

Abstract
Small unextended molecules based on the diamidophosphate structure with a covalent carbon-to-phosphorus bond to improve hydrolytic stability were developed as a novel group of inhibitors to control microbial urea decomposition. Applying a structure-based inhibitor design approach using available crystal structures of bacterial urease, N-substituted derivatives of aminomethylphosphonic and P-methyl-aminomethylphosphinic acids were designed and synthesized. In inhibition studies using urease from Bacillus pasteurii and Canavalia ensiformis, the N,N-dimethyl derivatives of both lead structures were most effective with dissociation constants in the low micromolar range (K i = 13 ± 0.8 and 0.62 ± 0.09 μM, respectively). Whole-cell studies on a ureolytic strain of Proteus mirabilis showed the high efficiency of N,N-dimethyl and N-methyl derivatives of aminomethane-P-methylphosphinic acids for urease inhibition in pathogenic bacteria. The high hydrolytic stability of selected inhibitors was confirmed over a period of 30 days using NMR technique.
Keywords: Urease inhibition, Transition state analogues, Diamidophosphate, Aminophosphonic acid, Aminophosphinic acid
Introduction
Urease (urea amidohydrolase, E.C. 3.5.1.5) catalyzes the hydrolysis of urea to ammonia and carbamate (Karplus et al. 1997; Krajewska 2009). Subsequently, carbamate decomposes spontaneously to carbon dioxide and a second molecule of ammonia (Fig. 1).
Fig. 1.
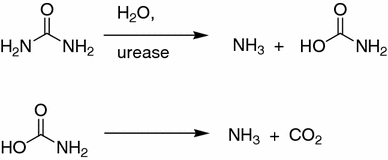
Enzymatic hydrolysis of urea
Ureolysis is a major virulence factor that enables Helicobacter pylori to colonize the highly acidic stomach environment due to the increase in pH caused by ammonia production (Burne and Chen 2000; Mobley et al. 1995; Mobley and Hausinger 1989). High ammonia concentration impairs hydrogen ion transport through mucosal tissue, which leads to ulcers and possibly cancer (Goodwin et al. 1986; Chen et al. 1986). Ureolytic bacteria infections of the urinary tract by Proteus species and Ureaplasma urealyticum with non-physiological alkalization of the urine induce stone formation, lead to chronic inflammatory disease combined with nephro- and ureolithiasis and cause a predisposition to opportunistic infections (Griffith 1979; Soriano and Tauch 2008; Worcester and Coe 2008).
The nitrogen cycle contributes to the ecosystem balance and includes nitrogen fixation, mineralization, nitrification and denitrification. Soil microorganisms play a crucial role in those mechanisms and maintaining balance is strongly dependent upon available nitrogen. Therefore, excessive urea fertilization and microbial enzymatic decomposition, which lead to uncontrolled ammonia release, are concerning (Mobley and Hausinger 1989). The use of urea in agriculture constitutes more that 50% of global N-fertilizer usage in addition to its growing application as an animal feed additive (Sahrawat 1980). Ammonia serves as the primary substrate in the two-step nitrification process that is conducted by autotrophic nitrifying bacteria. Enhanced ureolysis and nitrification in urea-fertilized soils results in N-losses due to ammonia volatilization and nitrate leaching. The local increase in pH due to high urease activity can damage plants in addition to the toxic effects of accumulating nitrate on seeds and germinating seedlings. Nitrogen losses resulting from these processes can amount to 50% of the fertilizer used (Gioacchini et al. 2002).
New strategies to regulate microbial urease activity both for therapeutic and agronomical purposes are currently being developed. Structurally diverse classes of urease inhibitors have been successfully characterized (Amtul et al. 2002). The most potent inactivators are phosphordiamides, which are classical transition state analogues (Faraci et al. 1995; Dominguez et al. 2008). Hydroxamates (Kobashi et al. 1962, 1971, 1975; Odake et al. 1992, 1994), imidazoles (Nagata et al. 1993; Kuehler et al. 1995), benzoquinones (Zaborska et al. 2002; Ashiralieva and Kleiner 2003), thiols (Ambrose et al. 1950; Kot et al. 2000), thioureas and selenoureas constitute other classes (Sivapriya et al. 2007). However, the most effective structures (particularly phosphordiamidates) lack stability in aqueous environments. A new class of compounds containing a hydrolytically stable C–P bond is one strategy for creating inhibitors with the desirable characteristics (Vassiliou et al. 2008, 2010). In our previous work, we used available crystal structures of bacterial urease for molecular modeling and refined chemical synthesis of peptidic derivatives of P-methyl- and P-hydroxymethyl-aminomethanephosphinic acids. We demonstrated the potential of this approach for the construction of active urease inhibitors.
In this paper, we describe the chemistry and biological activity of N-substituted derivatives of aminomethylphosphonic and P-methyl-aminomethylphosphinic acids towards plant and bacterial ureases. In comparison with previously studied compounds, the structure of these derivatives is less extended and less susceptible to hydrolysis. Molecular modeling studies helped to elucidate their mode of binding and to interpret kinetic data obtained in vitro.
Materials and methods
General
All reagents obtained from commercial suppliers (Aldrich, Fluka, Sigma, Merck, POCh) were of analytical grade and were used without further purification. All NMR spectra were recorded in D2O (99.8% D) with NaOD on Brucker Avance 300 or 600 MHz spectrometers. High-resolution mass spectra were measured on an LCT Premier XE (Waters) apparatus with electrospray ionization (positive ions). Preparative HPLC purifications were performed with Varian ProStar equipped with a C18 (21.4 mm × 250 mm) column (solvent A: 0.1% TFA in water, solvent B: 0.1% TFA in acetonitrile). The elute was monitored at both 210 and 254 nm. Gradient A (t [min], %B; flow rate: 10 mL/min): 0 min, 0%; 25 min, 18%; 35 min, 65%.
Chemistry
Compound 1 is commercially available (Aldrich). Compounds 2 (Rohovec et al. 1996), 5, 8, 10 (Tyka and Hagele 1984), 7 and 12 (Kudzin et al. 2005) were obtained based on literature protocols.
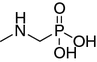
N,N-dimethylaminomethanephosphonic acid (4)
A mixture of N,N-dimethylamine (0.63 g, 10 mmol), diethyl phosphite (1.29 mL, 10 mmol) and paraformaldehyde (0.30 g, 10 mmol) was heated for 3 h at 80°C. The solution was diluted with diethylether (50 mL) and dried over magnesium sulfate. After evaporation of the solvent, the crude compound was purified using a preparative HPLC method. Pure diethyl N,N-dimethylaminomethanephosphonate was added to concentrated HCl (10 mL) and refluxed for 6 h. Evaporation of the reaction mixture in vacuo yielded crude product, which was dissolved in ethanol. Propylene oxide was added to the solution (to achieve pH 7) and pure compound 4 was precipitated. The compound was filtered and washed with acetone. Yield 0.97 g (70%). 1H NMR (D2O, ppm): δ 2.93 (s, 6H, 2×CH3, NCH3), 3.26 (d, 2H, J = 12.6 Hz, CH2P), 31P NMR (D2O, ppm): δ 7.35. HR-ESI MS (M + 1), expected 140.0477, found 140.0472.
N-Benzyl-N-methylaminomethanephosphonic acid (6)
Diethyl N-(benzyl-N-methyl)aminomethanophosphonate (2.1 g, 8 mmol) (Karp 1999) was hydrolyzed with concentrated HClaq using the above-described procedure for diethyl N,N-dimethylaminomethanephosphonate. Yield 1.46 g (85%). 1H NMR (D2O, ppm): δ 2.75 (s, 3H, NCH3), 3.13 (bs, 2H, CH2P), 4.39 and 4.17 (2*bs, 2H, CH2Ph), 7.34 (m, 5H, Ar), 31P NMR (D2O, ppm): δ 8.0. HR-ESI MS (M + 1), expected 216.0790, found 216.0790.
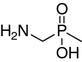
N-methylaminomethanephosphonic acid (3)
N-benzyl-N-methylaminomethanephosphonic acid (6) (2.75 g, 12.69 mmol) was dissolved in acetic acid (150 mL) and palladium catalyst (10% Pd/C) was added. Hydrogenation at 1 atm was performed for 12 h. After filtration of the catalyst, the solution was evaporated in vacuo. The residue was dissolved in water and evaporated again. Then, the residue was dissolved in HCl (20%, 80 mL) and evaporated. The crude hydrochloride product was dissolved in ethanol (100 mL) and propylene oxide was added to achieve a pH of 7. Precipitated compound 3 was filtered off and washed with acetone. Yield 0.93 g (44%). 1H NMR (D2O, ppm): δ 2.62 (s, 3H, CH3N), 2.96 (d, 2H, J = 12 Hz, CH2P), 31P NMR (D2O, ppm): δ 9.5. HR-ESI MS (M + 1), expected 126.0320, found 126.0309.
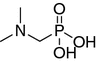
N,N-dimethylaminomethane-P-methylphosphinic acid (9)
(N-benzylamino)methane-P-methylphosphinic acid 10 (15 mmol, 3.0 g) and sodium carbonate (45 mmol, 4.8 g) were dissolved with stirring in 30 mL of water. The mixture was cooled in an ice-water bath. Next, iodomethane (39 mmol, 2.43 mL) in 30 mL of dioxane was added drop wise, and the mixture was stirred overnight. Precipitated solid (NaI) was filtered off and volatiles were removed in vacuo. Excess methyl iodide was removed by extraction with ether, and the residue was cautiously acidified with 2.0 M HCl until evolution of CO2 ceased. After evaporation of water, the solid residue was dissolved in methanol, and insoluble inorganic salts were filtered off. Methanol was then removed in vacuo to obtain 3.75 g (95%) of (N-benzyl-N,N-dimethylamino)methane-P-methylphosphinic acid hydrochloride.
Potassium carbonate (7.9 g) and 10% palladium on a carbon catalyst (0.9 g) were added to the stirred solution of crude (N-benzyl-N,N-dimethylamino)methane-P-methylphosphinic acid hydrochloride. Hydrogen was passed through the mixture under atmospheric pressure. After 3 h, NMR analysis of the mixture showed complete conversion of the substrate. The solids were filtered off and the solvent was removed in vacuo. The residue was dissolved in water and acidified with 2.0 M HCl (pH < 2). The resulting yellow–brown mixture was washed several times with chloroform to remove chloroform-soluble color contaminants. Water was then removed in vacuo and the residue was dissolved in acetonitrile. Insoluble inorganic salts were filtered off and the solvent of the filtrate was removed in vacuo. Finally, 1.9 g (80%) of 8 hydrochloride was obtained. 1H NMR (D2O, ppm): δ 1.43 (d, J = 14.4 Hz, 3H, PCH3); 3.00 (s, 6H, N(CH3)2); 3.37 (d, J = 8.6 Hz, 2H, PCH2). 31P NMR (D2O, ppm): δ 34.12. HR-ESI MS (M + 1), expected 138.0684, found 138.0661.
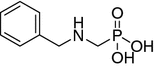
(N-benzyl-N-methylamino)methane-P-methylphosphinic acid (11)
A 100 mL flask equipped with a condenser was filled with benzylmethylamine (33 mmol, 4.25 mL) in 30 mL of water and heated on a steam bath. Next, 37% formalin (49 mmol 1.48 g) was added to the hot mixture. After 1 h, another portion of formalin was added followed by heating for an additional 2 h. Then the reaction mixture was cooled down to room temperature and the volatiles were removed in vacuo. The residue was dissolved in chloroform, washed with brine and dried over anhydrous sodium sulfate. The desiccant was filtered off and chloroform was removed in vacuo to yield crude N-hydroxymethyl-N-methylbenzylamine.
A portion of the crude N-hydroxymethyl-N-methylbenzylamine (12.6 mmol, 1.90 g) was dissolved in 30 mL of glacial acetic acid and added dropwise to dichloromethylphosphine (11.5 mmol, 1.0 mL). The mixture was magnetically stirred and cooled in an ice-water bath under inert atmosphere. After 15 min, the cooling bath was removed, and the mixture was heated under reflux for 40 min. When boiling ceased, 20% hydrochloric acid (40 mL) was added carefully to the reaction through the condenser. Hydrolysis was performed for an additional 30 min under reflux. The mixture was cooled, and the volatile products were removed in vacuo. The remaining residue was dissolved in 35 mL of ethanol and treated with propylene oxide until formation of precipitate could be observed. The precipitated white solid was filtered off, washed with acetone and dried. The product was separated with HPLC chromatography (gradient A) to yield 0.85 g (35% yield) of 4. 1H NMR (D2O, ppm): δ 1.25 (d, J = 14.4 Hz, 3H, PCH3); 2.85 (s, 3H, NCH3); 3.19 (bs, 2H, PCH2) 4.35 (s, 1H, PhCH2), 7.43 (s, 5H, Ph). 31P NMR (D2O, ppm): δ 28.80. HR-ESI MS (M + 1), expected 214.0997, found 214.0981.
Enzyme purification
Bacillus pasteurii CCM 2056T was grown in a nutrient media containing 20 g urea, 20 g/L of yeast extract with addition of 1 mM NiCl2, pH 8 at 30°C. The cultures were incubated for 48 h, yielding about 4.7 g/L of wet cells. The collected cells were resuspended in lysis buffer containing 50 mM phosphate, pH 7.5, 1 mM β-mercaptoetanol, and 1 mM EDTA and sonicated. Unbroken cells and cell debris were removed by centrifugation. The supernatant was clarified using a 0.22 μm filter (Rotilabo®, ROTH) and then desalted on a BioGel column (Bio-Rad). The obtained fractions were used as the starting material for the urease purification. The enzyme preparation procedure consisted of three steps: anion-exchange (Q Sepharose, GE Healthcare), hydrophobic (Phenyl Sepharose, GE Healthcare) and affinity chromatography (Cellufine Sulphate, Chisso Corporation). Initially the sample was loaded onto a Q Sepharose column equilibrated with 50 mM phosphate buffer at pH 7.5. Urease-containing fractions were eluted with a linear gradient of NaCl (0–1.5 M). The ionic strength of the obtained fractions was increased to 1 M (NH4)2SO4 and then applied onto a Phenyl Sepharose column. Urease was developed with a descending gradient of (NH4)2SO4 in 50 mM phosphate buffer, pH 7.5. The collected fractions were dialyzed against 20 mM phosphate buffer, pH 6.5 and then loaded onto a Cellufine Sulphate column, which had been equilibrated with the same buffer. Enzyme elution was performed with 20 mM phosphate buffer containing 0.5 M NaCl, pH 7.5. All of the purification steps were performed at 10°C (cold room) using an AKTA Prime system (Amersham Biosciences).
Partially purified urease from Bacillus pasteurii CCM 2056T exhibited Michaelis–Menten saturation kinetics with a K M of 17.6 ± 0.9 mM, V max and a specific activity of approximately 1,530.3 U/mg.
Urease activity was tested during purification by two different methods: a qualitative phenol red urease test based on the color change of the phenol red pH indicator as a result of urea hydrolysis (during the hydrophobic column chromatography step) and a quantitative indophenol assay measuring the amount of released ammonia (all other steps).
Inhibition test
The inhibitory properties of the analyzed compounds were evaluated using a colorimetric indophenol assay. Aliquots of the enzyme solution (10 μL) were incubated with commercially available jack bean urease (Sigma) with an estimated K M of 2.7 ± 0.6 mM or partially purified urease from Bacillus pasteurii CCM 2056T (K M 17.6 ± 0.9 mM) in 10 mM phosphate buffer, pH 7.0 in the presence of various inhibitor concentrations (0.001–1 mM) in 96-well plates. The IC50 determination was performed with 50 mM urea for bacterial urease and 20 mM for plant urease. For the K i measurements, the urea concentration varied from 10 to 100 mM and from 0.5 to 15 mM for the abovementioned enzymes, respectively. After 20 min of incubation at 37°C, the reaction was stopped by adding phenol-hypochlorite reagents and the absorbance of the formed indophenol blue complexes was measured at 650 nm with a Tecan Sunrise absorbance reader. The IC50 and K i parameters of the analyzed inhibitors were calculated with GraphPad Prism 5 software. All inhibitory tests were run in triplicate.
The qualitative red phenol assay was used during enzyme purification. The urease activity of the extracted fractions (10 μL) was monitored spectrophotometrically at 560 nm by measuring the color change of the mixture containing 0.2 mM phenol red and 100 mM urea in 10 mM phosphate buffer, pH 7.0 after incubation at 37°C.
Whole-cell urease activity assay
Proteus mirabilis CCM 1944 was routinely grown at 37°C on solidified medium containing 2% yeast extract, 2% urea and 1 mM NiCl2 (Benini et al. 1996). Christensen urea agar was used to confirm the ureolytic activity of the strain (Vuye and Pijck 1973).
Cells harvested in early log phase (20 h of culture) were suspended in saline at OD650 = 0.1 (Cfu = 1.2 × 105 mL−1). Urease was assayed in Eppendorf polypropylene microplates (50–350 μL well) in a 100 μL mixture containing modified Christensen liquid medium depleted of phosphate and urea, adjusted to pH 5.5 with citric acid. For IC50 estimation, 10 μL of the cell suspension were preincubated in the presence of a defined inhibitor concentration at 37°C for 15 min and the reactions were started with the addition of 5 mM urea. The IC50 values were calculated as described elsewhere (Vassiliou et al. 2008). The microplates were incubated in ELMI SkyLine thermostatic incubator at 37°C with gentle stirring (120 rpm). The reactions were stopped and ammonia quantified by subsequent addition of phenol/nitroprusside and NaOH/hypochlorite solutions (100 μL each) (Vassiliou et al. 2008). The plates were read at 650 nm using a TECAN Sunrise microplate reader. The progress curves were initiated by the addition of 20 μL of a cell suspension into 200 μL reactions in Eppendorf tubes in the presence of 10 mM urea at 37°C. Reactions were stopped by the addition of phenol and hypochlorite solutions (500 μL each) and then read at 650 nm.
Molecular modeling
The crystal structure of the phosphate-urease (Bacillus pasteurii) complex refined to 1.85 Å obtained from the Protein Data Bank (1IE7) was used as the starting point (Benini et al. 2001). All calculations were done with the use of the Insight 2000 (Accelrys) molecular modeling program package. Hydrogen atoms were added and the protonation states for all side chains were set for pH 7.0. The structure of the ligand was appropriately modified with the Builder module and the inhibitor-enzyme complex was optimized using the Discover program and cff97 force field.
Results and discussion
Urease is a large heteropolymeric enzyme with a variable subunit composition depending on the organism (Krajewska 2009; Ham et al. 2001; Benini et al. 2000). Despite the significant variability in the overall composition of the enzyme, the structure of its active site is highly conserved. The active site contains two nickel ions, has a relatively small volume, and is covered by a flap during the reaction. The most effective urease inhibitors are diamidophosphate (DAP) and its esters, which hydrolyze to the active molecule (DAP) in the active site (Benini et al. 1999). Diamidophosphate is a classical transition state analogue. Because the structure of DAP contains hydrolyzable (particularly at low pH) P–N bonds, we aimed to explore extended transition state analogues containing highly stable P–C bonds, such as aminomethylphosphonic and aminomethyl-P-methylphosphinic acids (compounds 1 and 2, Fig. 2). Previously, we showed that 2 is a moderate urease inhibitor and that some of its peptidic derivatives are very potent. Here, we used N-alkylated derivatives of 2 and its close analogue 1—structures 3-6 and 8-11. Although the proposed substituents do not offer the possibility of forming additional hydrogen bonds in contrast to the parent molecules, they allow for controlling the number of hydrogen bond donors (methyl moieties) and/or for the creation of aromatic interactions (benzyl groups). Moreover, this type of substitution provides highly stable derivatives.
Fig. 2.
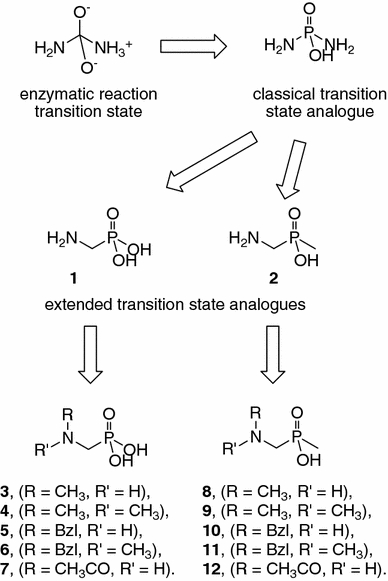
Structures of designed compounds
The parent molecules 1 and 2 are readily available—either commercially (1) or using standard protocols from the literature (2) (Rohovec et al. 1996). The use of the parent molecules in the synthesis of other designed compounds is not convenient with the exception of N-acetylated structures 7 and 12, which were obtained quantitatively by reaction of compounds 1 and 2, respectively, with excess acetic anhydride in acetic acid (Kudzin et al. 2005). Disubstituted phosphonic derivatives 4 and 6 were obtained from a three-component condensation of the appropriate amine, formaldehyde and diethylphosphite and by subsequent acidic hydrolysis (Scheme 1). Compound 6 was also used for the synthesis of the N-methyl analogue 3 by hydrogenolysis.
Scheme 1.
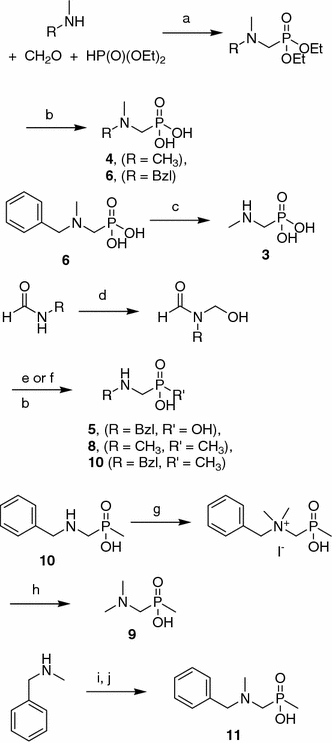
Reagents and conditions: a reflux, b HClaq, then popylene oxide, c H2, 10% Pd/C, d 37% formalin, steam bath, e PCl3, (for compound 5), f CH3PCl2,(for compounds 8, 10), g CH3I, Na2CO3, h H2, 10% Pd/C, K2CO3, i CH2Oaq, j CH3PCl2, then HClaq
Monosubstituted derivatives were synthesized using the known method with N-hydroxymethyl-N-alkylformamides as the starting material (Tyka and Hagele 1984). The reaction of an appropriate substrate with phosphorus thrichloride or dichloromethylphosphine and subsequent hydrolysis produced compounds 5, 8 and 10. N, N-dimethylaminomethane-P-methylphosphinic acid 9 was obtained using exhaustive methylation of the N-benzylated compound 10 and hydrogenolytic removal of the benzyl group under basic conditions (Hulme and Rosser 2002). Finally, we synthesized 11 by reaction of benzylmethylamine with formaline and fosfinylation of the hydroxymethyl intermediate with CH3PCl2.
All synthesized compounds were assayed against urease purified from Bacillus pasteurii CM2056T (Vassiliou et al. 2008) and commercially available enzyme from Canavalia ensiformis (Table 1). Kinetic data were obtained using the indophenol blue assay based on the Berthelot reaction (Chaney and Marbach 1962). Progress curves of the urease reaction in the presence of the studied compounds revealed the competitive reversible mode of inhibition within the micromolar range for most of the compounds. Both parent molecules (1 and 2) exhibited similar potency against the bacterial enzyme with inhibitory constants of 314 and 340 μM, respectively. Subsequent substitution of the nitrogen atom with a methyl group caused a significant increase in the activity (inhibitors 3 and 8). Introduction of a second methyl moiety (compounds 4 and 9) further reduced the inhibition constants, K i to 13 μM and 620 nM, respectively. The difference in the free energy of binding (ΔΔG) between the unsubstituted compound 2 and its N-methyl analogue 8 as well as between inhibitor 8 and its N,N-dimethyl counterpart 9 is similar (1.7 and 2.0 kcal/mol, respectively) and corresponds to the energy of hydrogen bond breakage. We assume that during the formation of the urease-inhibitor 2 complex, three hydrogen bonds between bulk water and inhibitor amine group are broken and replaced with only one interaction with the enzyme active residues.
Table 1.
Inhibitory activities of aminomethanephosphonic and aminomethyl-P-methylphosphinic acid derivatives (NI not inhibitory)
| No | Compound | Canavalia ensiformis urease | Bacillus pasteurii urease | ||
|---|---|---|---|---|---|
| IC50 [μM] | K i [μM] | IC50 [μM] | K i [μM] | ||
| 1 |
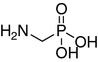
|
432 ± 37.8 | 240 ± 64.3 | 700 ± 23 | 314 ± 8.2 |
| 2 |
|
NI | 1100 ± 10.5a | 340 ± 22a | |
| 3 |
|
678 ± 48 | 204 ± 52 | 228 ± 15.6 | 70 ± 2,2 |
| 4 |
|
82 ± 26 | 21 ± 5.7 | 49 ± 1.7 | 13 ± 0.8 |
| 5 |
|
NI | NI | ||
| 6 |
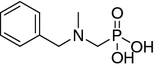
|
NI | 300 ± 6.5 | 115 ± 7.4 | |
| 7 |
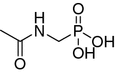
|
NI | 463 ± 34 | 148 ± 18.5 | |
| 8 |
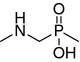
|
884 ± 14 | 224 ± 39 | 60.0 ± 0.3a | 18.0 ± 0.7a |
| 9 |
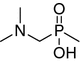
|
14.4 ± 4.8 | 3.7 ± 1.7 | 3.8 ± 0.4 | 0.62 ± 0.09 |
| 10 |
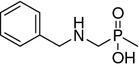
|
1 632 ± 226 | 571 ± 119 | 256 ± 12.8 | 72 ± 3.8 |
| 11 |
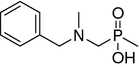
|
NI | NI | ||
| 12 |
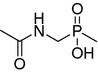
|
1 455 ± 189 | 295 ± 96 | 258 ± 12.7 | 95 ± 6.3 |
aValues reported previously (Vassiliou 2008)
In the case of inhibitor 8, two hydrogen bonds with its amine group are broken (with bulk water) and one is formed (with enzyme) upon complexation with urease. The energy balance is most favorable with inhibitor 9 because one hydrogen bond is broken and one is formed with its amine group during the complexation process. The modeled mode of binding of the most active inhibitor 9 shows that its amine group forms one hydrogen bond with the carbonyl moiety of Ala366, which is consistent with the experimental data for differences in the free energy of binding. The phosphinic acid group of this inhibitor forms two hydrogen bonds with His222 and Asp363 and interacts with the two nickel ions (Fig. 3).
Fig. 3.
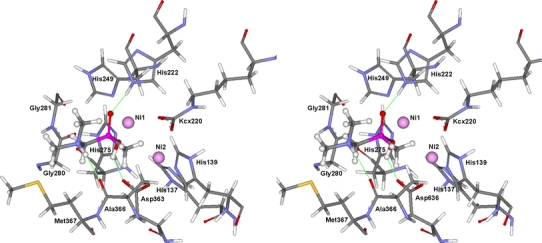
Stereo image of modeled mode of binding of compound 9 to the Bacillus pasteurii urease active site. Hydrogen bonds are marked as thin solid lines
In addition to the N-methyl compounds, we also evaluated the acetylated and N-benzylated compounds. Unfortunately, these modifications did not enhance the inhibitory potency of the compounds. Most likely, the space available within the active site of urease imposes steric requirements, which cannot be fulfilled by the additional aromatic substituents of compounds 5, 6, 10 and 11. The enzyme delivered from plant material was generally less susceptible to inhibition by the studied compounds consistent with data published on other urease inhibitors. Within the analyzed group of inhibitors, analogously to the bacterial enzyme, structures 4 and 9 were the most potent with K i values equal 21 and 3.7 μM, respectively.
We evaluated the hydrolytic stability of the most active compounds 3, 4, 8 and 9. NMR analysis of aqueous solutions (pH 2) of the compounds showed unchanged spectral characteristics after 30 days at room temperature. It is worth to note that, the class of compounds presented herein is considerably more stable than the widely studied phosphoramidates (Pope et al. 1998).
For whole-cell urease inhibition studies, we used the Proteus mirabilis strain, which is a common pathogen of the human urinary tract (Mobley et al. 1995). The inhibition studies using whole cells were all performed at pH 5.5, which corresponds to the acidity of physiological urine. Plate cell counts proved that the Proteus cells remained viable and actively multiplied under the assay conditions used.
The effectiveness of urease inhibition against intact cells was evaluated for compounds 8 and 9, and compared with well-established acetohydroxamic acid (AHA). In the presence of 5 mM urea using 1.2 × 103 cells, the IC50 values were 154 ± 6 μM for compound 9, 36 ± 3 μM for compound 8 and 64.6 ± 3 μM for AHA. Similarly to the results of the cell-free inhibition studies (Table 1), the values were in the micromolar range in the whole-cell assay and the mono-substituted derivative 8 was a more efficient inhibitor than the N,N-dimethyl compound 9.
Similar to acetohydroxamate, the time course of urease activity in the presence of compounds 9 and 8 in non-preincubated system showed a short lag phase of inhibition (Fig. 4). The non-preincubated system likely requires time to allow the interaction between the intracellular enzyme and inhibitor. Because the Proteus urease is located in the cell cytoplasm, the inhibitory efficiency is strongly dependent on the compound diffusivity. The obtained results show that the newly developed small structures containing the remarkably stable carbon-to-phosphorus bond are active against urease in intact bacterial cells and their effectiveness is comparable with acetohydroxamic acid.
Fig. 4.
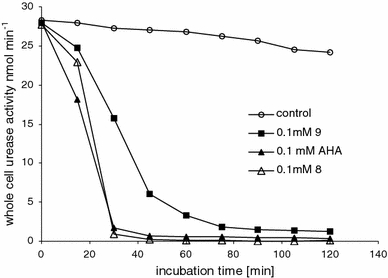
Time course showing the effects of 100 μM urease inhibitors acetohydroxamic acid (AHA), compound 8 and 9 on P. mrabilis urease activity in the whole-cell non-preincubated assay
Conclusions
Herein, we explored the potential of phosphonates and phosphinates as urease inhibitors. This is the first report on phosphonic acids designed for use against bacterial urease. However, the P-methylphosphinates were developed to enhance the inhibitory activity of previously published structures (Vassiliou et al. 2008). We investigated chemical structures based on the C–P bond as an attractive alternative to known phosphoramidates. Our studies led to the synthesis of structures with low structural complexity, high hydrolytic stability and satisfactory biological activity against purified cytoplasmic urease of pathogenic Proteus species. The most active compound 9 showed high inhibitory activity against bacterial urease with a K i of 0.62 μM, making it one of the most active reversible urease inhibitors (Kosikowska and Berlicki 2011; Follmer 2010; Amtul et al. 2002). The most widely studied and clinically used inhibitor acetohydroxamic acid showed a K i of 2.6 μM (against Klebsiella aerogenes enzyme, Todd 1989) and similar activity against whole cells. Such characteristics imply that these compounds could be used in therapeutic and agricultural applications. Moreover, these structures offer the possibility of various modifications, which might provide improved physicochemical and inhibitory properties.
Acknowledgments
This work was supported by a grant from the Polish Ministry of Science and Higher Education (no. PBZ-MEiN-5/2/2006 and 681/N-COST/2010/0). The calculations were performed using hardware and software resources (including the Accelrys programs) at the Supercomputing and Networking Center in Wrocław. P. Kafarski, Ł. Berlicki, A. Białas and P. Kosikowska thank the Foundation for Polish Science for financial support. P. Kosikowska is grateful for a fellowship that was co-financed by the European Union and the European Social Fund.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- Ambrose JF, Kistiakowsky GB, Kridl AG. Inhibition of urease by sulfur compounds. J Am Chem Soc. 1950;72:317–321. doi: 10.1021/ja01157a083. [DOI] [Google Scholar]
- Amtul Z, Atta-ur-Rahman, Siddiqui RA, Choudhary MI (2002) Chemistry and Mechanism of Urease Inhibition. Curr Med Chem 9:1323–1348 [DOI] [PubMed]
- Ashiralieva A, Kleiner D. Polyhalogenated benzo- and naphthoquinones are potent inhibitors of plant and bacterial urease. FEBS Lett. 2003;555:367–370. doi: 10.1016/S0014-5793(03)01289-4. [DOI] [PubMed] [Google Scholar]
- Benini S, Rypniewski WR, Wilson KS, Ciurli S, Mangani S. Structure-based rationalization of urease inhibition by phosphate: novel insights into the enzyme mechanism. J Biol Inorg Chem. 2001;6:778–790. doi: 10.1007/s007750100254. [DOI] [PubMed] [Google Scholar]
- Benini S, Rypniewski WR, Wilson KS, Miletti S, Ciurli S, Mangani S. The complex of Bacillus pasteurii urease with acetohydroxamate anion from X-ray data at 1.55 Å resolution. J Biol Inorg Chem. 2000;5:110–118. doi: 10.1007/s007750050014. [DOI] [PubMed] [Google Scholar]
- Benini S, Rypniewski WR, Wilson KS, Miletti S, Ciurli S, Mangani S. A new proposal for urease mechanism based on the crystal structures of the native and inhibited enzyme from Bacillus pasteurii: why urea hydrolysis costs two nickels. Struct Fold Des. 1999;7:205–216. doi: 10.1016/S0969-2126(99)80026-4. [DOI] [PubMed] [Google Scholar]
- Benini S, Gessa C, Ciurli S. Bacillus pasteurii urease: a heteropolymeric enzyme with a binuclear nickel active site. Soil Biol Biochem. 1996;28:819–821. doi: 10.1016/0038-0717(96)00017-X. [DOI] [Google Scholar]
- Burne RA, Chen YM. Bacterial ureases in infectious diseases. Microb Infect. 2000;2:533–542. doi: 10.1016/S1286-4579(00)00312-9. [DOI] [PubMed] [Google Scholar]
- Chaney AL, Marbach EP. Modified reagents for determination of urea and ammonia. Clin Chem. 1962;8:130–132. [PubMed] [Google Scholar]
- Chen XG, Correa P, Offerhaus J, Rodriguez E, Janney F, Hoffmann E, Fox J, Hunter F, Diavolitsis S. Ultrastructure of the gastric mucosa harboring Campylobacter-like organisms. Am J Clin Pathol. 1986;86:575–582. doi: 10.1093/ajcp/86.5.575. [DOI] [PubMed] [Google Scholar]
- Dominguez MJ, Sanmartin C, Font M, Palop JA, San Francisco S, Urrutia O, Houdusse F, Garcia-Mina JM. Design synthesis and biological evaluation of phosphoramide derivatives as urease inhibitors. J Agric Food Chem. 2008;56:3721–3731. doi: 10.1021/jf072901y. [DOI] [PubMed] [Google Scholar]
- Faraci WS, Yang BV, O’Rourke D, Spencer RW. Inhibition of Helicobacter pylori urease by phenyl phosphorodiamidates: mechanism of action. Bioorg Med Chem. 1995;3:605–610. doi: 10.1016/0968-0896(95)00043-G. [DOI] [PubMed] [Google Scholar]
- Follmer C. Ureases as a target for the treatment of gastric and urinary infections. J Clin Pathol. 2010;63:424–430. doi: 10.1136/jcp.2009.072595. [DOI] [PubMed] [Google Scholar]
- Gioacchini P, Nastri A, Marzadori C, Giovannini C, Vittori AL, Gessa C. Influence of urease and nitrification inhibitors on N losses from soils fertilized with urea. Biol Fert Soil. 2002;36:129–135. doi: 10.1007/s00374-002-0521-1. [DOI] [Google Scholar]
- Goodwin CS, Armstrong JA, Marshall BJ. Campylobacter pyloridis gastritis and peptic ulceration. J Clin Pathol. 1986;39:353–365. doi: 10.1136/jcp.39.4.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith DP. Urease stones. Urol Res. 1979;7:215–221. doi: 10.1007/BF00257208. [DOI] [PubMed] [Google Scholar]
- Ham NC, Oh ST, Sung JY, Cha KA, Lee MH, Oh BH. Supramolecular assembly and acid resistance of Helicobacter pylori urease. Nat Struct Biol. 2001;8:505–509. doi: 10.1038/88563. [DOI] [PubMed] [Google Scholar]
- Hulme AN, Rosser EM. An aldol-based approach to the synthesis of the antibiotic anisomycin. Org Lett. 2002;4:265–267. doi: 10.1021/ol017016u. [DOI] [PubMed] [Google Scholar]
- Karp GM. An expeditious route to novel 142-benzodiazaphosphepin-5-one 2-oxide analogues. J Org Chem. 1999;64:8156–8160. doi: 10.1021/jo9908400. [DOI] [PubMed] [Google Scholar]
- Karplus PA, Pearson MA, Hausinger RP. 70 years of crystalline urease: what have we learned? Acc Chem Res. 1997;30:330–337. doi: 10.1021/ar960022j. [DOI] [Google Scholar]
- Kobashi K, Hase J, Uehare K. Specific inhibition of urease by hydroxamic acids. Biochim Biophys Acta. 1962;65:380–383. doi: 10.1016/0006-3002(62)91067-3. [DOI] [PubMed] [Google Scholar]
- Kobashi K, Kumaki K, Hase J. Effect of acyl residues of hydroxamic acids on urease inhibition. Biochim Biophys Acta. 1971;227:429–441. doi: 10.1016/0005-2744(71)90074-x. [DOI] [PubMed] [Google Scholar]
- Kobashi K, Takebe S, Terashima N, Hase J. Inhibition of urease activity by hydroxamic acid derivatives of amino acids. J Biochem (Tokyo) 1975;77:837–843. doi: 10.1093/oxfordjournals.jbchem.a130791. [DOI] [PubMed] [Google Scholar]
- Kosikowska P, Berlicki Ł (2011) Urease inhibitors as potential drugs for gastric and urinary tract infections: a patent review. Expert Op Ther Pat. doi:10.1517/13543776.2011.574615 [DOI] [PubMed]
- Kot M, Zaborska W, Juszkiewicz A. Inhibition of jack bean urease by thiols: Calorimetric studies. Thermochim Acta. 2000;354:63–69. doi: 10.1016/S0040-6031(00)00451-2. [DOI] [Google Scholar]
- Krajewska B. Ureases I functional catalytic and kinetic properties: a review. J Mol Cat B. 2009;59:9–21. doi: 10.1016/j.molcatb.2009.01.003. [DOI] [Google Scholar]
- Kudzin ZH, Depczyński R, Andijewski G, Drabowicz J, Łuczak J. 1-(N-acylamino)alkanephosphonates Part IV. N-Acylation of 1-aminoalkanephosphonic Acids. Pol J Chem. 2005;79:499–513. [Google Scholar]
- Kuehler TC, Fryklund J, Bergman N, Weilitz J, Lee A, Larsson H. Structure-activity relationship of omeprazole and analogs as Helicobacter pylori urease inhibitors. J Med Chem. 1995;38:4906–4916. doi: 10.1021/jm00025a008. [DOI] [PubMed] [Google Scholar]
- Mobley HL, Hausinger RP. Microbial ureases: significance regulation and molecular characterization. Microbiol Rev. 1989;53:85–108. doi: 10.1128/mr.53.1.85-108.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobley HLT, Island MD, Hausinger RP. Molecular biology of microbial ureases. Microbiol Rev. 1995;59:451–480. doi: 10.1128/mr.59.3.451-480.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata K, Satoh H, Iwahi T, Shimoyama T, Tamura T. Potent inhibitory action of the gastric proton pump inhibitor lansoprazole against urease activity of Helicobacter pylori: unique action selective for H. pylori cells. Antimicrob Agents Chemother. 1993;37:769–774. doi: 10.1128/aac.37.4.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odake S, Morikawa T, Tsuchiya M, Imamura L, Kobashi K. Inhibition of Heliocobacter pylori urease activity by hydroxamic acid derivatives. Biol Pharm Bull. 1994;17:1329–1332. doi: 10.1248/bpb.17.1329. [DOI] [PubMed] [Google Scholar]
- Odake S, Nakahashi K, Morikawa T, Takebe S, Kobashi K. Inhibition of Urease activity by dipeptidyl hydroxamic acids. Chem Pharm Bull (Tokyo) 1992;40:2764–2768. doi: 10.1248/cpb.40.2764. [DOI] [PubMed] [Google Scholar]
- Pope AJ, Toseland CD, Rushant B, Richardson S, McVey M, Hills J. Effect of potent urease inhibitor, fluorofamide, on Helicobacter sp. in vivo and in vitro. Dig Dis Sci. 1998;43:109–119. doi: 10.1023/A:1018884322973. [DOI] [PubMed] [Google Scholar]
- Rohovec J, Luke I, Vojtíek P, Císaová I, Hermann P. Complexing properties of phosphinic analogs of glycine. J Chem Soc Dalton Trans. 1996;1996:2685–2691. doi: 10.1039/dt9960002685. [DOI] [Google Scholar]
- Sahrawat KL. Control of urea hydrolysis and nitrification in soil by chemicals. Prospects and problems. Plant Soil. 1980;57:335–352. doi: 10.1007/BF02211691. [DOI] [Google Scholar]
- Sivapriya K, Suguna P, Banerjee A, Saravanan V, Rao DN, Chandrasekaran S. Facile one-pot synthesis of thio and selenourea derivatives: a new class of potent urease inhibitors. Bioorg Med Chem Lett. 2007;17:6387–6391. doi: 10.1016/j.bmcl.2007.07.085. [DOI] [PubMed] [Google Scholar]
- Soriano F, Tauch A. Microbiology and Clinical Features of Corynebacterium urealyticum: urinary tract stones and genomics as the rosetta stone. Clin Microbiol Infect. 2008;14:632–643. doi: 10.1111/j.1469-0691.2008.02023.x. [DOI] [PubMed] [Google Scholar]
- Todd MJ, Hausinger RP. Competitive inhibitors of Klebsiella aerogenes urease. Mechanisms of interaction with the nickel active site. J Biol Chem. 1989;264:15835–15842. [PubMed] [Google Scholar]
- Tyka R, Hagele GA. convenient synthesis of N-alkylaminomethanephosphonic and N-alkylaminomethanephosphinic acids. Synthesis. 1984;1984:218–219. doi: 10.1055/s-1984-30776. [DOI] [Google Scholar]
- Vassiliou S, Grabowiecka A, Kosikowska P, Yiotakis A, Kafarski P, Berlicki Ł. Design synthesis and evaluation of novel organophosphorus inhibitors of bacterial urease. J Med Chem. 2008;51:5736–5744. doi: 10.1021/jm800570q. [DOI] [PubMed] [Google Scholar]
- Vassiliou S, Grabowiecka A, Kosikowska P, Yiotakis A, Kafarski P, Berlicki Ł. Computer-aided optimization of phosphinic inhibitors of bacterial ureases. J Med Chem. 2010;53:5597–5606. doi: 10.1021/jm100340m. [DOI] [PubMed] [Google Scholar]
- Vuye A, Pijck J. Urease activity of Enterobacteriaceae: which medium to choose. Appl Environ Microbiol. 1973;26:850–854. doi: 10.1128/am.26.6.850-854.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worcester EM, Coe FL. Nephrolithiasis. Prim Care. 2008;35:369–391. doi: 10.1016/j.pop.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaborska W, Kot M, Superata K. Inhibition of Jack Bean Urease by 14-benzoquinone and 25-dimethyl-14-benzoquinone. Evaluation of the inhibition mechanism. J Enzyme Inhib Med Chem. 2002;17:247–253. doi: 10.1080/1475636021000011670. [DOI] [PubMed] [Google Scholar]