

Abstract
We evaluate experimentally and computationally the membrane permeability of matched sets of peptidic small molecules bearing natural or bioisosteric unnatural amino acids. We find that the intentional introduction of hydrogen bond acceptor-donor pairs in such molecules can improve membrane permeability while retaining or improving other favorable drug-like properties. We employ an all-atom force-field based method to calculate changes in free energy associated with the transfer of the peptidic molecules from water to membrane. This computational method correctly predicts rank-order experimental permeability trends within congeneric series and is much more predictive than calculations (e.g. clogP) that do not consider three-dimensional conformation.
Keywords: Intramolecular hydrogen bonds, membrane permeability, unnatural amino acids
Introduction
Short peptides and peptidic small molecules are useful both as probes to interrogate biological pathways and as lead compounds for the discovery of new therapeutics. Unfortunately, the utility of such molecules is often limited by poor pharmacokinetic profiles resulting from poor membrane permeability, low solubility, and proteolytic or metabolic instability. Many of these undesirable features stem from physiochemical properties that can be altered through structural modification. The introduction of basic and/or hydrophilic atoms generally improves solubility and reduces metabolism, but these improvements often come at the expense of membrane permeability. Conversely, masking hydrogen bond donor (HBDa) functions by N- or O-alkylation increases lipophilicity and often improves permeability, but frequently at the expense of solubility and binding affinity to the intended biological target. Chemical strategies that can improve membrane permeability without negatively impacting solubility or target binding affinity are therefore of significant interest.
An attractive alternative to the permanent removal (e.g., alkylation) of HBD functionality is the introduction of complementary hydrogen bond acceptor (HBA) functionality. Through the formation of intramolecular hydrogen bonds, the HBD and HBA atoms are effectively shielded, thereby reducing the energetic penalty of desolvation required in moving from an aqueous environment to a lipophilic membrane interior. Importantly, the formation of such HBD/HBA contacts when the peptide is within the membrane does not preclude the adoption of very different conformations upon binding to the intended biological target.
Various reports of improved membrane permeability resulting from intramolecular hydrogen bonding have appeared in the recent literature.1-3 For example, in a series of small molecule neurokinin inhibitors, the introduction of an HBA-bearing side chain led to notably improved permeability across the blood-brain barrier in animals.4 A recent systematic analysis of orally available drugs suggested that those lying well outside rule-of-five chemical space (e.g., cyclosporine A) are often capable of forming intramolecular hydrogen bonding interactions.5 Lokey and Jacobson recently showed with diastereomeric cyclic peptides that those peptides capable of forming intramolecular hydrogen bonds were more permeable that those that cannot.3,6 These permeability trends could be predicted de novo using an all-atom force-field based method to calculate changes in free energy (ΔGtr) associated with the transfer of the peptides from water to membrane.2 This same computational approach has also been used to predict the permeability of a set of approved small molecule drugs for which experimental permeability values are available.7
Several predictive models to study permeability trends have been reported. In the Quantitative Structure Permeability Relationship models8-15, statistical relationships are derived between various molecular descriptors (molecular weight, polar surface area, hydrogen bond donor/acceptor counts, octanol-water partition coefficients, etc.) and the experimentally determined permeability for a training set of compounds. The performance of these empirical scoring models depends on the chemical similarity of the test compounds and the training set, and therefore the transferability of such methods is an issue. The physics-based models16-20, on the other hand, seek to employ the physics underlying the permeation process and generally do not employ empirical data from a compound training set. Physics-based models are therefore more general and transferable to diverse set of molecules. Depending on the particular task at hand, either of the two approaches can return favorable results.
To more systematically explore the possible role of intramolecular hydrogen bonding in facilitating permeability, we designed and synthesized a set of small molecules containing bioisosteric amino acids in which a HBA atom is positioned within intramolecular hydrogen bonding distance (5-6 atoms) of backbone N-H functions. Analogs containing either HBA-bearing or unmodified amino acids were tested for permeability employing MDR1-MDCK cell monolayers expressing the drug transporter P-gp. Blinded predictions of passive permeability were made using the previously described physics-based computational approach7 and these could be correlated with the experimental permeability values. Our results suggest that suitably positioned HBA atoms can in some cases improve the membrane permeability of peptidic small molecules without increasing overall lipophilicity or removing otherwise important HBD functions. Because the bioisosteric amino acids are typically less lipophilic than their natural counterparts, this chemical strategy can potentially afford peptidic molecules with improved membrane permeability and solubility.
Results and Discussion
The requisite HBA-containing amino acids (HBA-AA) were prepared using the general approach described by Vederas for the synthesis of beta amino acids.21 Briefly, the β-lactone derived from Cbz-L-serine was reacted with amine nucleophiles, resulting in the formation of the desired HBA-AA and varying amounts of undesired amide side-product resulting from reaction at the β-lactone carbonyl function. Consistent with earlier reports21, we found that the chemoselectivity of ring opening varies depending on the amine nucleophile and solvent employed. This issue was of little practical consequence however since only carboxylic acid containing products can participate in the subsequent coupling reaction to afford the desired C-terminal carboxamide or glycine nitrile analogs (Table 1). A variety of aliphatic, anilinic, and heteroaromatic amines participated in the ring opening reaction to afford novel amino acids that were converted to the final analogs 2, 4-5, 8-9, 11 and 13. The remaining analogs were prepared in a single step from known or commercially available Cbz-protected amino acids.
Table 1.
Experimental permeability in MDR1-MDCK cell monolayers and calculated ΔGtr and logS values for compounds 1-16.
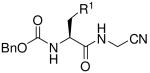 | |||||||
|---|---|---|---|---|---|---|---|
| R1 = |  |
 |
--- |  |
 |
 |
|
| Compound | 1 | 2 | 3 | 4 | 5 | ||
| Papp (nm/sec) |
A to B | 244 | 106 | 218 | 196 | 203 | |
| B to A | 296 | 167 | 310 | 318 | 364 | ||
| effluxx ratio | 1.2 | 1.6 | 1.4 | 1.6 | 1.8 | ||
| ΔGtr | 5.41 | 9.58 | 8.77 | 7.68 | 8.17 | ||
| −logS | 4.62 | 2.74 | 5.92 | 4.99 | 4.01 | ||
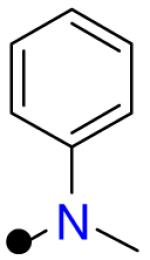
| R1 = |  |
 |
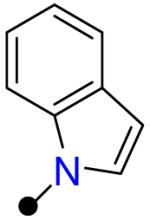 |
 |
 |
 |
|
|---|---|---|---|---|---|---|---|
| Compound | 6 | 7 | 8 | 9 | 10 | 11 | |
| Papp (nm/sec) |
A to B | 47.5 | 117 | 145 | 103 | 5.1 | 84.1 |
| B to A | 428 | 249 | 342 | 455 | 29.4 | 257 | |
| effluxx ratio | 9.0 | 2.1 | 2.4 | 4.4 | 5.8 | 3.1 | |
| ΔGtr | 12.6 | 10.1 | 7.82 | 7.97 | 13.0 | 8.17 | |
| −logS | 5.27 | 5.96 | 5.46 | 4.97 | 4.03 | 4.04 | |

| |||||||
| R2 = | 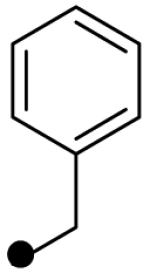 |
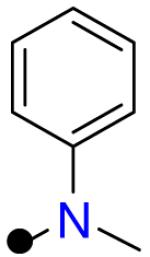 |
--- | 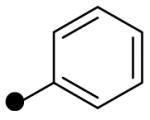 |
 |
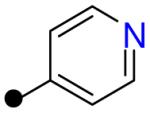 |
|
|---|---|---|---|---|---|---|---|
| Compound | 12 | 13 | 14 | 15 | 16 | ||
| Papp (nm/sec) |
A to B | 232 | 239 | 246 | 177 | 43.1 | |
| B to A | 198 | 192 | 211 | 200 | 134 | ||
| effluxx ratio | 0.85 | 0.81 | 0.86 | 1.1 | 3.1 | ||
| ΔGtr | 8.1 | 8.56 | 7.65 | 10.2 | 11.0 | ||
| −logS | 3.62 | 3.60 | 3.34 | 3.00 | 2.81 | ||
Permeability of test compound in the apical to basolateral (A to B) and basolateral to apical (B to A) directions was determined in MDR1-MDCK cell monolayers. The ratio of permeability values (the efflux ratio) provides a measure of the extent of active transport by the drug transporter P-glycoprotein (P-gp), which is expressed in this cell line. This additional information is useful insofar as P-gp mediated efflux impacts drug action, but in correlating experimental and predicted permeability it is the Papp(A->B) value that is most representative of intrinsic passive permeability.
In the prediction algorithm, a systematic conformational search is first conducted for each compound and the resulting low energy conformations clustered. The structure closest to the geometric center of the cluster is chosen as representative and the single point energy is calculated in an implicit solvent model meant to mimic chloroform (low dielectric medium to represent membrane). The single point energy of this same conformation is then calculated in water. The computational evidence suggests that treating these particular peptides with single conformations is a reasonable approximation in most cases (see Experimental Section). To the difference between the membrane energy and water energy is added a neutralization penalty calculated using the pKa values of any titratable groups, as estimated by Epik.22 The rationale for adding this penalty is that charged compounds permeate through membranes much slower than neutral species. The resulting value for transfer of the compound from membrane to water (ΔGtr) is employed as a predictor of relative permeability: lower ΔGtr values predict for higher permeability.
Calculations of ΔGtr were performed blind of the experimental results. A comparison of these predicted ΔGtr values and the experimentally determined Papp(A->B) values yielded a reasonable correlation coefficient r2 = 0.61 (Figure 1A). Accounting for the free energy cost of neutralization at pH 7.4 did improve the r2 value slightly (from 0.56 to 0.61). As a point of comparison, correlation of the experimental Papp(A->B) values with clogP (calculated in QikProp), molecular weight, SASA, or HBD/HBA count, produced r2 values respectively of 0.26 (Figure 1B), 0.00007, 0.01 and 0.005 (data not shown). Conformational sampling is an important aspect of our prediction protocol that improves the predictive power of the method as compared to clogP which does not consider conformational information. Not surprisingly, the calculated hydrophilic surface area of the low energy conformers used in the ΔGtr calculation was better correlated to experimental permeability than clogP (r2 = 0.41 vs. 0.26), but still inferior to ΔGtr (Figure 1). Thus, the best predictive methods include both conformational sampling and a measure of hydrophobicity that depends on the three dimensional structure. A comparison of predicted solubility (clogS, determined in QikProp) with ΔGtr values is also provided (Figure 2). From this plot it is apparent that analogs bearing HBA-AA (colored red in Figure 2) are in general predicted to be more permeable than unmodified comparators, while at the same time spanning a more favorable range of clogS values.
Figure 1.

Plots correlating predicted and measured values for compounds 1-16. A: experimentally derived permeabilities, −logPe(A->B) and the calculated ΔGtr; B: −logPe(A->B) and the calculated value logP(oct/wat) (QikProp); C: −logPe(A->B) versus calculated hydrophilic surface area (QikProp) of each low energy conformer. Analogs bearing HBA-AA are shown in red while those without such modifications are in green. Data points are colored red for analogs with HBA-AA and green for control analogs without HBA-AA. Data points are shaped according to chemotype as follows: compounds 1-2 (square), 3-5 (circle), 6-9 (inverted triangle), 10-11 (diamond), 12-13 (triangle), 14-16 (hexagon).
Figure 2.

A plot of predicted solubility (clogS calculated using QikProp) against the calculated ΔGtr values for the compounds 1-16. Data points are colored red for analogs with HBA-AA and green for control analogs without HBA-AA. Compound numbers are included alongside data points, which are colored and shaped as in Figure 1.
Examining the matched analog pairs, we see that the measured permeability rate of leucine analog 1 is about twice that of its bioisostereic analog 2; both analogs are highly permeable and neither appears to be subject to P-gp mediated efflux. Both experimental and computational prediction are in agreement concerning the greater permeability of 1, despite the fact that an intramolecular hydrogen bond is observed in the lowest energy conformer of 2 (Table 1). The presence of an intramolecular hydrogen bond is supported experimentally by the observed chemical shift of the amide N-H signal, which falls at 7.57 ppm for 1 and 9.09 ppm for 2 (both spectra recorded in CDCl3). Apparently, an intramolecular hydrogen bond in 2 is not sufficient to shield the excess polar surface area, as compared to 1 which lacks the hydrophilic nitrogen atom present in 2. Another important factor undoubtedly is the presence of a basic amine in 2 that would be significantly protonated under the aqueous conditions of the permeability assay.
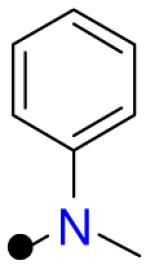
A fairer comparison is that of 3 with its anilinic HBA-AA analogs 4 and 5, neither of which should be significantly protonated under the assay conditions. Compounds 3-5 show very similar experimental permeability values and efflux ratios. Consistent with this finding are the similar ΔGtr values obtained for 3-5. Thus in the case of 3-5, a hydrophilic but non-basic HBA atom can be introduced (in 4 and 5) without negatively impacting permeability. As with analog 2, the low energy conformers of both 4 and 5 possess the expected intramolecular hydrogen bonding interaction. While no significant improvement in permeability is realized in this case, compounds 4 and 5 do possess improved clogS value as compared to compound 3 (Table 1 and Figure 2).
Much more compelling effects were observed in the compound set comprising tryptophan analogs 6 and 7 and bioisosteric congeners 8 and 9. Hence compound 6 has the lowest intrinsic permeability (A to B) in this set and also an unfavorable efflux ratio of ~9.0; in a separate experiment this compound was found to be a substrate for P-gp.23 N-methylation of the indole ring (compound 7) improves passive permeability and also reduces the efflux ratio to ~2.1. The HBA-AA bearing analogs 8 and 9 are both more permeable and exhibit improved efflux ratios as compared to 6. The relative permeability of 6 as compared to 7-9 are correctly predicted by the ΔGtr values and the expected intramolecular hydrogen bond is present in the low energy conformer of indazole 9 (Table 2). The increased permeability of 9 is similar to that achieved by N-methylation (as in 7), but is accompanied in 9 by a superior clogS value as compared to either 6 or 7 (Table 1).
Table 2.
Low energy conformations of selected analogs, calculated as described in the text and experimental section. Predicted intramolecular hydrogen bonds are indicated with dashed lines.
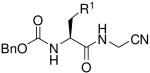 | ||
|---|---|---|
| Compound | R1 |
Low energy conformation |
| 2 |

|

|
| 4 |
|

|
| 5 |

|

|
| 9 |

|

|
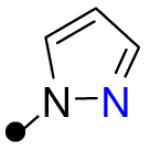
| 11 |
|

|
 | ||
|---|---|---|
| 14 |

|

|
| 15 |

|

|
| 16 |

|

|
The histidine analog 10 exhibited the poorest intrinsic permeability of all the compounds examined, and like tryptophan analog 6, was subject to P-gp mediated efflux. The isosteric pyrazole analog 11 was >15-fold more permeable than 10, and this large difference in Papp(A->B) values is reflected in the divergent ΔGtr values obtained for these analogs (8.7 and 13.0 kcal/mol, respectively). Importantly, the dramatically improved permeability of HBA-AA bearing analog 11 was achieved without a significant increase in clogS or clogP values as compared to 10.
The final set of analogs 12-16 possess a C-terminal carboxamide function and thus possess two additional HBD not present in 1-11. As with anilinic analogs 4 and 5, the anilinic side chain in 13 afforded no notable improvement in permeability over the comparator 12, and this was reflected in similar ΔGtr values. The 2-pyridyl and 4-pyridyl regioisomers 15 and 16 present an interesting comparison as only the former can possibly form an intramolecular hydrogen bond involving the pyridine nitrogen atom. Indeed, 2-pyridyl analog 15 was both predicted and found experimentally to be more permeable than 16. Unexpectedly, the unmodified phenylalanine comparator 14 was found experimentally (and also predicted) to be more permeable than either 15 or 16. Inspection of the predicted low energy conformations of 14-16 provides a possible explanation for this result (Table 2). The low energy conformer of 15 possesses the expected intramolecular hydrogen bond involving the pyridine nitrogen atom, an interaction that is not possible in its regioisomer 16. In the low energy conformation of 14, and to a lesser degree 16, the aryl ring is turned to face the N-H function in a putative N-H-π interaction that might partially shield the N-H function from solvent. In conformers 14, 15 and 16, we measure the distances between N-H hydrogen atom and the center of the aromatic ring to be 3.2 Å, 3.5 Å, 3.4 Å, respectively, and the angle between N, H, and the center of the ring to be 126.5°, 118.9°,121.5°, respectively. The values for compound 14 are in agreement with the typical distance and angle for an N-H-π interaction.24 Supporting this hypothesis, we find that the rank-order experimental permeability of 14-16 is correctly predicted by calculating the hydrophilic surface area of these low energy conformers.
Conclusions
In conclusion, the introduction of suitably positioned hydrogen bond acceptor functionality can in some cases improve membrane permeability in peptidic molecules that otherwise retain favorable clogP and clogS values. In other cases permeability rates are merely retained while clogP and clogS values can be nudged into more favorable ranges. Thus, this approach may prove useful in improving the drug-like properties of peptidic small molecules. We advise however that the approach be applied in combination with robust conformational analysis. The all-atom force-field based method applied herein provides accurate rank-order permeability prediction within congeneric analog sets and is much more predictive than simple clogP calculations. The appropriate shielding of hydrophilic functionality is an important factor to consider when addressing poor membrane permeability; the introduction of intramolecular hydrogen bonding interactions is one viable means to achieving this end.
Experimental Section
MDR1-MDCK monolayer permeability assay
MDR1-MDCK cells (obtained from Piet Borst at the Netherlands Cancer Institute) were seeded onto polyethylene membranes (PET) in 96-well BD insert systems at 2 × 105 cells/ cm2 for 4-6 days for confluent cell monolayer formation. Test compounds were diluted with the transport buffer (HBSS, pH 7.4) from a 10 mM stock solution to a concentration of 2 μM and applied to the apical or basolateral side of the cell monolayer. Permeation of the test compounds from A to B direction or B to A direction was determined in triplicate over a 150-minute incubation at 37°C and 5% CO2 with a relative humidity of 95%. In addition, the efflux ratio of each compound was also determined. Test and reference compounds were quantified by LC-MS/MS analysis based on the peak area ratio of analyte/IS.
The apparent permeability coefficient Papp (cm/s) was calculated using the equation:
Where dCr/dt is the cumulative concentration of compound in the receiver chamber as a function of time (μM/s); Vr is the solution volume in the receiver chamber (0.075 mL on the apical side, 0.25 mL on the basolateral side); A is the surface area for the transport, i.e. 0.084 cm2 for the area of the monolayer; C0 is the initial concentration in the donor chamber (μM).
The efflux ratio (ER) was calculated using the equation:
Percent recovery was calculated using the equation:
Where Vd is the volume in the donor chambers (0.075 mL on the apical side, 0.25 mL on the basolateral side); Cd and Cr are the final concentrations of transport compound in donor and receiver chambers, respectively. Cc is the compound concentration in the cell lysate solution (μM). Vc is the volume of insert well (0.075 mL in this assay).
Permeability determinations were performed in triplicate and are reported as mean values. The mean total recovery was greater than 90% in both directions for all compounds tested, with the exception of compound 7 where recovery was 85% (A to B) and 93% (B to A).
Calculation of ΔGtr values
All compounds studied in this work were constructed using Maestro ligand building panel and they were subsequently energy minimized using Ligprep (Schrodinger software, version 8.5). For each of the compounds parameters were generated using a utility named hetgrp_ffgen (a part of Schrodinger software). The forcefield was OPLS 2005.25,26 The neutralization penalty for each of the compounds was calculated using Epik22 at pH 7.4. Conformational sampling and calculation of all energies was done using Protein Local Optimization Program (PLOP) using OPLS-AA force field and the surface generalized Born implicit solvent model.27 The conformational sampling for each ligand was carried out using the ‘tether pred’ functionality of PLOP. This was done by systematically varying the rotatable dihedral angles. The ‘overlap factor’, a criterion defined by the user (set to 0.65, in this case) computes the distance between two atoms divided by the sum of their radii and eliminates any conformational clashes. The conformational search is performed in an exhaustive fashion with a maximal resolution of 10° (36 conformations per rotatable dihedral angle). Subsequently, the generated conformations are clustered. In this work 400 clusters were generated and the representative member was chosen as the one closest to the geometric center of the cluster. The lowest energy conformer is selected as the final conformer and the single point energy is calculated in chloroform28 (low dielectric medium to represent membrane). The single point energy of this structure is then calculated in water. To this difference between the membrane energy and the water energy, is added the neutralization penalty calculated earlier using the Epik. This is the ΔG of transfer (ΔGtr) of the compound from membrane to water. This is used as a predictor of relative permeability within a chemical series: the lower the ΔGtr, the higher the permeability.
Prompted by reviewer comments, we performed additional analysis to confirm that treating the peptides described herein as single conformations is indeed a reasonable approximation for the purposes of the ΔGtr calculation. For example, we found that in the low dielectric calculations the key hydrogen bonds are invariably conserved across the lowest energy clusters of conformations. The structures observed in these low-energy clusters differ slightly in their backbone and side chain conformations, but the hydrogen bond is still present. Other conformations that break these hydrogen bonds tend to be significantly higher in energy. Thus, selection of a single (centroid) conformation from the lowest energy cluster is justified.
While the ΔGtr calculation does not involve a conformational search in high dielectric (water), we thought this would be an interesting question to explore. Using the same protocol to perform a conformational search in implicit water, we found that of the eight analogs bearing HBA groups, six of them (2, 4, 5, 9, 11, 15) do possess an internal hydrogen bond in the lowest energy conformer in water, while analogs 8 and 13 do not form the hydrogen bond in the lowest energy conformer in water.
Synthesis
1H NMR spectra were recorded on a Varian INOVA-400 400 MHz spectrometer. Chemical shifts are reported in θ units (ppm) relative to the residual NMR solvent peak. Coupling constants (J) are reported in hertz (Hz). Compounds 1-6 and 8-11 were prepared as described previously.23 All other reagents and solvents were purchased from Aldrich Chemical or Acros Organics and used as received. Column chromatography was carried out using a Biotage SP1 flash chromatography system and silica gel cartridges from Biotage or Silicycle. Analytical TLC plates from EM Science (Silica Gel 60 F254) were employed for TLC analyses. Preparative HPLC purifications were performed using a Biotage Parallex Flex equipped with Waters Xbridge 19 × 50mm C18 5 μM OBD columns.
Compound Purity
Compounds 1-16 were judged to be of 95% or higher purity based on analytical LC/MS analysis. LC/MS analyses were performed on a Waters Micromass ZQ/Waters 2795 Separation Module/Waters 2996 Photodiode Array Detector system controlled by MassLynx 4.0 software. Separations were carried out on an XTerra® MS C18 5μm 4.6 × 50mm column at ambient temperature using a mobile phase of water-methanol containing 0.2% formic acid. Gradient elution was employed wherein the methanol-water ratio was increased linearly from 5 to 95% methanol over 8 minutes, then maintained at 95% methanol for 1.5 min., and then decreased to 5% methanol over 0.5 min, and maintained at 5% methanol for 0.5 min. Compound purity was determined by integrating peak areas of the liquid chromatogram, monitored at 254 nm.
(2S)-2-{[(Benzyloxy)carbonyl]amino}-3-(1-methyl-1H-indol-3-yl) propanoic acid (17)
1-Methyl-L-tryptophan (0.40 g, 1.8 mmol) was added to a suspension of NaHCO3 (0.38 g, 4.6 mmol) in THF/water (1:2, 4 mL) at ambient temperature. The mixture was stirred for 5 min and then benzyl chloroformate (0.26 mL, 1.8 mmol) was added dropwise. After stirring for 1 h, the reaction mixture extracted thrice with 15 mL of diethyl ether. The aqueous phase was acidified to pH 2 by addition of HCl (1N), and the slurry was extracted thrice with 15 mL of ethyl acetate. The combined organic phases were washed with 1N HCl (2 × 15 mL) and brine (25 mL). The crude mixture was dried (MgSO4), filtered, concentrated, and the resulting residue purified by silica gel chromatography to afford 180 mg of the title compound (0.51 mmol, 30%). 1H NMR (400 MHz, CDCl3) θ 8.69 (br s, 1 H), 7.59 (d, J = 7.87 Hz, 1 H), 7.34 (br s, 3 H), 7.19 - 7.31 (m, 2 H), 7.08 (m, 1 H), 6.86 (s, 1 H), 5.44 (d, J = 7.69 Hz, 1 H), 4.94 - 5.18 (m, 2 H), 4.75 (m, 1 H), 3.65 (s, 3 H), 3.24 - 3.43 (m, 2 H). MS (m/z): [M+H]+ = 352.
Benzyl N-[(1S)-1-[(cyanomethyl)carbamoyl]-2-(1-methyl-1H-indol-3-yl)ethyl]carbamate (7)
To a solution of 17 (50 mg, 140 μmol) in DMF (0.4 mL) was added aminoacetonitrile bisulfate (44 mg, 280 μmol), [dimethylamino(triazolo[4,5-b]pyridin-3-yloxy)methylene]-dimethyl-ammonium hexafluorophosphate (0.108 g, 280 μmol), and N,N-diisopropylethylamine (150 μl, 280 μmol). The reaction was stirred at ambient temperature overnight. The reaction mixture was then poured into ethyl acetate, and the resulting organic solution washed in succession with 1N HCl (25 mL), 50% saturated aqueous NaHCO3 (25 mL), and brine (25 mL). The organic layer was then dried (MgSO4), filtered, and concentrated via rotary evaporation. The crude product was purified by preparative HPLC to afford 32 mg of the title compound (82 μmol, 58%).
1H NMR (400 MHz, CDCl3) δ 7.55 (d, J = 7.51 Hz, 1H), 7.18 - 7.38 (m, 6H), 7.08 (m, 1H), 6.89 (s, 1H), 6.33 (br s, 1H), 5.44 (br s, 1H), 5.03 (s, 2H), 4.49 (d, J = 5.1 Hz, 1H), 3.97 (dd, J = 17.3, 5.4 Hz, 1H), 3.86 (dd, J = 17.3, 5.4 Hz, 1H), 3.71 (s, 3H), 3.32 (dd, J = 14.4, 5.2 Hz, 1H), 3.13 (dd, J = 14.4, 7.8 Hz, 1H). MS: m/z = 391 [M + H]+.
Benzyl [(2S)-1-amino-1-oxo-4-phenylbutan-2-yl]carbamate (12)
To a solution of N-α-Cbz-L-homophenylalanine (16 mg, 50 μmol) in THF (0.5 mL) were added sequentially 1-hydroxybenzotriazole (9.5 mg, 70 μmol) and 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride (12 mg, 60 μmol). The mixture was stirred at ambient temperature for 10 min. Ammonium hydroxide (30% aq., 0.2 mL) was then added, and the reaction was stirred at ambient temperature for 2.5 days. The reaction mixture was diluted in ethyl acetate, and washed sequentially with saturated aqueous Na2CO3, saturated aqueous NaHCO3, and brine. The organic layer was separated, dried over MgSO4, filtered, and concentrated via rotary evaporation. The resulting oil was purified by silica gel column chromatography eluting with ethyl acetate in hexanes to provide 6.3 mg of the title compound (20 μmol, 40%).
1H NMR (400 MHz, CDCl3) δ 7.09 - 7.39 (m, 10H), 5.94 (br s, 1H), 5.49 (br s, 1H), 5.34 (d, J = 7.5 Hz, 1H), 5.10 (m, 2H), 4.19 (m, 1H), 2.69 (app t, J = 7.8 Hz, 2H), 2.18 (dtd, J = 13.7, 7.9, 6.2 Hz, 1H), 1.95 (dq, J = 14.1, 7.1 Hz, 1H); MS: m/z = 313 [M + H]+.
Benzyl {(2S)-1-amino-3-[methyl(phenyl)amino]-1-oxopropan-2-yl}carbamate (13)
To a solution of (2S)-2-{[(benzyloxy)carbonyl]amino}-3-[methyl(phenyl)amino]propanoic acid potassium hydrogen sulfate23 (23 mg, 50 μmol) in THF (0.5 mL) were added sequentially 1-hydroxybenzotriazole (9.5 mg, 70 μmol) and 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride (12 mg, 60 μmol). The mixture was stirred at ambient temperature for 10 min. Ammonium hydroxide (30% aq., 0.2 mL) was then added, and the reaction was stirred at ambient temperature for 2.5 days. The reaction mixture was diluted in ethyl acetate, and washed sequentially with saturated aqueous Na2CO3, saturated aqueous NaHCO3, and brine. The organic layer was separated, dried over MgSO4, filtered, and concentrated via rotary evaporation. The resulting oil was purified by silica gel column chromatography eluting with ethyl acetate in hexanes to provide 6.0 mg of the title compound (18 μmol, 37%). 1H NMR (400 MHz, CDCl3) δ 7.19 - 7.41 (m, 7H), 6.84 - 6.96 (m, 2H), 6.79 (t, J = 7.1 Hz, 1H), 6.13 (br s, 1H), 5.66 (br s, 1H), 5.57 (br s, 1H), 5.11 (s, 2H), 4.44 (br m, 1H), 3.74 (br d, J = 13.7 Hz, 1H), 3.45 (dd, J = 14.5, 9.3 Hz, 1H), 2.95 (s, 3H); MS: m/z = 328 [M + H]+.
Benzyl [(2S)-1-amino-1-oxo-3-phenylpropan-2-yl]carbamate (14)
To a solution of N-α-Cbz-L-phenylalanine (100 mg, 0.33 mmol) in THF (2 mL) were added sequentially 1-hydroxybenzotriazole (63 mg, 0.47 mmol) and 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride (77 mg, 0.40 mmol). The reaction mixture was stirred at ambient temperature for 15 min. Ammonium hydroxide (30% aq, 0.4 mL) was then added, and the reaction was stirred at ambient temperature overnight. The reaction was diluted in ethyl acetate, and washed sequentially with KHSO4 (10% aq.), saturated aqueous Na2CO3, and brine. The organic layer was separated, dried over MgSO4, filtered and concentrated via rotary evaporation. The resulting oil was purified by silica gel column chromatography eluting with ethyl acetate in hexanes to provide 58 mg of the title compound (0.19 mmol, 58%). 1H NMR (400 MHz, CDCl3) δ 7.14 - 7.39 (m, 10H), 5.60 (br s, 1H), 5.29 (br s, 1H), 5.08 (s, 2H), 4.42 (m, 1H), 3.13 (dd, J = 13.9, 6.0 Hz, 1H), 3.04 (dd, J = 13.7, 7.3 Hz, 1H); MS: m/z = 299 [M + H]+.
Benzyl [(2S)-1-amino-1-oxo-3-(pyridine-2-yl)propan-2-yl]carbamate (15)
To a suspension of N-α-Cbz-3-(pyridine-2-yl)alanine (25 mg, 83 μmol) in THF (1 mL) was added N-methyl morpholine (30 μL, 92 μmol). The solution was cooled in a 0 oC water/ice bath. Isobutyl chloroformate (13 μL, 0.10 mmol) was added, and the reaction was stirred at 0 oC for 5 min. Ammonium hydroxide (30% aq, 0.4 mL) was then added, and the reaction was stirred at ambient temperature for 4 h. The reaction was diluted in ethyl acetate, and washed sequentially with saturated aqueous NH4Cl, saturated aqueous NaHCO3, and brine. The organic layer was separated, dried over MgSO4, filtered and concentrated via rotary evaporation. The resulting oil was purified by silica gel column chromatography eluting with acetone in dichloromethane to provide 6.0 mg of the title compound (20 μmol, 24%).
1H NMR (400 MHz, CDCl3) δ 8.46 (d, J = 4.2 Hz, 1H), 7.61 (t, J = 7.3 Hz, 1H), 7.34 (br s, 4H), 7.23 (m, 1H), 7.16 (app t, J = 5.3 Hz, 2H), 6.73 (br s, 1H), 5.35 (br s, 1H), 5.12 (d, J = 12.5 Hz, 1H), 5.09 (d, J = 12.5 Hz, 1H), 4.64 (m, 1H), 3.34 (br d, J = 14.7 Hz, 1H), 3.24 (dd, J = 14.8, 6.0 Hz, 1H); MS: m/z = 300 [M + H]+.
Benzyl [(2S)-1-amino-1-oxo-3-(pyridine-4-yl)propan-2-yl]carbamate (16)
To a solution of N-α-Cbz-3-(pyridine-4-yl)alanine (0.25 mmol) in DMF (2.5 mL) were sequentially added 1-hydroxybenzotriazole (36 mg, 0.27 mmol), N,N-diisopropylethyl amine (100 μL, 0.60 mmol), and [dimethylamino(triazolo[4,5-b]pyridin-3-yloxy)methylene]-dimethyl-ammonium hexafluorophosphate (103 mg, 0.271 mmol). After 10 min, NH4OH (30% aq, 0.8mL) was added, and the reaction was stirred overnight. The solution was diluted in ethyl acetate and washed sequentially with water, saturated aqueous NaHCO3, and water. The organic layer was separated, dried over MgSO4, filtered, and concentrated via rotary evaporation. The crude material was purified via preparative HPLC to provide 3.0 mg of the title compound (10 μmol, 4%).
1H NMR (400 MHz, CD3CN) δ 8.47 (d, J = 5.7 Hz, 2H), 7.25 - 7.51 (m, 7H), 6.02 (br s, 1H), 5.86 (br s, 1H), 5.04 (d, J = 12.6 Hz, 1H), 4.98 (d, J = 12.5 Hz, 1H), 4.42 (td, J = 9.1, 4.9 Hz, 1H), 3.26 (dd, J = 13.8, 4.5 Hz, 1H), 2.96 (dd, J = 13.6, 9.8 Hz, 1H); MS: m/z = 300 [M + H]+.
Acknowledgement
This work was supported by NIH grant R01-GM86602 (MPJ) and by a grant from the Sandler Foundation (ARR). We thank Drs. Siegfried Leung, Joseline Ratnam, and Michelle Arkin for helpful conversations. MPJ is an advisor to Schrodinger LLC.
Footnotes
Abbreviations: HBD, hydrogen bond donor; HBA, hydrogen bond acceptor; P-gp, P-glycoprotein; HBA-AA, amino acid containing a hydrogen bond acceptor atom in the side chain.
References
- 1.Kuhn B, Mohr P, Stahl M. Intramolecular hydrogen bonding in medicinal chemistry. J. Med. Chem. 2010;53:2601–2611. doi: 10.1021/jm100087s. [DOI] [PubMed] [Google Scholar]
- 2.Rezai T, Bock JE, Zhou MV, Kalyanaraman C, Lokey RS, Jacobson MP. Conformational Flexibility, Internal Hydrogen Bonding, and Passive Membrane Permeability: Successful in Silico Prediction of the Relative Permeabilities of Cyclic Peptides. J. Am. Chem. Soc. 2006;128:14073–14080. doi: 10.1021/ja063076p. [DOI] [PubMed] [Google Scholar]
- 3.Rezai T, Yu B, Millhauser GL, Jacobson MP, Lokey RS. Testing the conformational hypothesis of passive membrane permeability using synthetic cyclic peptide diastereomers. J. Am. Chem. Soc. 2006;128:2510–2511. doi: 10.1021/ja0563455. [DOI] [PubMed] [Google Scholar]
- 4.Ashwood VA, Field MJ, Horwell DC, Julien-Larose C, Lewthwaite RA, McCleary S, Pritchard MC, Raphy J, Singh L. Utilization of an intramolecular hydrogen bond to increase the CNS penetration of an NK(1) receptor antagonist. J. Med. Chem. 2001;44:2276–2285. doi: 10.1021/jm010825z. [DOI] [PubMed] [Google Scholar]
- 5.Millan DS, Alex A, Perez M, Wakenhut F, Whitlock GA. Intramolecular hydrogen bonding to improve membrane permeability and absorption in beyond rule of five chemical space. Med. Chem. Commun. 2011;2:669–674. [Google Scholar]
- 6.White TR, Renzelman CM, Rand AC, Rezai T, McEwen CM, Gelev VM, Turner RA, Linington RG, Leung SS, Kalgutkar AS, Bauman JN, Zhang Y, Liras S, Price DA, Mathiowetz AM, Jacobson MP, Lokey RS. On-resin N-methylation of cyclic peptides for discovery of orally bioavailable scaffolds. Nat. Chem. Biol. 2011;7:810–817. doi: 10.1038/nchembio.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kalyanaraman C, Jacobson MP. An atomistic model of passive membrane permeability: application to a series of FDA approved drugs. J. Comput. Aid. Mol. Des. 2007;21:675–679. doi: 10.1007/s10822-007-9141-z. [DOI] [PubMed] [Google Scholar]
- 8.Egan WJ, Merz KM, Jr., Baldwin JJ. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000;43:3867–3877. doi: 10.1021/jm000292e. [DOI] [PubMed] [Google Scholar]
- 9.Ekins S, Durst GL, Stratford RE, Thorner DA, Lewis R, Loncharich RJ, Wikel JH. Three-dimensional quantitative structure-permeability relationship analysis for a series of inhibitors of rhinovirus replication. J. Chem. Inf. Comput. Sci. 2001;41:1578–1586. doi: 10.1021/ci010330i. [DOI] [PubMed] [Google Scholar]
- 10.Ekins S, Waller CL, Swaan PW, Cruciani G, Wrighton SA, Wikel JH. Progress in predicting human ADME parameters in silico. J. Pharmacol. Toxicol. 2000;44:251–272. doi: 10.1016/s1056-8719(00)00109-x. [DOI] [PubMed] [Google Scholar]
- 11.Fujikawa M, Ano R, Nakao K, Shimizu R, Akamatsu M. Relationships between structure and high-throughput screening permeability of diverse drugs with artificial membranes: Application to prediction of Caco-2 cell permeability. Bioorg. Med. Chem. 2005;13:4721–4732. doi: 10.1016/j.bmc.2005.04.076. [DOI] [PubMed] [Google Scholar]
- 12.Fujiwara S, Yamashita F, Hashida M. Prediction of Caco-2 cell permeability using a combination of MO-calculation and neural network. Int. J. Pharm. 2002;237:95–105. doi: 10.1016/s0378-5173(02)00045-5. [DOI] [PubMed] [Google Scholar]
- 13.Acharya C, Seo PR, Polli JE, MacKerell AD. Computational model for predicting chemical substituent effects on passive drug permeability across parallel artificial membranes. Mol. Pharm. 2008;5:818–828. doi: 10.1021/mp800035h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krarup LH, Christensen IT, Hovgaard L, Frokjaer S. Predicting drug absorption from molecular surface properties based on molecular dynamics simulations. Pharm. Res. 1998;15:972–978. doi: 10.1023/a:1011905522110. [DOI] [PubMed] [Google Scholar]
- 15.Refsgaard HH, Jensen BF, Brockhoff PB, Padkjaer SB, Guldbrandt M, Christensen MS. In silico prediction of membrane permeability from calculated molecular parameters. J. Med. Chem. 2005;48:805–811. doi: 10.1021/jm049661n. [DOI] [PubMed] [Google Scholar]
- 16.Bemporad D, Luttmann C, Essex JW. Behaviour of small solutes and large drugs in a lipid bilayer from computer simulations. Bba-Rev.Biomembranes. 2005;1718:1–21. doi: 10.1016/j.bbamem.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 17.Alper HE, Stouch TR. Orientation and Diffusion of a Drug Analog in Biomembranes - Molecular-Dynamics Simulations. J. Phys. Chem. 1995;99:5724–5731. [Google Scholar]
- 18.Bemporad D, Luttmann C, Essex JW. Computer simulation of small molecule permeation across a lipid bilayer: Dependence on bilayer properties and solute volume, size, and cross-sectional area. Biophysical Journal. 2004;87:1–13. doi: 10.1529/biophysj.103.030601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bemporad D, Essex JW, Luttmann C. Permeation of small molecules through a lipid bilayer: A computer simulation study. J. Phys. Chem. B. 2004;108:4875–4884. [Google Scholar]
- 20.Marrink SJ, Berendsen HJC. Permeation process of small molecules across lipid membranes studied by molecular dynamics simulations. J. Phys. Chem. 1996;100:16729–16738. [Google Scholar]
- 21.Ratemi ES, Vederas JC. Reaction of trimethylsilylamines with N-Cbz-L-serine- [beta]-lactone: A convenient route to optically pure [beta]-amino-L-alanine derivatives. Tetrahedron Lett. 1994;35:7605–7608. [Google Scholar]
- 22.Shelley JC, Cholleti A, Frye LL, Greenwood JR, Timlin MR, Uchimaya M. Epik: a software program for pK(a) prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 2007;21:681–691. doi: 10.1007/s10822-007-9133-z. [DOI] [PubMed] [Google Scholar]
- 23.Dolghih E, Bryant C, Renslo AR, Jacobson MP. Predicting binding to p-glycoprotein by flexible receptor docking. PLoS Comput. Biol. 2011;7:e1002083. doi: 10.1371/journal.pcbi.1002083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bissantz C, Kuhn B, Stahl M. A medicinal chemist’s guide to molecular interactions. J Med. Chem. 2010;53:5061–5084. doi: 10.1021/jm100112j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jorgensen WL, Tiradorives J. The OPLS Potential Functions for Proteins - Energy Minimizations for Crystals of Cyclic-Peptides and Crambin. J. Am. Chem. Soc. 1988;110:1657–1666. doi: 10.1021/ja00214a001. [DOI] [PubMed] [Google Scholar]
- 26.Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B. 2001;105:6474–6487. [Google Scholar]
- 27.Ghosh A, Rapp CS, Friesner RA. Generalized born model based on a surface integral formulation. J. Phys. Chem. B. 1998;102:10983–10990. [Google Scholar]
- 28.Luo R, Head MS, Given JA, Gilson MK. Nucleic acid base-pairing and N-methylacetamide self-association in chloroform: affinity and conformation. Biophys. Chem. 1999;78:183–193. doi: 10.1016/s0301-4622(98)00229-4. [DOI] [PubMed] [Google Scholar]