quantification of TTR Concentration in Human Plasma.
Anti-human prealbumin (TTR) antibody (DiaSorin, Stillwater, MN) was diluted 1:100 in 15 mM Na2CO3/35 mM NaHCO3, and 100 μl of the solution was added to each well of an Immulon-4HBX microtiter plate (Thermal Labsystems, Helsinki, Finland) which absorbed the protein after incubation overnight at 4°C. The plate was washed with 200 μl of 4× TBST, followed by the addition of 200 μl of blocking buffer (TBST, 3% dried milk) to each well, which was incubated for 60 min at 37°C. The plate was again washed four times with 200 μl of TBST, and 100 μl of TTR standards (10–100 ng/ml) and plasma (1:4000 dilution in blocking buffer) were loaded into different wells of the plate and incubated for 75 min at 37°C. The plate was subsequently washed four times with 200 μl of TBST. One hundred microliters of alkaline phosphatase-conjugated goat anti-human prealbumin (TTR) antibody (EY Laboratories) (1:250 dilution in blocking buffer) was added to each well and incubated for 75 min at 37°C. ELISA substrate solution from the GIBCO/BRL ELISA amplification system (Life Technologies) was added in 50-μl aliquots to each well and incubated for 15 min at room temperature. This incubation was followed by the addition of 50 μl of ELISA amplifier solution (Life Technologies), which was incubated for an additional 10–15 min at room temperature. The absorbance of each well at 495 nm was then measured by using a Molecular Dynamics Spectramax 340PC microplate spectrophotometer. The absorbances of the TTR standards were fit by linear regression using Softmax Pro 2.4 (Molecular Dynamics), and the amount of TTR in the samples was quantified on the basis of this equation.
Immunoprecipitation of TTR and Bound Small Molecule Inhibitors.
A 2-ml Eppendorf tube was filled with 1.5 ml of human blood plasma and 7.5 μl of a 2.16 mM DMSO solution of the small molecule inhibitor under evaluation. This solution was incubated on an orbital shaker at 60 rpm and 37°C for 24 h. A 1:1 gel/TSA slurry (187 μl) of quenched Sepharose was added to the solution and incubated on a rocker at 18 rpm and 4°C for 1 h. The solution was centrifuged (16,000 × g) and the supernatant was divided into three aliquots of 400 μl each. These were each added to 200 μl of a 1:1 gel/TSA slurry of the anti-TTR antibody-conjugated Sepharose and slowly rocked at 4°C for 20 min. The samples were centrifuged (16,000 × g) and the supernatant was removed. The gel was washed with 1 ml of TSA/0.05% saponin (Fisher Scientific) (three times, 10 min each) at 4°C, and additionally with 1 ml of TSA (twice, 10 min each) at 4°C. The samples were centrifuged (16,000 × g), the final wash was removed, and 155 μl of 100 mM triethylamine, pH 11.5, was added to elute the TTR and bound small molecules from the antibodies. After gentle rocking at 4°C for 30 min, the elution sample was centrifuged (16,000 × g) and 145 μl of the supernatant, containing TTR and inhibitor, was removed.
HPLC Analysis and Quantification of TTR and Bound Small Molecule Inhibitors.
The supernatant elution samples from the TTR antibody beads (145 μl) were loaded onto a Waters 71P autosampler. A 135-μl injection of each sample was separated on a Keystone 3-cm C18 reverse-phase column using a 40–100% gradient of solution B over 8 min (solution A: 94.8% water/5% acetonitrile/0.2% trifluoroacetic acid; solution B: 94.8% acetonitrile/5% water/0.2% trifluoroacetic acid), controlled by a Waters 600E multisolvent delivery system. A 20–100% B gradient over 8 min was used for compound 12 to separate its peak from the void volume (see Tables 1 and 2 for structures of compounds 1–15). Detection was accomplished at 280 nm with a Waters 486 tunable absorbance detector, and the peaks were integrated to give the area of both TTR and the small molecule. To determine the quantity of each species, known amounts of tetrameric TTR or small molecule were injected onto the HPLC. The peaks were integrated to create calibration curves from linear regressions of the data by using Kaleidagraph (Synergy Software, Reading, PA). The calibration curves were used to determine the number of moles of each species present in the plasma samples. The ratio of small molecule to protein was calculated to yield the stoichiometry of small molecule bound to TTR in plasma.
Table 1.
TTR binding selectivity in human plasma of NSAIDs that inhibit amyloid formation in vitro
| Compound | Molar eq bound at 10.8 μM | Maximal therapeutic concentration, μM | Molar eq bound at max. therapeutic conc. |
|---|---|---|---|
| 1 Flufenamic acid | 0.20 ± 0.02 | 54 | 0.59 ± 0.05 |
| 2 Mefenamic acid | 0.20 ± 0.03 | 21 | 0.20 ± 0.03 |
| 3 Meclofenamic acid | 0.14 ± 0.02 | 15 | NA |
| 4 Diflunisal | 0.13 ± 0.02 | 224 | 0.86 ± 0.12 |
| 5 Diclofenac | 0.04 ± 0.01 | 5 | NA |
| 6 Flurbiprofen | No binding | 62 | No binding |
| 7 Fenoprofen | No binding | 96 | No binding |
| 8 Indomethacin | No binding | 6 | NA |
NA, not applicable.
Table 2.
TTR binding selectivity in human plasma of highly efficacious in vitro amyloid inhibitors
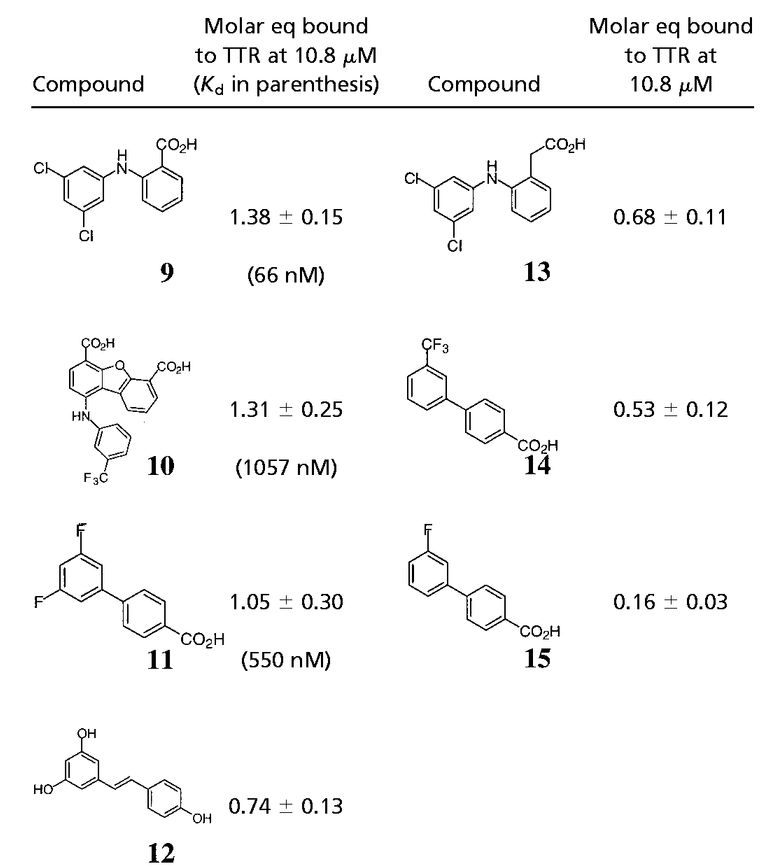 |
LC/MS Analysis of TTR and Small Molecules.
Samples were injected onto a Hewlett–Packard Series 1100 liquid chromatography/electrospray ionization mass spectrometer. The HPLC conditions were the same as listed above and the detector was in positive ion mode. Major peaks were multiplied by the net charge and averaged to yield the overall mass of the species. For the small molecules, the peaks were collected off the Waters HPLC and directly injected onto the Hewlett–Packard Series 1100 electrospray ionization mass spectrometer with the detector in negative ion mode.
T4 Displacement Determination of Small Molecule TTR Binding Affinity.
Nelson dialysis cells (Nichols Institute Diagnostics, San Juan Capistrano, CA) were used to carry out equilibrium dialysis at 60 rpm for 1 week at 25°C. The bottom reservoir was loaded with 2.4 ml of 10 mM sodium phosphate, pH 7.6/100 mM KCl/1 mM EDTA. The top reservoir was loaded with 2.4 ml of the same buffer containing 30 nM TTR, 55 nM 125I-labeled T4 (NEN/Perkin–Elmer), and the appropriate TTR amyloid inhibitor at a concentration between 1 and 5,000 nM. An additional set of TTR samples were incubated with labeled T4 at concentrations between 1 and 1,000 nM. After incubation, the radioactivity in 100 μl of each reservoir was measured in a Beckman 5001 γ counter. The fraction of total T4 bound to TTR was calculated and used to determine the concentration of the T4⋅TTR complex. Scatchard analysis (44) of the binding data of T4 alone yielded a dissociation constant (Kd) for T4 of 84 ± 17 nM. This was used to calculate the Kd for each inhibitor at every concentration through application of the following equation:
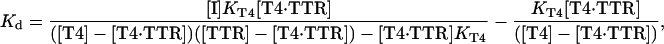 |
1 |
where [T4⋅TTR] is the concentration of the T4⋅TTR complex; [TTR], [T4], and [I] are the total TTR, T4, and inhibitor concentrations, respectively; KT4 is the dissociation constant for the first equivalent of T4, and Kd is the apparent dissociation constant for the inhibitor (45). Because the concentration of T4 in the experiment is well below the dissociation constant for the second T4 site (low μM) (46), we can treat the data as if only a single binding site for T4 is available. The geometric mean of each inhibitor's Kd was calculated to reach the reported value. Several Kd values at low inhibitor concentrations were discarded because the value was not physically realistic (i.e., negative).
Results
Assay Development.
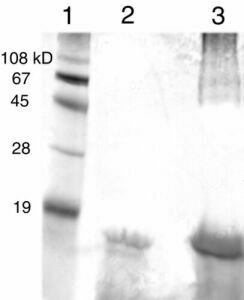
To minimize the immunoprecipitation of other plasma proteins, we generated rabbit polyclonal antibodies to purified recombinant human TTR. Western blots demonstrated that the antibodies had no significant reactivity with other plasma proteins (Fig. 1), including albumin, which typically cross-reacts with commercial TTR antibodies. TTR amyloid inhibitors showing efficacy in vitro were added to human blood plasma at a concentration of 10.8 μM (Fig. 2). The TTR concentration in plasma as determined by ELISA was ≈5.4 μM, implying that sufficient ligand was present to saturate the two available TTR-binding sites, assuming a submicromolar Kd. After a 24-h incubation, polyclonal anti-TTR antibodies conjugated to Sepharose were used to capture TTR and any bound small molecules. The carbohydrate saponin proved to be a necessary additive to the wash buffer to prevent the nonspecific interaction of the inhibitors with the antibody-Sepharose (see Fig. 5, which is published as supplemental data on the PNAS web site, www.pnas.org). High pH elution conditions released the TTR⋅inhibitor complex from the Sepharose-bound antibodies (Fig. 2). Silver-stained SDS/PAGE gels confirmed that the antibodies are selective for native TTR (Fig. 6 in the supplemental data), showing no significant binding to other plasma proteins. The HPLC solvents were sufficient to dissociate the TTR tetramer and the bound small molecule. To quantify the amount of TTR and inhibitor present, we created calibration curves by using stock solutions of recombinant tetrameric TTR and the small molecules to determine the integrated area/mole of each species.
Figure 1.

Rabbit polyclonal anti-TTR antibodies selectively bind TTR in human blood plasma. Lane 1, molecular mass standards; lane 2, isolated human blood plasma diluted 10-fold; lane 3, recombinant human TTR. The higher molecular mass band in lane 3 is due to an SDS-resistant dimer present when the recombinant protein is run on a gel.
Figure 2.

Summary of the procedure to evaluate inhibitor binding selectivity to TTR versus other plasma proteins. Human blood plasma is incubated with a small molecule inhibitor (black bars) for 24 h. The TTR tetramer (squares) and bound small molecules are precipitated with antibody-conjugated Sepharose (circle). The Sepharose is washed to remove unbound proteins (oval and diamond) and small molecules (triangle). TTR with (shown here) or without ligands bound is eluted at high pH. The supernatant is loaded onto an HPLC column for analysis and quantification.
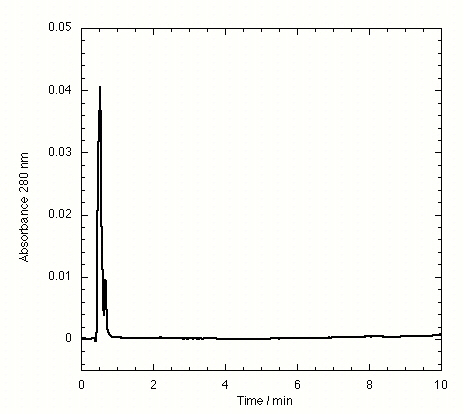
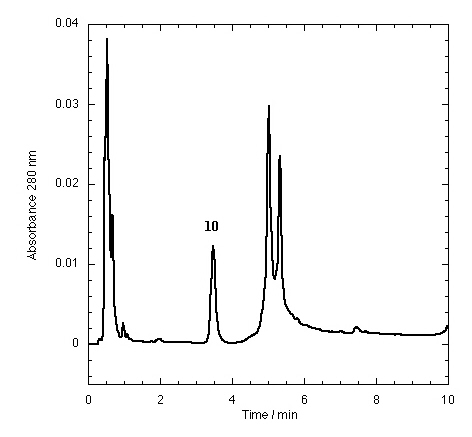
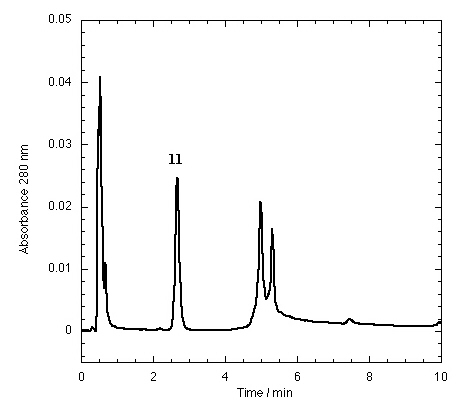
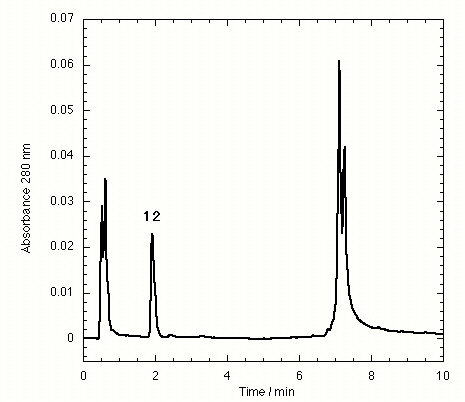
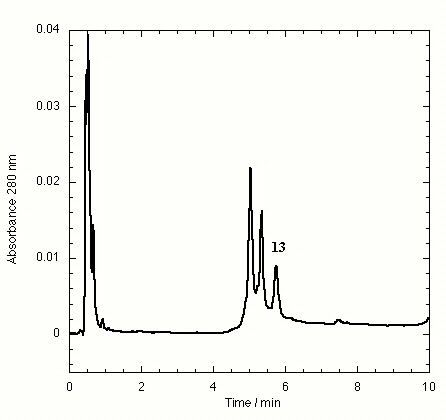
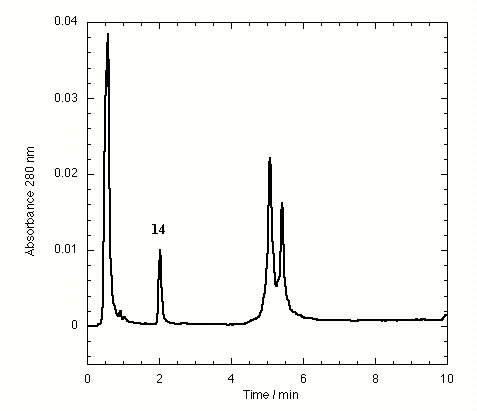
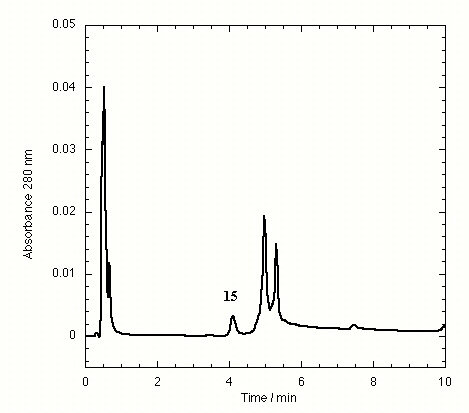
The TTR isolated from freshly drawn human plasma yielded two separate peaks on the HPLC chromatogram (Fig. 3A). Liquid chromatography coupled to electrospray mass spectrometry revealed that the two species had masses of 13,879 ± 4 and 13,758 ± 3 Da. The early peak corresponds to a TTR monomer disulfide linked through its single cysteine residue to a single cysteine amino acid, which has been observed previously (47–49), whereas the second peak corresponds to the expected TTR monomer. Inhibitors that bind to TTR in plasma elute separately on the chromatogram, e.g., inhibitor 9 (Fig. 3B) and inhibitors 10–15 (Figs. 7–12 in the supplemental data).
Figure 3.

HPLC chromatogram of immunoprecipitated plasma TTR in the absence of small molecule inhibitors (A) and in the presence of compound 9, a small molecule inhibitor of TTR fibril formation exhibiting selective TTR binding in plasma (B). TTR appears as two peaks, the first peak being monomer disulfide linked to cysteine and the second peak being unconjugated monomer.
Analysis of Nonsteroidal Antiinflammatory Drugs (NSAIDs).
The first compounds evaluated for selective binding to TTR in plasma were the NSAIDs previously identified to be potent TTR amyloid inhibitors in vitro (37, 42). Because these compounds are already approved by the Food and Drug Administration, they could easily be evaluated in human clinical trials for another indication if they proved to be selective TTR binders in human plasma. However, none of the NSAIDs exhibited significant selectivity for binding TTR in human plasma at a concentration of 10.8 μM (Table 1), although most exhibit a submicromolar Kd (37, 50). The most selective NSAIDs, flufenamic acid (1) and mefenamic acid (2), only had 0.2 eq of a maximum of 2 molar eq bound to TTR. However, fenoprofen (7), flurbiprofen (6), flufenamic acid (1), mefenamic acid (2), and diflunisal (4) all have maximal therapeutic plasma concentrations exceeding 20 μM (51, 52). When these compounds were incubated with plasma at their maximal therapeutic concentrations, flufenamic acid and diflunisal showed increased binding selectivity (stoichiometry) to TTR (Table 1). Diflunisal (4) is notable in that its 224 μM maximal therapeutic concentration leads to 0.85 eq of drug bound to TTR. Increasing the concentration of all other NSAIDs to their maximal therapeutic dose did not result in dramatic increases in binding selectivity, likely because of binding to other plasma proteins.
Analysis of the Remaining Lead Compounds.
Approximately 40 additional small molecule amyloid inhibitors were evaluated for their ability to bind to TTR in plasma by using our immunoprecipitation/HPLC approach (Fig. 2). These compounds were derived from screening or structure-based design and identified as promising by an in vitro fibril formation assay (refs. 40–42; V. H. Oza, H. M. Petrassi, and J.W.K., unpublished data). Lead compounds having diverse structures including biaryls, biarylamines, stilbenes, and dibenzofurans showed promising selectivity (Table 2). Three compounds from this group (9-11) possess excellent TTR-binding selectivity in plasma. At a concentration of 10.8 μM, they exhibit saturation of >1 of the two possible binding sites in the TTR tetramer. Determination of the TTR-binding affinities of these three compounds in buffer shows that the Kd values do not correlate with the plasma binding selectivity (Table 2). For example, inhibitors 9 and 10 have greater than an order of magnitude difference in Kd but nearly identical binding selectivity (stoichiometry) to TTR in plasma. Moreover, compounds with modest binding selectivity, such as flufenamic acid (1), have been previously determined to have Kd values very similar to those exhibiting excellent selectivities (37). Mass spectrometry confirmed the identity of inhibitors 9-12 isolated by immunoprecipitation/HPLC as the compounds that were initially incubated with plasma. Interestingly, compound 9, synthesized by our laboratory, was initially synthesized and evaluated by Bristol–Myers and Parke–Davis as a possible antiinflammatory compound (53, 54). Its modest activity never warranted pursuit beyond initial animal testing. However, this compound exhibits the highest binding selectivity (stoichiometry) for TTR in plasma of any compound evaluated thus far at a concentration of 10.8 μM. It is a potent TTR amyloid inhibitor in vitro, thus making it a candidate for further evaluation in animals and possibly in humans.
Discussion
Binding to plasma proteins is an important factor in determining the overall distribution, metabolism, activity, and toxicity of a drug (55). In this particular case, we desire small molecules that bind to the plasma protein TTR over protein competitors whose identities are not known and likely change with the small molecule under evaluation. This binding is known to stabilize TTR's normally folded state, thus preventing the conformational changes required for amyloidogenicity (14, 36). The immunoprecipitation/HPLC-based binding selectivity method outlined above allows quantification of the stoichiometry of small molecule binding to TTR in human plasma in the presence of all other plasma proteins and numerous competing small molecules without the use of tags, such as radiolabels, on the small molecule. Immunoprecipitation has been used previously to determine the stoichiometry of metal ion binding to specific plasma proteins (56, 57). However, to our knowledge, this is the first use of the technique to determine the binding selectivity of a small molecule to a single protein in human plasma. In principle, this approach is applicable to evaluate the binding selectivity of small molecules to any plasma protein, provided that a highly selective antibody for the protein can be generated, binding does not block the epitope or destroy it by conformational change, and appropriate wash steps can be introduced to avoid nonspecific binding of the small molecule and other proteins to the resin.
The protein target(s) of small molecule drugs have been established in the presence of all plasma components or in whole blood in surprisingly few instances. Typically, binding is evaluated either to all plasma proteins or to an isolated suspected protein target in a standard buffer in vitro. Equilibrium dialysis and ultrafiltration are commonly used to evaluate drug binding to whole plasma (55). These methods provide information on the amount of compound bound to total plasma protein, but they do not identify the specific target. Drug binding to isolated or recombinant plasma proteins can be characterized by numerous methods such as fluorescence (58), NMR (59), CD or related spectroscopies (60), chromatography (61, 62), calorimetry (37), or surface plasmon resonance (63). While these methods provide the binding affinity to the suspected plasma protein target, typically albumin for acidic drugs, or α1-acid glycoprotein for basic drugs (64), they do not predict the extent of binding in a biological fluid because the competitive proteins and small molecules are generally unknown. In contrast, the immunoprecipitation approach presented here allows one to determine the amount of small molecule bound to a specific protein in the presence of all plasma proteins and competitive ligands, without the requirement of identifying the appropriate competitor protein(s).
The effect of small molecule structure on TTR binding selectivity in plasma is dramatically demonstrated in a series of biarylamines of nearly identical chemical structure (Fig. 4). Notably, biarylamines 5, 9, and 13 show equal amyloid fibril inhibition efficacy in vitro at 1 eq of inhibitor bound per TTR tetramer (65–70% fibril inhibition) (V. H. Oza and J.W.K., unpublished data). However, these compounds exhibit dramatically different TTR binding selectivity in human plasma. The NSAID diclofenac (5) has low binding selectivity for TTR. Moving the Cl substituents from the 2 and 6 positions (5) to the 3 and 5 positions (13) results in a significant increase in TTR binding selectivity. Additional shortening of the carboxylic acid substituent by one methylene group (9) yields a further dramatic increase in binding selectivity. The structural changes that increase selectivity most likely interfere with binding to competitor plasma proteins rather than decreasing the affinity to TTR. We have not yet attempted to identify the competitor(s), but this could be done by using the immunoprecipitation/HPLC approach with antibodies specific for the suspected competitors, but this could prove difficult.
Figure 4.

Modest structural changes of the small molecule dramatically alter TTR plasma binding selectivity of chlorinated biarylamines possessing equal in vitro amyloid inhibition efficacy.
The highly selective TTR-binding compounds (e.g., 9-11, Table 2) identified through this immunoprecipitation procedure are now the primary candidates for further evaluation in mouse models of TTR amyloid disease (65). Because these compounds display binding selectivity for TTR in human plasma, they should partition primarily into human TTR in transgenic mice, which will be confirmed. In addition, diflunisal (4) may hold promise as a Food and Drug Administration-approved drug that displays moderate selectivity for TTR at its maximal therapeutic plasma concentration. It is essential that amyloid inhibitors that function by binding and stabilizing the native state of TTR exhibit high binding selectivity in vivo at reasonable concentrations.
These selective inhibitors now provide us with the tools to directly test the amyloid hypothesis in transgenic mice and possibly in humans. If small molecule inhibitor binding to TTR prevents the formation of amyloid fibrils in vivo as well as the organ dysfunction and/or neurodegeneration characteristic of these diseases, we will have direct evidence that the process of amyloid fibril formation is the cause of TTR amyloid diseases.
Supplementary Material
Acknowledgments
We thank H. Michael Petrassi, Vibha Oza, and Prakash Raman for synthesizing the compounds used in this study, Kyle Chiang and H. Michael Petrassi for initial development of the equilibrium binding competition assay, and Joel Buxbaum for helpful discussions on immunoprecipitation approaches and for providing the TTR ELISA protocol. This work was supported by National Institutes of Health Grant R01 DK 46335, the Lita Annenberg Hazen Family, the Skaggs Family, the Harold L. Dorris Neurological Research Center, and a San Diego Achievement Rewards for College Scientists Foundation Fellowship (H.E.P.).
Abbreviations
- TTR
transthyretin
- T4
thyroxine
- NSAID
nonsteroidal antiinflammatory drug
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Pepys M B. In: Immunological Diseases. Samter M, editor. Vol. 1. Boston: Little Brown; 1988. pp. 631–674. [Google Scholar]
- 2.Sipe J D. Annu Rev Biochem. 1992;61:947–975. doi: 10.1146/annurev.bi.61.070192.004503. [DOI] [PubMed] [Google Scholar]
- 3.Tan S Y, Pepys M B. Histopathology. 1994;25:403–414. doi: 10.1111/j.1365-2559.1994.tb00001.x. [DOI] [PubMed] [Google Scholar]
- 4.Sipe J D. Crit Rev Clin Lab Sci. 1994;31:325–354. doi: 10.3109/10408369409084679. [DOI] [PubMed] [Google Scholar]
- 5.Stine W B, Snyder S W, Ladror U S, Wade W S, Miller M F, Perun T J, Holzman T F, Krafft G A. J Protein Chem. 1996;15:193–203. doi: 10.1007/BF01887400. [DOI] [PubMed] [Google Scholar]
- 6.Harper J D, Wong S S, Lieber C M, Lansbury P T., Jr Chem Biol. 1997;4:119–125. doi: 10.1016/s1074-5521(97)90255-6. [DOI] [PubMed] [Google Scholar]
- 7.Kelly J W. Proc Natl Acad Sci USA. 1997;95:930–932. doi: 10.1073/pnas.95.3.930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kelly J W. Curr Opin Struct Biol. 1998;8:101–106. doi: 10.1016/s0959-440x(98)80016-x. [DOI] [PubMed] [Google Scholar]
- 9.Cohen A S, Shirahama T, Sipe J D, Skinner M. Lab Invest. 1983;48:1–4. [PubMed] [Google Scholar]
- 10.Kirschner D A, Abraham C, Selkoe D J. Proc Natl Acad Sci USA. 1986;83:503–507. doi: 10.1073/pnas.83.2.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sunde M, Serpell C S, Bartlam M, Fraser E P, Pepys B M, Blake C F C. J Mol Biol. 1997;273:729–739. doi: 10.1006/jmbi.1997.1348. [DOI] [PubMed] [Google Scholar]
- 12.Jimenez J L, Guijarro J I, Orlova E, Zurdo J, Dobson C M, Sunde M, Saibil H R. EMBO J. 1999;18:815–821. doi: 10.1093/emboj/18.4.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Serpell L C, Sunde M, Benson M D, Tennent G A, Pepys M B, Fraser P E. J Mol Biol. 2000;300:1033–1039. doi: 10.1006/jmbi.2000.3908. [DOI] [PubMed] [Google Scholar]
- 14.Colon W, Kelly J W. Biochemistry. 1992;31:8654–8660. doi: 10.1021/bi00151a036. [DOI] [PubMed] [Google Scholar]
- 15.Hurle M R, Helms L R, Li L, Chan W, Wetzel R. Proc Natl Acad Sci USA. 1994;91:5446–5450. doi: 10.1073/pnas.91.12.5446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McCutchen S L, Lai Z, Miroy G, Kelly J W, Colon W. Biochemistry. 1995;34:13527–13536. doi: 10.1021/bi00041a032. [DOI] [PubMed] [Google Scholar]
- 17.Lai Z, Colon W, Kelly J W. Biochemistry. 1996;35:6470–6482. doi: 10.1021/bi952501g. [DOI] [PubMed] [Google Scholar]
- 18.Kelly J W. Curr Opin Struct Biol. 1996;6:11–17. doi: 10.1016/s0959-440x(96)80089-3. [DOI] [PubMed] [Google Scholar]
- 19.Booth D R, Sunde M, Bellotti V, Robinson C V, Hutchinson W L, Fraser P E, Hawkins P N, Dobson C M, Radford S E, Blake C C F, Pepys M B. Nature (London) 1997;385:787–793. doi: 10.1038/385787a0. [DOI] [PubMed] [Google Scholar]
- 20.Kelly J W. Structure. 1997;5:595–600. doi: 10.1016/s0969-2126(97)00215-3. [DOI] [PubMed] [Google Scholar]
- 21.Blake C C F, Geisow M J, Swan I D A, Rerat C, Rerat B. J Mol Biol. 1974;88:1–12. doi: 10.1016/0022-2836(74)90291-5. [DOI] [PubMed] [Google Scholar]
- 22.Ferguson R N, Edelhoch H, Saroff H A, Robbins J. Biochemistry. 1975;14:282–289. doi: 10.1021/bi00673a014. [DOI] [PubMed] [Google Scholar]
- 23.Blake C C F, Geisow M J, Oatley S J, Rerat B, Rerat C. J Mol Biol. 1978;121:339–356. doi: 10.1016/0022-2836(78)90368-6. [DOI] [PubMed] [Google Scholar]
- 24.Nilsson S F, Rask L, Peterson P. J Biol Chem. 1975;250:8554–8563. [PubMed] [Google Scholar]
- 25.Cornwell G C, Sletten K, Johansson B, Westermark P. Biochem Biophys Res Commun. 1988;154:648–653. doi: 10.1016/0006-291x(88)90188-x. [DOI] [PubMed] [Google Scholar]
- 26.Westermark P, Sletten K, Johansson B, Cornwell G G. Proc Natl Acad Sci USA. 1990;87:2843–2845. doi: 10.1073/pnas.87.7.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saraiva M J M, Costa P P, Goodman D S. J Lab Clin Med. 1983;102:590–603. [PubMed] [Google Scholar]
- 28.Saraiva M J M, Costa P P, Goodman D S. Adv Neurol. 1988;48:189–200. [PubMed] [Google Scholar]
- 29.Benson M D, Wallace M R. In: The Metabolic Basis of Inherited Disease. 6th Ed. Scriver C R, Beaudet A L, Sly W S, Valle D, editors. Vol. 2. New York: McGraw Hill; 1989. pp. 2439–2460. [Google Scholar]
- 30.Jacobson D R, Buxbaum J N. Adv Hum Genet. 1991;20:69–123. doi: 10.1007/978-1-4684-5958-6_2. [DOI] [PubMed] [Google Scholar]
- 31.Saraiva M J M. Hum Mutat. 1995;5:191–196. doi: 10.1002/humu.1380050302. [DOI] [PubMed] [Google Scholar]
- 32.Sousa A, Coelho T, Barros J, Sequeiros J. Am J Med Genet. 1995;60:512–521. doi: 10.1002/ajmg.1320600606. [DOI] [PubMed] [Google Scholar]
- 33.Coelho T. Curr Opin Neurol. 1996;9:355–359. [PubMed] [Google Scholar]
- 34.Jacobson D R, Pastore R D, Yaghoubian R, Kane I, Gallo G, Buck F S, Buxbaum J N. N Engl J Med. 1997;336:466–473. doi: 10.1056/NEJM199702133360703. [DOI] [PubMed] [Google Scholar]
- 35.Connors L H, Richardson A M, Theberge R, Costello C E. Amyloid. 2000;7:54–69. doi: 10.3109/13506120009146826. [DOI] [PubMed] [Google Scholar]
- 36.Miroy G J, Lai Z, Lashuel H, Peterson S A, Strang C, Kelly J W. Proc Natl Acad Sci USA. 1996;93:15051–15056. doi: 10.1073/pnas.93.26.15051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peterson S A, Klabunde T, Lashuel H A, Purkey H, Sacchettini J C, Kelly J W. Proc Natl Acad Sci USA. 1998;95:13407–13412. doi: 10.1073/pnas.95.22.12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baures P W, Peterson S A, Kelly J W. Bioorg Med Chem. 1998;6:1389–1401. doi: 10.1016/s0968-0896(98)00130-8. [DOI] [PubMed] [Google Scholar]
- 39.Baures P W, Oza V H, Peterson S A, Kelly J W. Bioorg Med Chem. 1999;7:1339–1347. doi: 10.1016/s0968-0896(99)00066-8. [DOI] [PubMed] [Google Scholar]
- 40.Oza V B, Petrassi H M, Purkey H E, Kelly J W. Bioorg Med Chem Lett. 1999;9:1–6. doi: 10.1016/s0960-894x(98)00696-9. [DOI] [PubMed] [Google Scholar]
- 41.Petrassi H M, Klabunde T, Sacchettini J, Kelly J W. J Am Chem Soc. 2000;122:2178–2192. [Google Scholar]
- 42.Klabunde T, Petrassi H M, Oza V B, Raman P, Kelly J W, Sacchettini J C. Nat Struct Biol. 2000;7:312–321. doi: 10.1038/74082. [DOI] [PubMed] [Google Scholar]
- 43.Anonymous. CNBr-activated Sepharose 4B Instructions. Uppsala, Sweden: Amersham Pharmacia Biotech; 1998. [Google Scholar]
- 44.Scatchard G. Ann NY Acad Sci. 1949;51:660–675. [Google Scholar]
- 45.Horovitz A, Levitzki A. Proc Natl Acad Sci. 1987;84:6654–6658. doi: 10.1073/pnas.84.19.6654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Robbins J, Cheng S-Y, Gershengorn M C, Glinoer D, Cahnmann H J, Edelhoch H. Rec Prog Horm Res. 1978;34:477–519. doi: 10.1016/b978-0-12-571134-0.50017-x. [DOI] [PubMed] [Google Scholar]
- 47.Terazaki H, Ando Y, Suhr O, Ohlsson P-I, Obayashi K, Yamashita T, Yoshimatsu S-I, Suga M, Uchino M, Ando M. Biochem Biophys Res Commun. 1998;249:26–30. doi: 10.1006/bbrc.1998.9097. [DOI] [PubMed] [Google Scholar]
- 48.Suhr O B, Ando Y, Ohlsson P I, Olofsson A, Andersson K, Lundgren E, Ando M, Holmgren G. Eur J Clin Invest. 1998;28:687–692. doi: 10.1046/j.1365-2362.1998.00345.x. [DOI] [PubMed] [Google Scholar]
- 49.Theberge R, Connors L, Skinner M, Skare J, Costello C E. Anal Chem. 1999;71:452–459. doi: 10.1021/ac980531u. [DOI] [PubMed] [Google Scholar]
- 50.Munro S L, Lim C-F, Hall J G, Barlow J W, Craik D J, Topliss D J, Stockigt J R. J Clin Endocrinol Metab. 1989;68:1141–1147. doi: 10.1210/jcem-68-6-1141. [DOI] [PubMed] [Google Scholar]
- 51.Angelucci L, Petrangeli B, Celletti P, Favilli S. J Pharm Sci. 1976;65:455–456. doi: 10.1002/jps.2600650340. [DOI] [PubMed] [Google Scholar]
- 52.Sifton D W, editor. Physicians' Desk Reference. 54th Ed. Montvale, NJ: Medical Economics; 2000. [Google Scholar]
- 53.Juby P F, Hudyma T W, Brown M. J Med Chem. 1968;11:111–116. doi: 10.1021/jm00307a025. [DOI] [PubMed] [Google Scholar]
- 54.Kaltenbronn J S, Scherrer R A, Short F W, Jones E M, Beatty H R, Saka M M, Winder C V, Wax J, Williamson W R N. Arzneim-Forsch. 1983;33:621–627. [PubMed] [Google Scholar]
- 55.Oravcova J, Boehs B, Lindner W. J Chromatogr B Biomed Appl. 1996;677:1–28. doi: 10.1016/0378-4347(95)00425-4. [DOI] [PubMed] [Google Scholar]
- 56.Weiblen B J, Melaragno A J, Catsimpoolas N, Valeri C R. J Immunol Methods. 1983;58:73–81. doi: 10.1016/0022-1759(83)90264-8. [DOI] [PubMed] [Google Scholar]
- 57.Huebers H A, Eng M J, Josephson B M, Ekpoom N, Rettmer R L, Labbe R F, Pootrakul P, Finch C A. Clin Chem. 1987;33:273–277. [PubMed] [Google Scholar]
- 58.Epps D E, Raub T J, Caiolfa V, Chiari A, Zamai M. J Pharm Pharmacol. 1999;51:41–48. doi: 10.1211/0022357991772079. [DOI] [PubMed] [Google Scholar]
- 59.Jenkins B G. Life Sci. 1991;48:1227–1240. doi: 10.1016/0024-3205(91)90517-f. [DOI] [PubMed] [Google Scholar]
- 60.Otagiri M, Nakamura H, Imamura Y, Matsumoto U, Fleitman J, Perrin J H. Chem Pharm Bull. 1989;37:1401–1403. doi: 10.1248/cpb.37.1401. [DOI] [PubMed] [Google Scholar]
- 61.Noctor T A G, Diaz-Perez M J, Wainer I W. J Pharm Sci. 1993;82:675–676. doi: 10.1002/jps.2600820629. [DOI] [PubMed] [Google Scholar]
- 62.Hage D S, Tweed S A. J Chromatogr B Biomed Sci Appl. 1997;699:499–525. doi: 10.1016/s0378-4347(97)00178-3. [DOI] [PubMed] [Google Scholar]
- 63.Frostell-Karlsson A, Remaeus A, Roos H, Andersson K, Borg P, Hamalainen M, Karlsson R. J Med Chem. 2000;43:1986–1992. doi: 10.1021/jm991174y. [DOI] [PubMed] [Google Scholar]
- 64.Herve F, Urien S, Albengres E, Duche J C, Tillement J P. Clin Pharmacokinet. 1994;26:44–58. doi: 10.2165/00003088-199426010-00004. [DOI] [PubMed] [Google Scholar]
- 65.Teng M-H, Buxbaum J N. Amyloid. 1996;3:187–208. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}