

Abstract
Quantum mechanical calculations reveal the origin of diastereo- and enantioselectivities of aldol reactions between aldehydes catalyzed by histidine, and differences between related reactions catalyzed by proline. A stereochemical model that explains both the sense and the high levels of the experimentally observed stereoselectivity is proposed. The computations suggest that both the imidazolium and the carboxylic acid functionalities of histidine are viable hydrogen-bond donors that can stabilize the cyclic aldolization transition state. The stereoselectivity is proposed to arise from minimization of gauche interactions around the forming C–C bond.
Introduction
Aldol reactions1 constitute an important class of carbon–carbon bond formation reactions amenable to enantioselective organocatalysis.2–10 Intramolecular aldol reactions were found to be catalyzed by proline in the 1970s.11,12 Proline was also used in the first examples of intermolecular organocatalyzed aldol reactions, starting from acetone and a variety of aldehydes.13,14 Since then, the use of non-enolizable aldehydes and ketones as the acceptor and the donor components in enamine catalysis, respectively, has been amply demonstrated. On the other hand, it was recognized early on that the use of enolizable aldehydes as acceptor components was challenging due to competing side reactions, especially aldehyde self-condensation.14 The search for general, efficient methods for the metal- or organocatalyzed aldol reactions between two different, readily enolizable aldehydes remains an intensively investigated topic.15–17 Proline18–28 and its derivatives, as well as chiral imidazolidinones,29 also catalyze the aldol reactions between two enolizable aldehydes with high enantioselectivities, although the lack of differentiation of reaction roles in the case of two dissimilar aldehydes necessitates either the use of careful and cumbersome syringe pump techniques19 or a large excess of one of the aldehyde reactants.30
We recently reported31 that the readily available histidine is a superior catalyst to facilitate the onepot, cross-aldol reaction of two dissimilar enolizable aldehydes, without the need for any specialized experimental setup. As reviewed recently by List,8 α-branched aldehydes such as isobutyraldehyde, are typically used as electrophiles in asymmetric enamine catalysis including organocatalyzed aldol reactions. A diamine containing two pyrrolidine moieties, in conjunction with an acid cocatalyst, was identified by Barbas32 to promote highly enantioselective aldol reactions of isobutyraldehyde and other α,α-dialkyl aldehydes as donors, although only non-enolizable, aromatic aldehydes were used as acceptors. We found,31 however, that in the presence of histidine as catalyst, α-branched aldehydes react as donor components with a variety of enolizable aldehydes such as 1a–g, including electron-deficient aldehydes, forging quaternary stereogenic centers with defined configurations in the aldol products with remarkable ease (Scheme 1). The enantioselectivities in the isolated β-hydroxyaldehydes 2a–g were good to excellent. Moreover, with the α-chiral aldehydes 1f and 1g as the electrophilic components, the aldol products 2f and 2g were obtained with exclusive syn diastereoselectivity (Scheme 1). These transformations also pose interesting mechanistic problems due to the presence of both an imidazole and a carboxylic acid in the amino acid. Although catalytic motifs containing imidazole or imidazolium ions have been extensively studied in relation to natural and artificial enzymes,33 much less has been explored about their potential involvement in small-molecule catalysis.34 We now present further experimental and computational results that uncover the origin of the asymmetric induction in these histidine-catalyzed aldehyde–aldehyde cross-aldol reactions.
Scheme 1. Asymmetric (S)-Histidine-Catalyzed Aldol Addition Reactions of Isobutyraldehyde as Donor Component to Enolizable Aldehydes as Acceptor Components.

a aldehydes 2d and 2e were isolated as their corresponding 1,3-dioxolanes
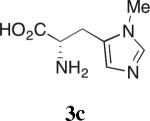
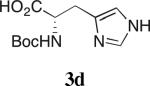
Our studies on direct amine-catalyzed aldol additions showed that tertiary amines are effective catalysts,35 giving yields and reaction rates comparable to a variety of secondary and primary amines in some cases,36 for which the participation of enamine intermediates in the catalytic cycle is conceivable. In light of these results, we felt it important to establish whether enamine formation from histidine is involved in the aldol additions studied here. Thus, using the aldol addition of 1c and isobutyraldehyde (Table 1), we additionally tested various derivatives of histidine 3a–d for their catalytic performance. While histidine gave rise to the aldol product in 70% yield with 77% ee (entry 1), the methyl ester (3a) gave a substantially lower yield and enantioselectivity (entry 2). Replacing histidine by either of the two analogues N-methylated on the imidazole ring (3b and 3c, entries 3 and 4) also resulted in lower yields with further eroded enantioselectivities. These results suggest that the both the imidazole ring and the carboxylic acid are important in catalyzing product formation. Importantly, no reaction was observed in the presence of Boc-L-histidine (3d, entry 5), suggesting that the aldol addition is likely to proceed with enamine formation at the primary amine of histidine.
Table 1.
Aldol Additions of 1c and Isobutyraldehyde in the Presence of Histidine Derivatives
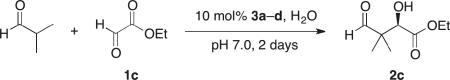
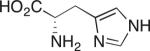
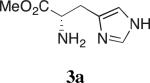
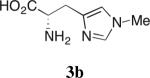
Isolated yield.
Determined by 1H NMR after derivatization of 2c to the Mosher's ester.
No reaction.
The mechanism and origin of the stereochemical induction in proline-catalyzed aldol reactions are well established.37,38 Proline serves as a bifunctional catalyst as the amine functionality forms an enamine with the donor carbonyl component while the carboxylic acid maintains a hydrogen bond with the oxygen atom of the acceptor carbonyl.39 Thus, the stereochemistry-determining, carbon–carbon bond formation step involves a partial Zimmerman–Traxler-like transition state,40 featuring a chair-like arrangement of the enamine and the carbonyl atoms (Fig. 1A). The nucleophilic addition of the enamine proceeds with proton transfer from the carboxylic acid functional group to the developing alkoxide. This so-called List–Houk stereochemical model, featuring a (+)-synclinal arrangement of the carbonyl group and enamine double bond (when the configuration of the proline is S), has seen widespread use in understanding the origin of asymmetric induction observed with different proline-derived organocatalysts. The intermediacy of the proline-enamine in aldol additions has also been demonstrated crystallographically41 and spectroscopically.42–44
Figure 1.

Stereochemical models for the aldol reaction of cyclohexanone and benzaldehyde catalysed by (A) (S)-proline, (B) (S)-alanine, and (C) (S)-tryptophan. (D) Stereochemical model for the Hajos–Parrish reaction.
While proline and its derivatives have dominated research on asymmetric organocatalysis, relatively little is known about other proteinogenic amino acids with regard to their efficiency and mechanism of asymmetric induction in the catalysis of aldol reactions. Alanine45 (Fig. 1B) and tryptophan46 (Fig. 1C) have been studied by Himo and Cordova, and Lu, respectively, with regard to their catalysis of the aldol reaction of cyclohexanone and benzaldehyde, whereas our group previously computed the stereoselectivities of intramolecular aldol cyclizations catalyzed by proline, glycine and phenylalanine (Fig. 1D).47
Histidine differs from proline in three respects. Firstly, the enamine formed from the acyclic histidine can rotate about the Cα–N bond, in contrast to proline-enamines, which contain a pyrrolidine ring. In this regard, the cyclic transition structures computed for the aldol additions catalyzed by the primary amino acids alanine45 and tryptophan46 were found to feature arrays of reacting atoms analogous to those in the proline-catalyzed aldolizations.39 Secondly, the enamine formed from a primary amino acid possesses an N–H bond which could be donated to the developing alkoxide in lieu of the carboxylic acid. Although our early computations on the addition reaction of N-methylvinylamine and acetaldehyde reveals the stabilizing role of hydrogen-bond donation from the N–H bond to the developing alkoxide,48 previous results on the Hajos–Parrish transition states47 established that this proton transfer is energetically unfavorable compared with that from the carboxylic acid. Most importantly, in water, histidine is protonated at the imidazolium group, and can, therefore, potentially act as the hydrogen-bond donor during the aldol additions. The role of a functionalized side chain in primary amino acid organocatalysts in influencing stereoselectivities has seldom been investigated, although in the case of tryptophan, an N–H…π interaction between the primary enamine and the indole ring on the side chain was suggested46 to stabilize the aldolization transition states.
For histidine, using quantum mechanical calculations, we have evaluated the relative importance of the imidazolium ring and the carboxylic acid in stabilizing the aldol addition transition state through hydrogen bonding. This allows us to propose a model to rationalize the stereoselectivity of the series of histidine-catalyzed aldehyde–aldehyde cross-aldol reactions.
Computational Methods
The stationary points (reactant and transition structure geometries) were optimized and characterized by frequency computations at the M06-2X/6-31+G(d,p) level of theory, in the presence of the SMD continuum solvation model49,50 with water as the solvent. Truhlar's M06-2X functional51,52 was developed for computations involving main-group thermochemistry, kinetics and noncovalent interactions, and has been found53 to give good energetics for the types of reactions studied here. All of the computations were performed using Gaussian 09.54
Results and Discussion
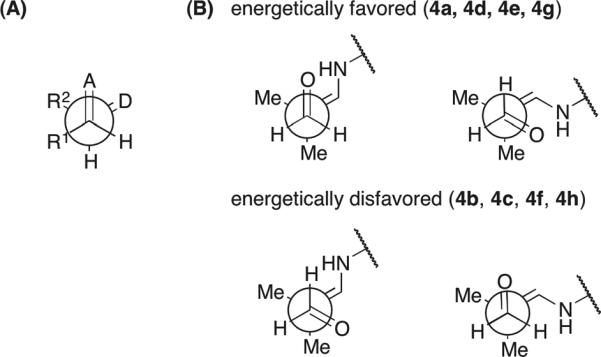
In view of the importance of hydrogen bonding in the List–Houk model and the conformational flexibility of histidine, we first studied the C–C bond forming transition structures using formaldehyde and the histidine-enamine of isobutyraldehyde, as model reactants. The transition structures with either the carboxylic acid or the imidazolium group as the hydrogen bond donor were computed. Only transition structures in which the enamine adopts an s-trans conformation about the Cα–N bond were considered, as it has been shown that the s-cis analogues are at least 3 kcal/mol higher in energy in the transition states of aldol and other electrophilic additions catalyzed by proline39 and alanine.45 For the developing alkoxide to be associated with a hydrogen-bonding group of histidine during the aldol addition, the acceptor carbonyl group must be placed at dihedral angles within ±60° relative to the enamine C=C bond, and only these transition structures were considered. With each hydrogen-bond donor, two arrangements differing in the conformation of the Zimmerman–Traxler ring and, therefore, the face of the enamine exposed for electrophilic attack were investigated. The computed transition structures with the carboxylic acid (4a–4d) and the imidazolium group (4e–4h) as the hydrogen bond donor are illustrated in Fig. 2 and 3, respectively.
Figure 2.

Relative free energies (kcal/mol) of the carboxylic acid-hydrogen-bonded transition structures 4a–4d for the addition of histidine-enamine of isobutyraldehyde and formaldehyde. The Newman projections along the forming C–C bond (inset) show that the disposition of the carbonyl and enamine groups in 4a and 4d is analogous to that of the carbonyl and the enolate in the Zimmerman–Traxler transition state.
Figure 3.

Relative free energies (kcal/mol) of the imidazolium-hydrogen-bonded transition structures 4e–4h for the addition of histidine-enamine of isobutyraldehyde and formaldehyde. The Newman projections along the Cα–Cβ of the histidine moiety (blue) and along the forming C–C bond (red) are shown in the inset.
Among the carboxylic-acid-bonded transition structures, structure 4a, in which the carbonyl group is (+)-synclinal to the C=C double bond, has the lowest energy. This is the original Zimmerman–Traxler conformation,40 although here the enamine N–H does not coordinate with the developing alkoxide, in contrast to the metal in a Zimmerman–Traxler transition state. Both the imidazolium N–H bond and the carboxylic acid O–H bond are better hydrogen-bond donors than the enamine N–H bond due to their higher acidities. Structure 4d, the second most stable, is 1.7 kcal/mol higher in energy than 4a, and contains a (−)-synclinal disposition of the reacting double bonds. The geometry of 4a also corresponds to that of the transition structure in proline-catalyzed aldol reactions. Its (−)-synclinal rotamer analogue, 4b, is 6.6 kcal/mol higher in energy. After optimization, the hydrogen bond from the carboxylic acid changes the ideal staggered substituents to more eclipsed arrangements. The (+)-synclinal rotamer, 4c, is the least stable in this series. 4b and 4c are non-Zimmerman–Traxler structures, in which the enamine N–H and the carbonyl C=O bond, pointing approximately anti as shown by the inset in Fig. 2, are positioned too far to interact with each other.
In the series of transition structures featuring the imidazolium group as the hydrogen-bond donor (4e–4h, Fig. 3), the most stable rotamer was found to be 4e, in which the acceptor carbonyl group is positioned (−)-synclinal to the enamine double bond, again corresponding to a Zimmerman–Traxler-like arrangement. Importantly, 4e is isoenergetic to (+)-synclinal 4a, in which the carboxylic acid functions as the hydrogen bond donor. The (+)-synclinal analogue in the imidazolium-hydrogen-bonding 4f, is 4.3 kcal/mol higher in free energy. The alternative (−)- and (+)-synclinal rotamers, 4g and 4h, are 2.0 and 6.1 kcal/mol less stable, respectively. Structures 4e–4h possess a ten-membered ring including the hydrogen bond and the partially formed C–C bond. In contrast to the nine-membered ring involving the carboxylic acid functionality in 4a–4d, the larger ring size in 4e–4h can accommodate a hydrogen bond from the imidazolium N–H to the developing alkoxide without much distortion of the largely staggered arrangement of substituents on the partial C–C bond, as shown in the red inset. A staggered arrangement is also maintained about the Cα–Cβ bond in the side chain of histidine (blue inset). Nevertheless, the relative free energies show that only 4e is significant among the imidazolium-hydrogen-bonded transition structures.
It is noteworthy that the lower-energy transition structures are consistent with Seebach's topological rule55 in describing the gross structures of transition states for a variety of carbon–carbon bond formation reactions (Fig. 4). One feature of this rule is that all of the bonds around the newly formed bond are staggered. Indeed, as discussed above, the carboxylic acid-bonded TSs 4a and 4d are more stable than the eclipsed structures 4b and 4c (Fig. 2). This staggered arrangement is found in all of the imidazolium-bonded TSs 4e–4h (Fig. 3). However, 4e and 4g are lower in energy due to a synclinal disposition of the donor C=N and the acceptor C=O bonds rather than an anticlinal arrangement in 4f and 4h, consistent with Seebach's rule.55 Furthermore, the geometries of 4e and 4g position the donor nitrogen and the acceptor oxygen close together, minimizing the charge separation. The role of electrostatic stabilization in addition transition structures has also been shown by our recent computations on the asymmetric Stetter reaction between an NHC-enol intermediate and a nitroalkene.56
Figure 4.
(A) Seebach's topological rule for C–C bond forming reactions. A = electron acceptor. D = electron donor. (B) Seebach's rule applied to TSs 4a–4h.
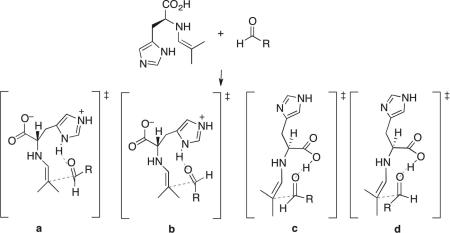
The systematic survey of transition structure conformers above established that both the imidazolium ring and the carboxylic acid are plausible alternatives in stabilizing the aldol transition states through hydrogen bond donation. On the basis of this, we computed the relative free energies of the transition structures for the experimental aldolization reactions to explain the enantioselectivities observed. The aldol addition of isobutyraldehyde to chloroacetaldehyde (1a) was found to give product (R)-2a in 90% ee (Scheme 1). The four transition structures 5a–5d, featuring a hydrogen bond from either the imidazolium group or the carboxylic acid and leading to either enantiomer for this reaction, are shown in Fig. 5. Transition structure 5a, which involves the addition of the enamine to the Re face of chloroacetaldehyde associated by a hydrogen bond to the imidazolium ring, is the lowest in energy. The lowest-energy transition structure in which the electrophilic aldehyde reacts from the Si face is the carboxylic-acid-bonded 5c, which contains a pseudoequatorial chloromethyl group and is 0.8 kcal/mol less stable than 5a. This energy difference corresponds to an enantiomeric excess of 75% in favor of the (R)-enantiomer. Transition structures 5b and 5d also lead to the (S)- and the (R)-enantiomers, respectively, but they are at least 2.5 kcal/mol higher in energy and are negligible.
Figure 5.

Transition structures for the aldolization reaction of the histidine-enamine of isobutyraldehyde and chloroacetaldehyde, in which a hydrogen bond is donated by the imidazolium ring (5a, 5b) or the carboxylic acid (5c, 5d). 5a and 5d lead to the major, (R)-enantiomer, while 5b and 5c give rise to the (S)-isomer.
The aldol addition transition structures for several other aldehydes reacting as the electrophilic component with the histidine-enamine of isobutyraldehyde were also computed (Table 2). As shown in Fig. 6 for the self-aldolization reaction involving isobutyraldehyde to give (S)-2e, the geometries of the transition structures resemble those found for chloroacetaldehyde. The preferred transition structures are invariably the imidazolium-hydrogen-bonded transition structures involving attack of the Re face of the electrophilic aldehyde (4a, 6a), while the carboxylic acid-hydrogen-bonded structures involving attack of the Si face of the electrophile (4c, 6c), which lead to the minor enantiomer, are also significant. In all of the examples studied, the sense of enantioselectivities was successfully reproduced, although the levels were underestimated in some cases.
Table 2.
Relative Free Energies (in kcal/mol) of Transition Structures with Imidazolium or Carboxylic Acid Group as Hydrogen Bond Donor
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | R | ΔΔG‡ (a) | ΔΔG‡ (b) | ΔΔG‡ (c) | ΔΔG‡ (d) | ee computed | ee found |
| 1 | CH2Cl (1a) | 0 | 2.5 | 0.8 | 2.8 | 75% (R) | 90% (R) |
| 2 | CH(OMe)2 (1b) | 0 | 3.9 | 0.6 | 3.4 | 47% (R) | 84% (R) |
| 3 | CO2Et a (1c) | 0 | – a | 1.3 | – a | 80% (R) | 77% (R) |
| 4 | CH2SBn b (1d) | 0 | 1.7 | 0.6 | 1.0 | 47% (R) | 71% (R) |
| 5 | CH2i-Pr (1e) | 0 | 2.0 | 0.7 | 1.3 | 54% (S) | 56% (S) |
Methyl glyoxylate (R = CO2Me) was used as a model reactant. Transition structures of types b and c could not be located.
2-(Methylthio)acetaldehyde (R = CH2SMe) was used as a model reactant.
Figure 6.

Transition structures for the aldolization reaction of the histidine-enamine of isobutyraldehyde and isovaleraldehyde, in which a hydrogen bond is donated by the imidazolium ring (6a, 6b) or the carboxylic acid (6c, 6d). 6a and 6d lead to the major (S)-enantiomer, while 6b and 6c give rise to the (R)-isomer.
The energy differences of the transition structures for a given reaction can be understood by a closer analysis of their geometries. The imidazolium-bonded transition structures of type a, in which the substituent of the acceptor aldehyde is pseudoaxial, are at least 1.7 kcal/mol more stable than those of type b with the substituent pseudoequatorial. Similarly, type c transition structures with pseudoaxial aldehydic substituents are more stable than those of type d. The pseudoaxial preference of the aldehyde substituent is contrary to the Houk–List model of proline-catalyzed aldol reactions,38 in which the aldehydic group prefers to be pseudoequatorial.
Most proline-catalyzed aldol reactions employ α-unbranched ketones as the donor components,48 forming predominantly the (E)-enamines43 as the key intermediates. Thus, the Zimmerman–Traxler-like ring will be monosubstituted at C2 and C3, as illustrated in Fig. 1A. Whether the substituent on the electrophilic aldehyde is pseudoaxial or pseudoequatorial, it will suffer one gauche interaction with the enamine substituent at C2. The pseudoequatorial preference of the aldehydic group can then be understood as a result of minimization of its 1,3-diaxial interactions with the C3-substituent. In the current series of aldehyde–aldehyde aldol transition states, on the other hand, the Zimmerman-Traxler-like rings are disubstituted at C2 and unsubstituted at C3. It is conceivable that the pseudoaxial preference might arise from the minimization of the number of gauche interactions between the aldehydic group (the C1-substituent) and the terminal enamine substituents (the C2-substituents). The transition structures for the pair of imidazolium-bonded transition structures 5a and 5b are illustrated in Fig. 7, emphasizing the chair-like arrangement of the reacting enamine, carbonyl and the hydrogen-bonding group, annotated with short H–H distances. These perspectives show that in the disfavored TS 5b, the chloromethyl group occupies a pseudoequatorial position, coming into two pairs of gauche interactions with the terminal methyl groups of the enamine. The pseudoaxial site is sterically less demanding, as the chloromethyl group suffers from only one gauche interaction and negligible 1,3-diaxial interaction from the C3-hydrogen.
Figure 7.

Newman projections along the forming C-C bond, and transition structures 5a–5d showing the Zimmerman-Traxler-like cyclic arrangement of the reacting enamine, carbonyl and hydrogen-bonding group. Close hydrogen-hydrogen distances are annotated.
This pseudoaxial preference of the substituent of the electrophilic aldehyde can be compared to the the transition structures for the allylation reactions of aldehydes by 3,3-disubstituted allylzinc reagents, which give homoallylic alcohols featuring a quaternary stereocenter with high levels of diastereoselectivity (Fig. 8A).57 The computed zinc-containing Zimmerman–Traxler TSs show that with gem-dimethyl groups on the terminus of the nucleophilic C=C bond, the bulky isopropyl or an aryl group on the electrophilic aldehyde prefers a pseudoaxial position to avoid unfavorable gauche interactions with the terminal methyl groups. Only when the allylzinc reagent carries an additional substituent at C2 will the pseudoaxial site be disfavored due to increased 1,3-diaxial strain. Previous work from our group also found58 that high exo selectivity was observed in Diels–Alder reactions when the termini of the acyclic diene and dienophile involved in the shorter of the forming C–C bonds were both monoalkyl-substituted (Fig. 8B). In the exo-selective reactions, the endo TSs, but not the exo TSs, were shown to be destabilized by significant gauche interactions around the partial bond between the methyl groups on the reacting partners. A series of highly diastereoselective, intramolecular cycloadditions involving nitrones and nitrile oxides forming 6,5-fused rings (Fig. 8C) was also rationalized by a pseudoaxial preference of the alkyl substituent on an incipient six-membered ring.59–61 This arrangement was presumed to reduce the gauche repulsive interaction with the neighboring N-substituent.
Figure 8.

Reported examples of influence of gauche interactions on reaction diastereoselectivities. The unfavorable gauche interactions are indicated by the red double-headed curved arrows.
This model also explains the diastereocontrol observed with chiral aldehydes used as acceptors. The aldol addition of isobutyraldehyde to the chiral aldehyde (R)-1f as the acceptor carbonyl occurs in 87% yield with exclusive stereoselectivity for the (R,R)-diastereomer of 2f (Scheme 1). The transition structures of types a (imidazolium-hydrogen-bonded) and c (carboxylic acid-hydrogen-bonded) were computed (Fig. 9), using 7 as the model reactant. The α-methoxy group of 7 makes it necessary to take two rotameric states of this moiety into account when locating the TSs. In the Felkin–Anh rotamer, the optimized geometry is consistent with the electronegative group being antiperiplanar to the incoming nucleophile, while the chelated rotamer features a chelated hydrogen bond formed by the developing alkoxide and the α-oxygenated substituent on the acceptor aldehyde.62 The computations show that only the chelated rotamer of the imidazolium-bonded TS 8a is significant, while the Felkin–Anh rotamer of TS 8a is 4.5 kcal/mol higher in free energy. Both rotamers of 8b with the carboxylic acid as the hydrogen bond donor are 3 kcal/mol higher than 8a. These calculations are consistent with the sense and the high level of selectivity for the formation of the (R,R) diastereomer of 2f found experimentally. Similarly, the stereoselectivity of the aldol addition of isobutyraldehyde to isopropylideneglyceraldehyde (R)-1g was also reproduced by the computed TSs illustrated in Fig. 10.
Figure 9.

Transition structures for the aldolization reaction of the histidine-enamine of isobutyraldehyde and 7, in which a hydrogen bond is donated by the imidazolium group (8a) or the carboxylic acid (8c). Two rotamers of the C–Cα bond for shown for 8a and 8c. 8a leads to the experimentally observed (R,R)-diastereomer, while 8c gives rise to the (S,R)-isomer.
Figure 10.

Transition structures for the aldolization reaction of the histidine-enamine of isobutyraldehyde and (R)-1g, in which a hydrogen bond is donated by the imidazolium group (9a) or the carboxylic acid (9c). 9a leads to the experimentally observed (R,R)-diastereomer, while 9c gives rise to the (S,R)-isomer.
Conclusion
In summary, we have computationally studied the origin of stereocontrol of histidine-catalyzed aldehyde–aldehyde cross-aldol reactions. The imidazolium and the carboxylic acid groups can both stabilize the aldol addition through hydrogen bonding in a Zimmerman–Traxler-type transition state. The computations also identified an infrequent pseudoaxial preference of the substituent of the electrophilic aldehyde in the presence of a terminal dimethyl substituted histidine enamine. This is rationalized by a minimization of the gauche interaction around the forming C–C bond. We envisage that this work will inspire future efforts in the design of organocatalysts that employ hydrogen bonding from the imidazolium group as the catalytic principle, a possibility that has been relatively overlooked to date.
Supplementary Material
Acknowledgment
We are grateful to the National Institute of General Medical Sciences, National Institute of Health (GM36700) for financial support of this research. We also thank UCLA Institute for Digital Research and Education, the Shared Research Computing Services Pilot Project for the UC systems. This work also used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number OCI-1053575.
Footnotes
Supporting Information Available. NMR data for all of the synthesized compounds, full characterization of novel compounds, Cartesian coordinates and energies of the transition structures, and the full citation for ref. 54. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Mahrwald R, editor. Modern Aldol Reactions. Vol. 1 and 2. Wiley-VCH; 2004. [Google Scholar]
- 2.Nielsen M, Worgull D, Zweifel T, Gschwend B, Bertelsen S, Jørgensen KA. Chem. Commun. 2010:632–649. doi: 10.1039/c0cc02417a. [DOI] [PubMed] [Google Scholar]
- 3.Trost BM, Brindle CS. Chem. Soc. Rev. 2010;39:1600–1632. doi: 10.1039/b923537j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pihko PM, Majander I, Erkkilä A. Top. Curr. Chem. 2009;291:29–74. doi: 10.1007/978-3-642-02815-1_21. [DOI] [PubMed] [Google Scholar]
- 5.Melchiorre P, Marigo M, Carlone A, Bartoli G. Angew. Chem. Int. Ed. 2008;47:6138–6171. doi: 10.1002/anie.200705523. [DOI] [PubMed] [Google Scholar]
- 6.Dondoni A, Massi A. Angew. Chem. Int. Ed. 2008;47:4638–4660. doi: 10.1002/anie.200704684. [DOI] [PubMed] [Google Scholar]
- 7.Guillena G, Nájera C, Ramón DJ. Tetrahedron: Asymmetry. 2007;18:2249–2293. [Google Scholar]
- 8.Mukherjee S, Yang JW, Hoffmann S, List B. Chem. Rev. 2007;107:5471–5569. doi: 10.1021/cr0684016. [DOI] [PubMed] [Google Scholar]
- 9.List B, Yang JW. Science. 2006;313:1584–1586. doi: 10.1126/science.1131945. [DOI] [PubMed] [Google Scholar]
- 10.Notz W, Tanaka F, Barbas CF., III Acc. Chem. Res. 2004;37:580–591. doi: 10.1021/ar0300468. [DOI] [PubMed] [Google Scholar]
- 11.Hajos ZG, Parrish DR. J. Org. Chem. 1974;39:1615–1621. [Google Scholar]
- 12.Eder U, Sauer G, Wiechert R. Angew. Chem. Int. Ed. Engl. 1971;10:496–497. [Google Scholar]
- 13.List B, Lerner RA, Barbas CF., III J. Am. Chem. Soc. 2000;122:2395–2396. [Google Scholar]
- 14.List B, Pojarliev P, Castello C. Org. Lett. 2001;3:573–575. doi: 10.1021/ol006976y. [DOI] [PubMed] [Google Scholar]
- 15.Scheffler U, Mahrwald R. Synlett. 2011:1660–1667. [Google Scholar]
- 16.Marigo M, Melchiorre P. ChemCatChem. 2010;2:621–623. [Google Scholar]
- 17.Alcaide B, Almendros P. Angew. Chem. Int. Ed. 2003;42:858–860. doi: 10.1002/anie.200390232. [DOI] [PubMed] [Google Scholar]
- 18.Northrup AB, MacMillan DWC. J. Am. Chem. Soc. 2002;124:6798–6799. doi: 10.1021/ja0262378. [DOI] [PubMed] [Google Scholar]
- 19.Northrup AB, Mangion IK, Hettche F, MacMillan DWC. Angew. Chem. Int. Ed. 2004;43:2152–2154. doi: 10.1002/anie.200453716. [DOI] [PubMed] [Google Scholar]
- 20.Northrup AB, MacMillan DWC. Science. 2004;305:1752–1755. doi: 10.1126/science.1101710. [DOI] [PubMed] [Google Scholar]
- 21.Storer RI, MacMillan DWC. Tetrahedron. 2004;60:7705–7714. [Google Scholar]
- 22.Mangion IK, MacMillan DWC. J. Am. Chem. Soc. 2005;127:3696–3697. doi: 10.1021/ja050064f. [DOI] [PubMed] [Google Scholar]
- 23.Córdova A, Engqvist M, Ibrahem I, Casas J, Sundén H. Chem. Commun. 2005:2047–2049. doi: 10.1039/b500589b. [DOI] [PubMed] [Google Scholar]
- 24.Córdova A, Ibrahem I, Casas J, Sundén H, Engqvist M, Reyes E. Chem. Eur. J. 2005;11:4772–4784. doi: 10.1002/chem.200500139. [DOI] [PubMed] [Google Scholar]
- 25.Smith AB, III, Tomioka T, Risatti CA, Sperry JB, Sfouggatakis C. Org. Lett. 2008;10:4359–4362. doi: 10.1021/ol801792k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chowdari NS, Ramachary DB, Córdova A, Barbas CF., III Tetrahedron Lett. 2002;43:9591–9595. [Google Scholar]
- 27.Zhao G-L, Liao W-W, Córdova A. Tetrahedron Lett. 2006;47:4929–4932. [Google Scholar]
- 28.Casas J, Engqvist M, Ibrahem I, Kaynak B, Córdova A. Angew. Chem. Int. Ed. 2005;44:1343–1345. doi: 10.1002/anie.200461400. [DOI] [PubMed] [Google Scholar]
- 29.Mangion IK, Northrup AB, MacMillan DWC. Angew. Chem. Int. Ed. 2004;43:6722–6724. doi: 10.1002/anie.200461851. [DOI] [PubMed] [Google Scholar]
- 30.Hayashi Y, Aratake S, Itoh T, Okano T, Sumiya T, Shoji M. Chem. Commun. 2007:957–959. doi: 10.1039/b613262f. [DOI] [PubMed] [Google Scholar]
- 31.Markert M, Scheffler U, Mahrwald R. J. Am. Chem. Soc. 2009;131:16642–16643. doi: 10.1021/ja907054y. [DOI] [PubMed] [Google Scholar]
- 32.Mase N, Tanaka F, Barbas CF., III Angew. Chem. Int. Ed. 2004;43:2420–2423. doi: 10.1002/anie.200353546. [DOI] [PubMed] [Google Scholar]
- 33.Noujeim N, Leclercq L, Schmitzer R. Curr. Org. Chem. 2010;14:1500–1516. [Google Scholar]
- 34.Yang H, Wong MW. J. Org. Chem. 2011;76:7399–7405. doi: 10.1021/jo2011413. [DOI] [PubMed] [Google Scholar]
- 35.Markert M, Mulzer M, Schetter B, Mahrwald R. J. Am. Chem. Soc. 2007;129:7258–7259. doi: 10.1021/ja071926a. [DOI] [PubMed] [Google Scholar]
- 36.Rohr K, Mahrwald R. Org. Lett. 2011;13:1878–1880. doi: 10.1021/ol200412r. [DOI] [PubMed] [Google Scholar]
- 37.Cheong PH-Y, Legault CY, Um JM, Çelebi-Ölçüm N, Houk KN. Chem. Rev. 2011;111:5042–5137. doi: 10.1021/cr100212h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Allemann C, Gordillo R, Clemente F, Cheong P, Houk KN. Acc. Chem. Res. 2004;37:558–569. doi: 10.1021/ar0300524. [DOI] [PubMed] [Google Scholar]
- 39.Bahmanyar S, Houk KN, Martin HJ, List B. J. Am. Chem. Soc. 2003;125:2475–2479. doi: 10.1021/ja028812d. [DOI] [PubMed] [Google Scholar]
- 40.Zimmerman HE, Traxler MD. J. Am. Chem. Soc. 1957;79:1920–1923. [Google Scholar]
- 41.Bock DA, Lehmann CW, List B. Proc. Nat. Acad. Sci. 2010;107:20636–20641. doi: 10.1073/pnas.1006509107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marquez C, Metzger JO. Chem. Commun. 2006:1539–1541. doi: 10.1039/b518288c. [DOI] [PubMed] [Google Scholar]
- 43.Schmid MB, Zeitler K, Gschwind RM. Angew. Chem. Int. Ed. 2010;49:4997–5003. doi: 10.1002/anie.200906629. [DOI] [PubMed] [Google Scholar]
- 44.Schmid MB, Zeitler K, Gschwind RM. J. Org. Chem. 2011;76:3005–3015. doi: 10.1021/jo200431v. [DOI] [PubMed] [Google Scholar]
- 45.Bassan A, Zou W, Reyes E, Himo F, Córdova A. Angew. Chem. Int. Ed. 2005;44:7028–7032. doi: 10.1002/anie.200502388. [DOI] [PubMed] [Google Scholar]
- 46.Jiang Z, Yang H, Han X, Luo J, Wong MW, Lu Y. Org. Biomol. Chem. 2010;8:1368–1377. doi: 10.1039/b921460g. [DOI] [PubMed] [Google Scholar]
- 47.Clemente FR, Houk KN. J. Am. Chem. Soc. 2005;127:11294–11302. doi: 10.1021/ja0507620. [DOI] [PubMed] [Google Scholar]
- 48.Bahmanyar S, Houk KN. J. Am. Chem. Soc. 2001;123:11273–11283. doi: 10.1021/ja011403h. [DOI] [PubMed] [Google Scholar]
- 49.Marenich AV, Cramer CJ, Truhlar DG. J. Phys. Chem. B. 2009;113:6378–6396. doi: 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- 50.Ribeiro RF, Marenich AV, Cramer CJ, Truhlar DG. J. Phys. Chem. B. 2011;115:14556–14562. doi: 10.1021/jp205508z. [DOI] [PubMed] [Google Scholar]
- 51.Zhao Y, Truhlar DG. Acc. Chem. Res. 2008;41:157–167. doi: 10.1021/ar700111a. [DOI] [PubMed] [Google Scholar]
- 52.Zhao Y, Truhlar D. Theor. Chem. Acc. 2008;120:215–241. [Google Scholar]
- 53.Wheeler SE, Moran A, Pieniazek SN, Houk KN. J. Phys. Chem. A. 2009;113:10376–10384. doi: 10.1021/jp9058565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Frisch MJ, et al. Gaussian 09, Revision A.02. Gaussian, Inc.; Wallingford CT: 2009. [Google Scholar]
- 55.Seebach D, Goliński J. Helv. Chim. Acta. 1981;64:1413–1423. [Google Scholar]
- 56.Um JM, DiRocco DA, Noey EL, Rovis T, Houk KN. J. Am. Chem. Soc. 2011;133:11249–11254. doi: 10.1021/ja202444g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gilboa N, Wang H, Houk KN, Marek I. Chem. Eur. J. 2011:8000–8004. doi: 10.1002/chem.201101049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lam Y.-h., Cheong PH-Y, Blasco Mata J, Stanway SJ, Gouverneur V, Houk KN. J. Am. Chem. Soc. 2009;131:1947–1957. doi: 10.1021/ja8079548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Noguchi M, Tsukimoto A, Kadowaki A, Hikata J, Kakehi A. Tetrahedron Lett. 2007;48:3539–3542. [Google Scholar]
- 60.Kadowaki A, Nagata Y, Uno H, Kamimura A. Tetrahedron Lett. 2007;48:1823–1825. [Google Scholar]
- 61.Tanaka M, Hikata J, Yamamoto H, Noguchi M. Heterocycles. 2001;55:223–226. [Google Scholar]
- 62.Barbas and coworkers studied the syn-selective aldol reactions of unprotected hydroxyacetone and dihydroxyacetone catalyzed by tryptophan, serine, and threonine, and their derivatives. The high degree of syn selectivity was proposed to arise from hydrogen bonding between the oxygen on the donor component and the enamine N–H. Ramasastry SSV, Zhang H, Tanaka F, Barbas CF., III J. Am. Chem. Soc. 2007;129:288–289. doi: 10.1021/ja0677012.. Ramasastry SSV, Albertshofer K, Utsumi N, Tanaka F, Barbas CF., III Angew. Chem. Int. Ed. 2007;46:5572–5575. doi: 10.1002/anie.200701269.. Ramasastry SSV, Albertshofer K, Utsumi N, Barbas CF., III Org. Lett. 2008;10:1621–1624. doi: 10.1021/ol8002833.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.