

Summary
Enteric bacterial pathogens have evolved sophisticated strategies to evade host immune defences. Some pathogens deliver anti-inflammatory effector molecules into the host cell cytoplasm via a type III secretion system (T3SS). Enteropathogenic Escherichia coli (EPEC) inhibits inflammation by an undefined, T3SS-dependent mechanism. Two proteins encoded outside of the EPEC locus of enterocyte effacement (LEE) pathogenicity island, non-LEE-encoded effector H1 (NleH1) and H2 (NleH2), display sequence similarity to Shigella flexneri OspG, which inhibits activation of the pro-inflammatory transcription factor NF-κB. We hypothesized that the anti-inflammatory effects of EPEC were mediated by NleH1 and NleH2. In this study, we examined the effect of NleH1/H2 on the NF-κB pathway. We show that NleH1/H2 are secreted via the T3SS and that transfection of cells with plasmids harbouring nleH1 or nleH2 decreased IKK-β-induced NF-κB activity and attenuated TNF-α-induced degradation of phospho-IκBα by preventing ubiquitination. Serum KC levels were higher in mice infected with ΔnleH1H2 than those infected with WT EPEC, indicating that NleH1/H2 dampen pro-inflammatory cytokine expression. ΔnleH1H2 was cleared more rapidly than WT EPEC while complementation of ΔnleH1H2 with either NleH1 or NleH2 prolonged colonization. Together, these data show that NleH1 and NleH2 function to dampen host inflammation and facilitate EPEC colonization during pathogenesis.
Introduction
The mammalian intestine constantly encounters a wide range of commensal and pathogenic organisms. Members of the Toll-like and NOD-like receptor proteins recognize signature microbial molecular structures such as flagellin and, when appropriate, activate signalling pathways that lead to the production of pro-inflammatory and antimicrobial compounds (Neish, 2004). A number of these signalling systems activate NF-κB, a member of the Rel family of transcription factors (Tato and Hunter, 2002). In the absence of stimulation, NF-κB is retained in the cytoplasm by members of IκB family of proteins. Phosphorylation and ubiquitination of IκB proteins targets them for degradation by the proteasome, thereby allowing free NF-κB to enter the nucleus and activate transcription. NF-κB induces the expression of a wide range of proteins including those with pro-inflammatory and anti-bacterial activities.
Bacterial pathogens, on the other hand, have developed sophisticated strategies to interfere with the effects of host inflammatory pathways and compounds. These include stealth strategies such as expression of flagellar variants that are not recognized by TLRs, production of virulence factors that directly interfere with host signalling pathways, and elaboration of structures that resist killing by host antimicrobial compounds (Neish, 2004). Several Gram-negative pathogens including Yersinia sp., Salmonella sp., Shigella flexneri and Vibrio cholerae utilize a type III secretion system (T3SS) to deliver specific anti-inflammatory effector molecules into host cytosol thereby subverting signal transduction cascades and cellular communications (Neish, 2004). For instance, S. flexneri OspG interacts with ubiquitin conjugating enzymes whose targets include phospho-IκBα (Kim et al., 2005). This blocks IκBα degradation, consequently inhibiting cytokine-dependent NF-κB activation. Salmonella typhimurium SseL is a deubiquitinase that suppresses IκBα ubiquitination and therefore NF-κB activation (Le Negrate et al., 2008). Similarly, Salmonella AvrA and Yersinia pseudotuberculosis YopJ proteins have been implicated in inhibiting the NF-κB pathway (Collier-Hyams et al., 2002; Jones et al., 2008; Mukherjee and Orth, 2008). Thus, inhibition of NF-κB signalling represents a general theme in microbial pathogenesis.
Enteropathogenic Escherichia coli (EPEC) causes diarrhoea particularly in children in developing countries. EPEC produces various pro-inflammatory molecules including flagellin (Sharma et al., 2006). In addition, EPEC injects a repertoire of virulence effector proteins into host cells via a T3SS. While some secreted effector proteins are encoded on a pathogenicity island known as LEE (locus of enterocyte effacement), several effector molecules are encoded outside the LEE (non-LEE encoded, or Nle). It has been reported previously that EPEC limit the pro-inflammatory effects of flagellin and host cytokines by repressing NF-κB activation through a T3SS-mediated mechanism that may involve several effectors (Hauf and Chakraborty, 2003; Maresca et al., 2005; Sharma et al., 2006; Ruchaud-Sparagano et al., 2007).
The NleH family of proteins is conserved among EPEC and other A/E pathogens including enterohemorrhagic E. coli (EHEC) and the murine pathogen Citrobacter rodentium (CR). EPEC and EHEC each contain two homologues of nleH, i.e. nleH1 and nleH2; in addition EPEC carries a third pseudogene encoding only the C-terminal part of NleH1/H2. CR encodes one orthologue of NleH that is type III-secreted (Tobe et al., 2006; Garcia-Angulo et al., 2008). EPEC nleH1 is encoded in a putative operon with cif while nleH2 and nleF appear to constitute a distinct operon (Deng et al., 2005; Tobe et al., 2006; Loukiadis et al., 2008; Hemrajani et al., 2010). A detailed discussion of the genome organization of nleH sequences in A/E pathogens and sequence similarities between NleH homologues has been published (Garcia-Angulo et al., 2008; Hemrajani et al., 2010).
The C-termini of EPEC NleH1 and NleH2 proteins display significant amino acid sequence similarity with the S. flexneri T3SS anti-inflammatory effector OspG (Kim et al., 2005) (30% and 27% identity, respectively, to OspG; 80% sequence identity between NleH1 and NleH2; Fig. S1A). Despite significant sequence identity, a 10-amino-acid sequence present in NleH2 (residues 127–136) is absent in NleH1 (Fig. S1A; Hemrajani et al., 2010). NleH effectors contain an N-terminal 95-amino-acid fragment that is missing in OspG. OspG encodes a kinase domain where lysine residue (K53) functions as the catalytic site of kinase activity. The corresponding lysine residues K159 and K169 contained in NleH1 and NleH2, respectively, are also conserved in the putative kinase domain of these proteins and may function as the catalytic site of kinase activity (Fig. S1A). The secondary structures of these proteins obtained using PRALINE (Simossis and Heringa, 2005) are also conserved (Fig. S1B).
In EHEC and CR, contradictory results regarding the influence of NleH proteins on the inflammatory response have been reported and there are variable in vivo data concerning the role of these proteins in pathogenesis (Garcia-Angulo et al., 2008; Hemrajani et al., 2008; Gao et al., 2009). Diverse roles for EPEC NleH proteins in modulating apoptosis (Hemrajani et al., 2010; Robinson et al., 2010) were reported recently; however, no data are available regarding the role of NleH proteins in EPEC pathogenesis. In this study, we performed transfection studies to evaluate the effect of EPEC NleH1 and NleH2 on NF-κB activation. An EPEC ΔnleH1H2 double mutant was generated and compared with the isogenic parent in vitro and in vivo. Finally, we assessed the role of NleH1/H2 in colonization and inflammation using a murine model of infection.
Results
NleH1 and NleH2 are secreted via the EPEC T3SS
To determine whether NleH1/H2 are secreted via the T3SS, PCR-amplified EPEC nleH1 and nleH2 were cloned into the IPTG-inducible vector pTrc2HisTOPO (Invitrogen) in-frame with the C-terminal Myc and His tags. Wild-type (WT) EPEC and the T3SS-defective ΔescN were transformed with these constructs (Table S1). Following growth in IPTG-supplemented DMEM, bacterial pellets and TCA-precipitated supernatants were immunoblotted for Myc. Expression of NleH1/H2 in bacteria was IPTG dose-dependent in both EPEC and ΔescN complemented strains (Fig. 1, top panel). The corresponding proteins were detected in supernatants of EPEC, but not ΔescN mutants (Fig. 1, bottom panel), demonstrating requirement of the T3SS for NleH1 and NleH2 secretion consistent with other reports (Garcia-Angulo et al., 2008). Overexpression of NleH2 with up to 200 μM of IPTG does not affect secretion (Fig. 1, bottom panel). In contrast, the secreted levels of NleH1 reached saturation with 50 μM of IPTG and decreased with higher concentrations (Fig. 1, bottom panel). Overexpression of NleH1 thus appears to inhibit secretion of NleH1, as well as EspF (data not shown). Reduced NleH1 secretion could be attributed to a reduced intracellular pool of NleH1 to be secreted or, less likely, due to an accessory function in secretion. Similar results have been documented previously for EspF (Elliott et al., 2000; Viswanathan et al., 2004). It is equally plausible that overexpression of NleH1 results in inclusion bodies or an overload of the secretion apparatus or chaperones reducing its secretion.
Fig. 1.

NleH1 and NleH2 secretion is dependent on a functional T3SS. EPEC nleH1 and nleH2 were cloned into the pTrcHis2-TOPO vector and expression of the gene was induced by IPTG. EPEC and the T3SS-deficient strain ΔescN were transformed with these constructs (designated as EPEC/pNleH1, EPEC/pNleH2, EPEC ΔescN/pNleH1 and EPEC ΔescN/pNleH2). Whole organisms (bacteria) and filter-sterilized supernatants (SN) were run on SDS-PAGE and blotted for Myc-tagged NleH1/H2. NleH1/H2 expression was detected in all bacteria organisms except EPEC/ΔescN (top panel). NleH1 and NleH2 were identified in SN from EPEC/pNleH1 and EPEC/pNleH2 but not in EPEC/ΔescN, EPEC/ΔescN/pNleH1 and EPEC/ΔescN/pNleH2 (bottom panel), indicating that NleH1/H2 secretion is dependent on the T3SS.
Deletion of nleH1 and nleH2 does not affect pedestal formation in mammalian cells
Since NleH1 and NleH2 are secreted in a T3SS-dependent manner, the ability of ΔnleH1H2 to form actin pedestals was examined. Murine intestinal epithelial CMT-93 cells were infected for 1 h with WT EPEC or ΔnleH1H2 and immunofluorescent staining for actin performed using BODIPY 558/568 phalloidin (Invitrogen). There was no discernible difference in actin pedestal formation in the absence of NleH1/H2 (Fig. S2A). Epithelial adhesion and A/E lesion formation was also examined in CMT-93 cells by transmission electron microscopy. In both WT EPEC and ΔnleH1H2-infected cells, intimately attached bacteria were identified, defined by the characteristic membrane cup formation and electron-dense cytoskeletal material directly beneath adherent bacteria (Fig. S2B, arrows). Thus, deletion of nleH1 and nleH2 does not alter pedestal formation in infected murine intestinal epithelial cells.
Effect of WT EPEC and ΔnleH1H2 infection on TNF-induced NF-κB activation
Enteropathogenic E. coli suppresses the host inflammatory response to cytokines by an undefined T3SS-dependent mechanism (Sharma et al., 2006; Ruchaud-Sparagano et al., 2007). Deletion of the ospG homologue in S. flexneri increases IκBα degradation in infected epithelial cells (Kim et al., 2005). To determine if NleH1/H2 affects the NF-κB pathway, a synchronized approach was used whereby uninfected cells or those infected with WT EPEC or ΔnleH1H2 were challenged with TNF-α as the pro-inflammatory stimulus. HEK293T cells were infected with WT EPEC or ΔnleH1H2 for 1 h then subjected to TNF-α and IκBα degradation was assessed by immunoblot analysis (Fig. 2A). TNF-α induced IκBα degradation in uninfected control cells by 15 min (Fig. 2A, top panel). Infection with EPEC delayed this response (Fig. 2A, centre panel), although as analysed by densitometry not to a statistically significant degree (Fig. 2B). The trend towards protection afforded by the EPEC strain was abolished with deletion of nleH1H2 (Fig. 2A, bottom panel; Fig. 2B). Infection with T3SS-defective ΔescN displayed no protection against IκBα degradation, indicating that T3S-effectors are responsible for this phenotype (Fig. 2C, bottom panel; Fig. 2D). While statistical analysis of densitometry showed no significant difference in the rate of IκBα degradation, we were impressed by the trend of a protective effect by EPEC and loss of this trend in absence of NleH1/H2. These data justified further pursuit of the effects of NleH1/H2 especially in view of the fact that EPEC infection alone does not induce the degree of TNF expression used in these in vitro experiments. Also these data are derived from a system where EPEC induces both pro- and anti-inflammatory effects (Hauf and Chakraborty, 2003; Chen and Frankel, 2005; Sharma et al., 2006), providing justification for transfection experiments where the action of individual NleH proteins could be defined.
Fig. 2. Effect of WT EPEC and ΔnleH1/H2 on TNF-α induced IκBα degradation.

A. HEK293T cells were challenged with TNF-α or infected with EPEC or ΔnleH1H2 for 1 h prior to challenge and IκBα degradation was monitored by immunoblot. β-Actin is shown as a loading control. Experiments were performed three times and a representative blot is shown.
B. Densitometric analysis of IκBα was measured using AlphaView (Alpha Innotech Corporation). Data represent the mean of three separate experiments and are shown as relative IκBα abundance compared with uninfected cells stimulated with TNF-α alone at 0 min. Error bars represent SEM. While EPEC infection resulted in a protective trend against TNF-induced IκBα degradation that was lost upon deletion of nleH1/H2, the differences were not statistically significant.
C. HEK293T cells were challenged with TNF-α or infected for 1 h with ΔescN prior to challenge and IκBα degradation was monitored by immunoblot. β-Actin is shown as a loading control. Three separate experiments were performed and a representative blot is shown.
D. Data from three experiments were evaluated by densitometry using AlphaView and are shown as relative IκBα abundance compared with uninfected cells stimulated with TNF-α alone at 0 min. Error bars represent SEM. There is no significant difference between uninfected and ΔescN-infected responses.
NleH1 and NleH2 repress the NF-κB pathway downstream of IKK and upstream of p65
In order to gain insight as to where in the NF-κB signalling pathway NleH1/H2 exert their effects, HEK293T cells constitutively expressing NleH1, NleH2 or OspG (Table S1) were transfected with a plasmid expressing IKK-β or p65 to activate NF-κB at different points in the pathway, and NF-κB activity was determined by a NF-κB-dependent reporter plasmid (NF-κB-Luc). Both NleH1 and NleH2 significantly decreased IKK-β-induced NF-κB activation as compared with OspG (Fig. 3). In contrast, neither NleH1, NleH2 nor OspG affected NF-κB activity when stimulated by NF-κB-p65 expression (Fig. 3), suggesting that NleH1/H2 influence on the NF-κB pathway occurs at or downstream of IKK-β but upstream of p65.
Fig. 3.

NleH1 and NleH2 inhibit the NF-κB pathway downstream of IKK-β but upstream of p65. Luciferase assays were performed on extracts from HEK293T cells co-transfected with a NF-κB-Luc reporter plasmid, expression plasmid for IKK-β or p65 as inducers, a pCMV-lacZ plasmid for normalization, and plasmids harbouring nleH1, nleH2 or ospG respectively. Data are the mean of three independent experiments and are shown as per cent induced activity compared with cells transfected with vector alone. β-Galactosidase activity from pCMV-lacZ was assayed and used to normalize all reactions for transfection efficiency. Error bars represent SEM. **P < 0.005.
NleH1 and NleH2 inhibit ubiquitination of phospho-IκBα
The reporter assays indicated that NleH1/H2 influence the NF-κB pathway at or downstream of the IKK complex. We thus hypothesized that NleH1/H2 prevents proteasomal degradation of IκBα by interfering with its phosphorylation or ubiquitination. IκBα phosphorylation was examined in HEK293T cells transfected with either an empty vector (pCMV-Myc), plasmids harbouring WT NleH1 or NleH2. In Shigella OspG, replacement of a conserved lysine residue at position 53 with alanine abolishes its kinase activity (Kim et al., 2005). To test if conserved lysine residues at position K159 and K169 of NleH1 and NleH2, respectively, have a similar function, these residues were mutated to alanine yielding NleH1K159A (mNleH1) or NleH2K169A (mNleH2), which were transfected into HEK293T cells. Transfected cells were stimulated with TNF-α and lysates analysed by immunoblot. Transfection efficiency was estimated at > 80% by co-transfection of a GFP expressing plasmid (data not shown). TNF-α-induced phosphorylation of IκBα was not inhibited by expression of NleH1 or NleH2 but rather phosphorylated IκBα was stabilized in these cells as compared with cells transfected with vector alone or mNleH1 and mNleH2 (Fig. 4A and B). The differences in IκBα phosphorylation between NleH1/H2 and their mutant forms were statistically significant as shown in Fig. 4C. Additionally, immunoblotting for total IκBα revealed that degradation of total IκBα was delayed in cells expressing NleH1/H2 when compared with controls (Fig. 4A). Thus these data demonstrate that NleH1 and NleH2 suppress TNF-α-induced IκBα degradation.
Fig. 4. NleH1 and NleH2 block TNF-α induced ubiquitination but not phosphorylation of IκBα.

A. Immunoblot analysis of cell lysates from cultured HEK293T cells transfected with vector alone, pCMV-myc-NleH2, pCMV-myc-mNleH2, pCMV-myc-NleH1 or pCMV-myc-mNleH1, and stimulated with TNF-α for up to 1 h, using antibodies against phospho-IκBα or total IκBα. Bacterial protein expression was confirmed using an anti-myc antibody and β-Actin is shown as a loading control. Experiments were performed in triplicate and a representative blot is shown.
B. Relative intensities of pIκB band (A, top panel) at 0–45 min were measured using Scion Image beta analysis program (Scion Corporation). Data are the mean of three experiments and error bars represent SEM.
C. Relative intensities of pIκB band (A, top panel) at 45 min were evaluated using Scion Image beta analysis program (Scion Corporation). Data are the mean of three separate experiments and error bars represent SEM. **P < 0.005.
D. Immunoblot analysis of IκBα from lysates from cultured HEK293T cells transfected with plasmids encoding WT and mutant forms of NleH1, NleH2 and OspG and treated with the proteasome inhibitor MG262 for 1 h (to preserve ubiquitinated IκBα) and stimulated with TNF-α for 30 min and probed using antibody against total IκBα. Bacterial protein expression was confirmed using an anti-myc antibody showing NleH1 and NleH2 at 30 kDa and OspG at 20 kDa. Experiments were performed three separate times and a representative blot is shown.
To assess the effect of NleH1/H2 on IκBα ubiquitination, HEK293T cells transfected as above were pretreated for 1 h with MG262 (a proteasome inhibitor that stabilizes ubiquintinated IκBα) and stimulated with TNF-α for 30 min. Ubiquitination of the phospho-IκBα species was inhibited in cells expressing NleH1 or NleH2, compared with cells transfected with vector alone or mutant forms, which showed strong IκBα ubiquitination at 30 min post TNF-α stimulation (Fig. 4D). In cells expressing OspG or OspGK53A (mOspG) (used as a positive control), OspG also suppressed IκBα ubiquitination compared with transfection with vector alone or its mutant form, consistent with previous reports (Kim et al., 2005; Fig. 4D). Together, these data indicate that NleH1 and NleH2 affect the NF-κB pathway at the level of phospho-IκBα ubiquitination and that like its orthologue OspG, the presence of conserved lysine residues K159 and K169 in the catalytic sites of NleH1 and NleH2, respectively, is required for NleH1/H2-mediated inhibitory effects on NF-κB signalling.
Mouse model of EPEC infection
In an effort to optimize EPEC colonization of the mouse intestine (Savkovic et al., 2005; Shifflett et al., 2005), we examined EPEC colonization of streptomycin-free versus streptomycin pretreated C57BL/6J mice (Fig. 5). EPEC colonization was assessed by determining viable EPEC in the stool 1–4 days post infection. Colonization levels reached 104 cfu g−1 stool at day 1 post infection and decreased to about 103 cfu g−1 stool at days 3–4 post infection in the streptomycin-free mice, while streptomycin pre-treatment increased the level of colonization to 108 cfu g−1 stool by day 3 post infection. Mice administered streptomycin therefore were used in these experiments.
Fig. 5.

Streptomycin pre-treatment improves EPEC colonization of C57BL/6J mice. C57BL/6J mice were orally gavaged with 2 × 109 EPEC and faeces were monitored daily for the presence of EPEC for 4 days. Horizontal bars denote median of cfu per gram of faeces for infected mice that were streptomycin-free (closed circles) and for mice pretreated with streptomycin (open circles) prior to EPEC infection. *P < 0.05 for EPEC cfu from streptomycin-free mice versus streptomycin-pretreated mice.
Deletion of nleH1H2 increases EPEC-induced serum KC levels in mice
Mouse KC is a functional homologue of human IL-8 and increased KC serum levels correlate with inflammatory conditions (Lira et al., 1994; Tani et al., 1996). Since KC expression is NF-κB dependent, we determined KC levels in the serum of EPEC and ΔnleH1H2 mutant infected mice at days 1, 3 and 5 post infection (Fig. 6). Although infection with EPEC increased serum KC levels significantly at day 1 post infection, KC was significantly greater in mice infected with ΔnleH1H2. At day 3 post infection, KC levels in animals infected with EPEC had fallen to those seen in uninfected mice while in the ΔnleH1H2-infected mice, KC remained significantly elevated. These data suggest a role for NleH1 and NleH2 in dampening the inflammatory response early in the course of EPEC infection in vivo, and support the hypothesis that modulation of the host inflammatory response by EPEC contributes to pathogenesis.
Fig. 6.
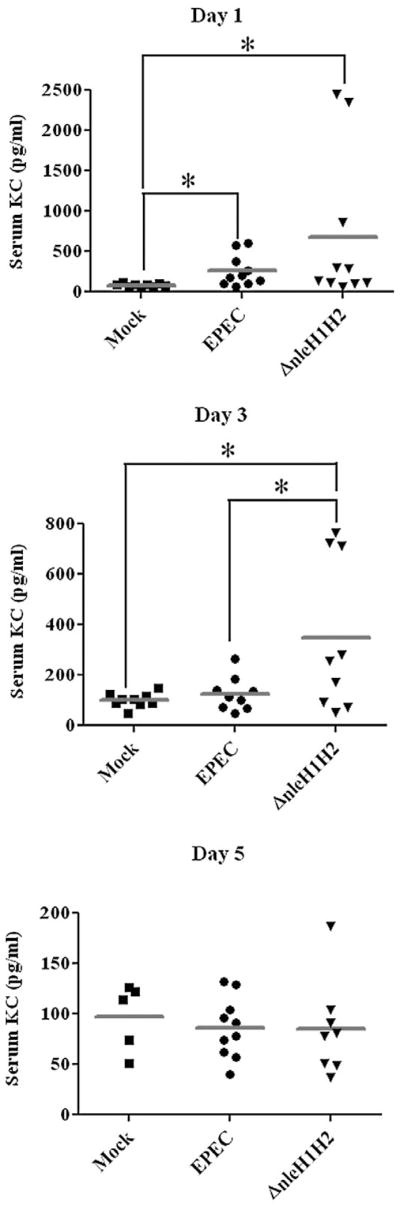
Deletion of nleH1H2 increases and prolongs EPEC-induced serum KC levels in mice. C57BL/6J mice were infected with EPEC or ΔnleH1H2 for up to 5 days and serum KC levels were analysed by ELISA on days 1, 3 and 5. Ten mice were analysed for each experimental group. Horizontal bars denote median. *P < 0.05. Note the scale differences for each panel.
NleH1/H2 dampen inflammation in the colon of EPEC-infected mice
We examined the degree of intestinal inflammation in colonic tissues from mice infected for 1–5 days with EPEC or ΔnleH1H2 by haematoxylin and eosin (H&E) staining (Fig. 7). Inflammatory changes were seen earlier and were moderately greater in animals infected with ΔnleH1H2 as compared with those infected with EPEC. At days 1–3 post infection with ΔnleH1H2, greater numbers of inflammatory cells, primarily neutrophils, and apoptotic cells (those with halos) were present in the surface epithelium (Fig. 7, arrows). In addition, decreased mucin in goblet cells was seen at day 3 in tissues from ΔnleH1H2 animals but not those infected with WT EPEC (Fig. 7). At days 5, inflammation persisted in WT EPEC-infected tissues but had diminished in ΔnleH1H2-infected tissues. These data suggest that NleH1/H2 dampen the inflammatory response in the colon of EPEC-infected mice.
Fig. 7.

Histology of colonic tissues from C57BL/6J mice infected with WT EPEC or ΔnleH1H2. C57BL/6J mice were infected for 1–5 days with EPEC or ΔnleH1H2 and distal colonic tissues examined by H&E staining. Mice infected with ΔnleH1H2 showed increased inflammatory cells, primarily neutrophils, and apoptotic cells at days 1 and 3 with greater loss of goblet cell mucin at day 3 compared with those infected with WT EPEC. By day 5, tissues from ΔnleH1H2 infected mice appeared normal while those from WT-infected animals displayed increased inflammatory cells. Arrows indicate PMN and apoptotic cells (characterized by halos).
NleH1/H2 promote bacterial colonization
The role of NleH1/H2 in EPEC pathogenesis was next addressed. Streptomycin-treated mice were infected with EPEC or ΔnleH1H2 and bacterial colonization determined from the stool (Fig. 8A). EPEC and ΔnleH1H2 mutants and complements colonized to similar levels at day 1 post infection. However, by day 3 colonization levels of the ΔnleH1H2 mutants were significantly less than for EPEC (Fig. 8A). By day 5 post infection, the differences in colonization were even greater with values for ΔnleH1H2 being 2-logs less than that of EPEC. More importantly, only 3 of 20 mice infected with EPEC had totally cleared the infection by day 5, while 8 of 20 infected with ΔnleH1H2 showed no evidence of infection (Fig. 8A), suggesting that NleH1H2 promotes colonization of EPEC in the mouse intestine.
Fig. 8. Deletion of nleH1H2 decreases persistence of infection in C57BL/6J mice.

A. C57BL/6J mice were orally gavaged with 2 × 109 of EPEC (open circles) or ΔnleH1H2 (closed triangles). Stool was monitored for the presence of EPEC for 5 days. Horizontal bars denote median.
*P < 0.05 for cfu of EPEC versus ΔnleH1H2.
B. C57BL/6J mice were orally gavaged with ΔnleH1H2 (closed triangles) or ΔnleH1H2/pNleH1 (closed squares) or ΔnleH1H2/pNleH2 (stars). Stool was monitored for the presence of EPEC for 5 days. Horizontal bars denote median. *P < 0.05 of cfu of ΔnleH1H2 versus ΔnleH1H2/pNleH1.
Next, colonization by ΔnleH1H2 was compared with strains complemented for NleH1 and NleH2. Complementation of ΔnleH1H2 with either NleH1 or NleH2 increased colonization and persistence levels above those seen with ΔnleH1H2 (Fig. 8B) and even beyond those seen with WT EPEC (Fig. 8A), likely due to the overexpression of proteins in the complemented strains. Furthermore at day 3, colonization levels of the ΔnleH1H2 mutants were significantly less than for its H1 complement (Fig. 8B). Thus these data demonstrate that NleH1H2 aid in colonization and persistence of EPEC in the mouse intestine.
Discussion
NF-κB plays an important role in activating the innate immune response and is a crucial factor in maintaining intestinal inflammation during host defence (Neurath et al., 1998; Li and Verma, 2002). However, some enteric pathogens encode type III secreted virulence factors that interfere with NF-κB activation, subverting signalling events involved in host defence (Angot et al., 2007; Munro et al., 2007). In polarized cell culture models, EPEC attenuates flagellin- and cytokine-mediated inflammation by inactivation of the host innate immune response in a T3SS-dependent manner (Savkovic et al., 1997; Zhou et al., 2003; Sharma et al., 2006; Ruchaud-Sparagano et al., 2007; Khan et al., 2008). Several effector(s) are likely involved in this process and their identities are just beginning to be elucidated. It was reported recently that the EPEC non-LEE-encoded effector NleE prevented TNF-α- or IL-1β-induced phosphorylation of both IKKβ and IκBα, thereby inhibiting NF-κB activation (Nadler et al., 2010). Effectors may also function in parallel, for example, NleE activity is enhanced but not dependent on NleB (Dean and Kenny, 2009; Nadler et al., 2010).
The NleH family of non-LEE encoded effector proteins is conserved in the A/E pathogens EPEC, EHEC and CR. It has been suggested that NleH effectors are multifunctional proteins (Gao et al., 2009). A novel mechanism for subverting NF-κB-dependent transcription was reported for EHEC and CR where NleH1 was found to bind human ribosomal protein S3 (RPS3), a newly identified sub-unit of NF-κB reducing its nuclear abundance (Gao et al., 2009). EPEC NleH also displays a broad range of anti-apoptotic activities attributed to its interaction with the ER protein Bax inhibitor-1 (Hemrajani et al., 2010) and inhibits apoptosis induced by Clostridium difficile toxin B (Robinson et al., 2010).
Intriguingly, EPEC NleH1 and NleH2 share sequence identity with the S. flexneri T3S-effector OspG, which binds various ubiquitin conjugating enzymes belonging to the SCFβ–TrCP complex, thereby preventing IκBα ubiquitination and dampening NF-κB activation (Kim et al., 2005). We therefore hypothesized that NleH1 and NleH2 contribute to the anti-inflammatory properties of EPEC. We attempted to determine if OspG and NleH1/H2 are functionally redundant but found that Shigella OspG expressed in EPEC was not secreted (data not shown).
It is important to recognize that EPEC induces both pro-and anti-inflammatory effects in vitro. Although some EPEC T3S-effectors dampen the host inflammatory response to infection, EPEC still induces inflammation. In order to differentiate between pro- and anti-inflammatory effects of EPEC on NF-κB signalling, we employed an ectopic expression system in mammalian cells whereby the effects of NleH1/H2 could be examined independent of the pro-inflammatory stimuli. In this system, TNF-α-induced IκBα phosphorylation was stabilized by NleH1 and NleH2, suggesting that its degradation was blocked. Subsequent studies confirmed that ubiquitination of phosphorylated IκBα was prevented by the expression of NleH1 and NleH2. Furthermore, replacement of lysine residues K159 and K169 of NleH1 and NleH2, respectively, with alanine ablated the protective effects of these proteins on IκBα ubiquitination. Similar results were obtained by replacing OspG K53 with alanine, confirming a previous report that this conserved residue is responsible for the kinase activity of OspG (Kim et al., 2005). Therefore, like its orthologue OspG, the presence of conserved lysine residues K159 and K169 in the catalytic sites of NleH1 and NleH2, respectively, is required for NleH1/H2-mediated inhibitory effects on NF-κB signalling.
Studies from various laboratories suggest that NleH1/H2 function(s) may depend on the context of infection, and on specific cell lines used. A recent study reported that EHEC NleH1/H2 differentially regulate NF-κB-dependent transcriptional activity in mammalian cells stimulated with TNF-α. NleH1 inhibited NF-κB activity while ectopic expression of NleH2 stimulated NF-κB activity (Gao et al., 2009). Our studies demonstrate that NleH1/H2 function similarly to Shigella OspG in blocking NF-κB activation by inhibiting IκB ubiquitination. Other studies failed to see an effect of NleH1/H2 on NF-κB activity (Ruchaud-Sparagano et al., 2007; Gao et al., 2009; Nadler et al., 2010). The discrepancies between the studies contained herein and those published previously could be due to the use of different cell lines and assay systems. It is also possible that NleH may function differently in various cell lines. While our data show that NleH1/H2 stabilize phospho-IκBα and prevent IκB ubiquitination, we cannot rule out the possibility that EPEC possesses additional mechanisms of interfering with other constituents of the NF-κB pathway, such as blocking phosphorylation of IκBα, p65 and potentially targeting other host signal transduction networks like MAPK pathways.
Interference with NF-κB activation and ubiquitination by other bacterial effector proteins has been reported (Mittal et al., 2006; Jones et al., 2008). For example, Yersinia YopJ hydrolyses ubiquitin and SUMO, acts as an acetyl-transferase blocking phosphorylation sites on the IKK-β kinase involved in NF-κB activation and/or interferes with ubiquitination of the TNF receptor-associated factor (TRAF) proteins thus effectively inhibiting NF-κB activation (Mukherjee and Orth, 2008). Similar effects were observed with S. typhimurium SseL, which acts as a deubiquitinase, inhibiting ubiquitin-mediated proteolysis of IκBα and impairing NF-κB activation in TNF-α-stimulated HEK293T cells (Ye et al., 2007; Jones et al., 2008; Le Negrate et al., 2008). Overexpression of S. typhimurium AvrA in eukaryotic cells blocks NF-κB translocation and transcriptional activation of inflammatory effector genes (Collier-Hyams et al., 2002). Salmonella effector SspH1 localizes to the mammalian nucleus and inhibits NF-κB-dependent gene expression (Haraga and Miller, 2003). Other effectors like Shigella IpaH9.8, a bacterial E3 ubiquitin ligase, targets NEMO/IKKγ to dampen the host NF-κB mediated inflammatory response (Ashida et al., 2010).
Modulation of the host immune response, particularly inflammation at mucosal surfaces, by pathogenic bacteria at early stages of infection is a crucial prerequisite for establishment of disease and the nature of the adaptive immunity that follows. Attenuation of NF-κB signalling is an emerging theme in enteric pathogenesis, highlighting the importance of this pathway in regulating the host response. In Yersinia spp., downregulation of the host innate response reduces or prevents inflammatory responses that would otherwise clear the organism (Isberg and Normark, 2000). The NF-κB p50 subunit is of critical importance in activation and regulation of the immune response and it has been demonstrated that mice lacking the NF-κB p50 subunit harbour deficiencies in immune response and are prone to infection (Sha et al., 1995; Li and Verma, 2002). Consequently, p50−/− mice are unable to clear C. rodentium as compared with p50+/+ control mice (Dennis et al., 2008).
There are limited, but conflicting reports concerning the in vivo role of NleH in the pathogenesis of A/E infections (Tobe et al., 2006; Garcia-Angulo et al., 2008). While there were no significant differences in clearance of WT CR and ΔnleH mutant, in a mixed infection, ΔnleH mutants were cleared more rapidly (Hemrajani et al., 2008). Another study showed reduced colonization of CR ΔnleH in the mouse colon at day 6 post infection, while at day 10 colonization levels were comparable, suggesting a role for NleH in promoting CR colonization of the mouse colon at early stages of infection, although the mechanism was not determined (Tobe et al., 2006; Garcia-Angulo et al., 2008). Surprisingly, a reduced TNF-α response was reported in colons of mice infected with ΔnleH as compared with WT (Hemrajani et al., 2008). It is important to note, however, that these data were obtained at the end of the course of infection, day 14, when both the WT and mutant strains had essentially cleared from the intestine leading one to question if effector molecules were still present in host cells. The timing of data acquisition in this study may well account for the conflicting results. These results are also in direct contrast to data regarding the S. flexneri homologue OspG, which was shown to inhibit NF-κB activity and infection with ΔospG significantly increased intestinal inflammation as compared with WT in a rabbit model of Shigella infection (Kim et al., 2005). Different phenotypes for calves and lambs infected with EHEC O157:H7 ΔnleH mutants have also been reported. In single infection studies, there was increased colonization of ΔnleH1nleH2 compared with WT EHEC in calves but similar colonization in lambs. However, the ΔnleH1nleH2 mutant was cleared more rapidly than WT EHEC in the lambs when co-infected (Hemrajani et al., 2008). More important, a hypervirulent phenotype was noted in 1-day-old gnotobiotic piglets infected with EHEC ΔnleH1, along with reduced colonization as compared with WT EHEC. Although piglets infected with ΔnleH2 displayed no clinical phenotype, colonization was reduced 10-fold (Gao et al., 2009). The variability in ΔnleH phenotypes among these A/E pathogens has been attributed to differences in strains, variation in host specificity and animal models and the possibility that effector proteins might have variable roles in different hosts.
The current study investigated the role of EPEC NleH1/H2 in colonization and suppression of inflammation in vivo. Consistent with the data derived from gnotobiotic piglets, the ΔnleH1H2 mutants were cleared more rapidly than WT EPEC and ΔnleH1H2-infected tissues displayed more inflammation. Furthermore, complementation of ΔnleH1H2 with either NleH1 or NleH2 prolonged infection even beyond that of WT, likely due to the overexpression of these proteins by the complemented strains. We further evaluated NleH1/H2 anti-inflammatory activity in vivo by quantifying the levels of pro-inflammatory cytokine KC in serum of infected mice. Deletion of nleH1H2 not only increased serum KC levels in infected mice as compared with WT infection but this response was prolonged as well, correlating with the histology and clearance of the infection.
In conclusion, our studies have clearly demonstrated a role for EPEC NleH1 and NleH2 in dampening the host inflammatory response by suppressing IκBα ubiquitination and degradation thereby attenuating NF-κB activation. Our data also provide evidence that conserved lysine residues in the catalytic site of NleH1/H2 are responsible for function of these proteins. Our in vivo data suggest that NleH1/H2 modulate the host inflammatory response at early stages of EPEC infection and play a role in promoting colonization and persistence of this pathogen and provide justification for the presence of multiple nleH genes in the EPEC chromosome.
Experimental procedures
Strain construction
EPEC E2348/69 (strain O127:H6) (Taylor, 1970; Iguchi et al., 2009) used in these studies; the T3SS-deficient ΔescN derivative CVD452 has been described previously (Jarvis et al., 1995).
The EPEC/ΔnleH1Δcif::cat mutant was generated in strain E2348/69 as follows: primers nleH-1 and nleH-2 (Table S2), which were tailed with 50 bp of sequence from the 5′ and 3′ ends of nleH1 and the downstream gene cif respectively, were used to amplify the cat gene from pKD3 (Datsenko and Wanner, 2000). This linear PCR product was electroporated into EPEC (carrying the Red-expressing plasmid pTP223) to generate EPEC/ΔnleH1Δcif::cat.
The ΔnleH2::kan deletion mutant was constructed as follows: cross-over PCR fusion products of the nleH2 upstream (generated using primers No and Ni) and downstream regions (generated using primers Co and Ci) were cloned into pTOF24 (Merlin et al., 2002), and a kanamycin resistance cassette from pTOF2 was subsequently inserted into the central NotI site to generate a plasmid containing ΔnleH2::kan (pTOF-NleH2). However, due to initial difficulties with this method, we used the Lambda Red Gam method (Datsenko and Wanner, 2000; Murphy and Campellone, 2003) to generate this mutant. The PCR fragment containing ΔnleH2::kan was amplified from pTOF-NleH2 using the No and Co primers. This linear PCR product was introduced into the chromosome of EPEC as described above to generate EPEC/ΔnleH2::kan.
For construction of the ΔnleH1H2 double mutant, the nleH-3 and nleH-4 primers (Table S2), designed to amplify a 1kb flank on either side of the chromosomal ΔnleH1Δcif::cat mutation, were employed. This PCR product was electroporated into EPEC/ΔnleH2::kan/pTP223 to generate EPEC/ΔnleH1Δcif::cat ΔnleH2::kan (ΔnleH1H2).
Plasmids
For expression of Myc-tagged proteins, EPEC nleH1 and nleH2 were PCR amplified (using primer pairs nleH-5/nleH-6 and nleH-7/nleH-8 respectively, Table S2), and cloned into pTrcHis2-TOPO vector (Invitrogen) in-frame with the C-terminal Myc and His sequences. The resulting plasmids (Table S1) were transformed into EPEC or ΔescN or ΔnleH1H2 and induced with varying concentrations of IPTG for expression of recombinant proteins. Myc-tagged NleH1 and NleH2 were detected by SDS-PAGE and immunoblotting.
The pCMV-Myc-NleH1 and pCMV-Myc-NleH2 plasmids were constructed as follows: nleH1 and nleH2 were amplified from EPEC chromosomal DNA by PCR with the nleH-9/ nleH-10 and nleH-11/nleH-12 primer pairs containing restriction sites for SalI and NotI respectively (Table S2). The purified PCR products were digested with SalI and NotI and cloned into the corresponding sites of pCMV-Myc to generate plasmids containing NleH1 and NleH2 with N-terminal Myc tags (Table S1).
To generate plasmid pCMV-Myc-OspG (Table S1), the ospG coding region was amplified from S. flexneri chromosomal DNA by PCR. Restriction sites for EcoRI in the ospG-1 forward primer and KpnI in the ospG-2 reverse primer were introduced to facilitate cloning (Table S2). The purified PCR product was digested with EcoRI and KpnI and cloned into the corresponding sites of the eukaryotic expression vector pCMV-Myc (Clonetech) to yield a construct containing OspG with an N-terminal Myc tag.
The site-directed mutants pCMV-Myc-NleH1-K159A, pCMV-Myc-NleH2-K169A and pCMV-Myc-OspG-K53A (Table S1) were generated with the primer pairs nleH-13/ nleH-14, nleH-15/nleH-16 and ospG-3/ospG4 respectively (Table S2), using the QuikChange II Site-Directed Mutagenesis Kit (Stratagene) following the manufacturer’s instructions.
Cell culture
HEK293T and CMT-93 cells were grown in high-glucose (25 mM) Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen) with 10% fetal bovine serum (FBS) (Invitrogen), 20 mM HEPES, 100 IU ml−1 penicillin and 100 μg ml−1 streptomycin at 37°C in 5% CO2. Cells employed in these studies were between passages 5 and 20 and used 5–7 days post plating. Before infection, the cells were placed in antibiotic-free, serum-free DMEM medium for 3 h and maintained in 5% CO2 at 37°C for the specified lengths of time.
Transfection
HEK293T cells were used for all transfections. Plasmids were transfected using Lipofectamine 2000 Reagent (Invitrogen). For reporter gene assays the transfected DNA quantities were as follows: 40 ng of pNF-κB-Luc (Stratagene), 25 ng of pcDNA-p65, 400 ng of pcDNA-IKK-β and 100 ng of pCMV-myc-NleH1, pCMV-myc-NleH2, pCMV-myc-OspG, pCMV-myc-mNleH1, pCMV-myc-mNleH2 or pCMV-myc-mOspG. pCMV-lacZ (200 ng) was included to normalize results for transfection efficiency. Luciferase and β-galactosidase activities were measured as described previously (Collier-Hyams et al., 2002). For signalling studies, 200 ng of plasmid DNA was used to transfect HEK293T cells grown in six-well plates.
Growth of bacteria and infection of host cells
Bacteria were grown overnight at 37°C in Luria–Bertani (LB) broth with appropriate antibiotics where required (ampicillin, 100 μg ml−1; chloramphenicol, 50 μg ml−1; kanamycin, 50 μg ml−1; tetracycline, 15 μg ml−1) and then inoculated 1:33 into serum-free and antibiotic-free DMEM containing 0.5% mannose. Bacteria were grown at 37°C with aeration until the culture reached mid-exponential phase (OD600 0.4~5 × 108 cells ml−1). Bacteria corresponding to a multiplicity of infection of 100 were then added to confluent HEK293T monolayers seeded in six-well plates. Infected monolayers were incubated at 37°C in a 0.5% CO2 for the specified lengths of time. Additionally, cytokine-mediated pathways were stimulated by treatment of monolayers with TNF-α (10 ng ml−1) for the indicated times. Proteins were extracted and analysed by immunoblot as described below. Bacterial supernatants were prepared as described previously (Sharma et al., 2006).
Preparation of protein extracts and SDS-PAGE immunoblot analysis
Infected monolayers were washed 3× in cold PBS and scraped in 500 μl of 2× SDS sample buffer and sonicated for 10–20 s. Transfected monolayers were washed once in HBSS and scraped in 200 μl of 2× SDS sample buffer. Cell lysates prepared from both infected and transfected monolayers were boiled for 10 min and total protein lysates were resolved by SDS-PAGE and transferred to nitrocellulose membranes. Immunoblot analysis was undertaken according to standard procedures. Membranes were incubated with primary antibodies against phospho-IκBα, anti-IκBα (Santa Cruz Biotechnology), anti-myc (Clontech) and β-actin (Sigma-Aldrich). Immunoreactive species were detected using anti-rabbit HRP or anti-mouse HRP followed by visualization of protein bands by ECL Chemiluminescence Detection Reagent (Amersham).
Actin immunofluorescence
To assess actin pedestal formation by immunofluorescence, subconfluent monolayers of CMT-93 cells grown on 12 mm glass coverslips were infected with EPEC or ΔnleH1H2 for 1 h, washed three times with cold phosphate-buffered saline, fixed and permeabilized with cold 3.7% paraformaldehyde at room temperature for 10 min. Cells were then extracted in ice cold acetone at −20°C for 4–5 min and air dried. The cells were incubated with BODIPY 558/568 phalloidin (Invitrogen) diluted 1:50 in phosphate-buffered saline, incubated with for 30 min in the dark, rinsed in phosphate-buffered saline and air dried. Coverslips were mounted on slides with ProLong Gold Antifade (Invitrogen) and examined using a Nikon Opti-Phot microscope. Images were captured using the Spot-RT digital imaging system.
Transmission electron microscopy
CMT-93 cells were grown on Transwell permeable supports (Corning) until fully differentiated (4 days) and infected with EPEC or ΔnleH1H2 for 6 h. Monolayers were then rinsed twice in warm PBS and fixed in 2.5% glutaraldehyde in 0.1 M Na cacodylate buffer, pH 7.4, overnight at 4°C. Specimens were post-fixed in 1% osmium tetroxide in cacodylate buffer and dehydrated with a series of ethanol up to 100% absolute. Samples were then embedded in LX112 epoxy resin and microtome sections mounted on 200 mesh copper grids stained with 2% aqueous uranyl acetate and lead citrate. Grid sections were imaged using a JEM-1220785 Erlangshen ES100W transmission electron microscope at 40–120 kV ACC voltage.
Murine infections
Six-week-old male C57BL/6J mice were obtained from Jackson Laboratory and housed in a specific pathogen-free facility at the University of Illinois-Chicago for 5–7 days with free access to food and water. Our initial experiments used antibiotic-free mice. For subsequent experiments, mice were pretreated with streptomycin to improve colonization. Mice were given drinking water containing streptomycin sulphate (5 g l−1) to reduce the normal facultative intestinal flora (Wadolkowski et al., 1990) 36 h prior to infection. Streptomycin water was replaced with sterile water 12 h prior to infection. EPEC or ΔnleH1H2 were grown in DMEM as described and resuspended in PBS. Mice were orally gavaged with ~2 × 109 cfu (in 200 μl) of EPEC or ΔnleH1H2 or mock infected with 200 μl of PBS. For complementation studies, after treatment with streptomycin water for 24 h, mice were given IPTG (25 mM) in the drinking water 12 h prior to infection. Mice were orally gavaged with ~2 × 109 cfu (in 200 μl) of ΔnleH1H2 or ΔnleH1H2/pNleH1 or ΔnleH1H2/ pNleH2 and maintained on IPTG water for the course of the experiment. All animal procedures were approved by the University of Illinois-Chicago Animal Care and Use Committee.
Bacterial colonization
Stool from mice infected with EPEC or ΔnleH1H2 or ΔnleH1H2/pNleH1 was resuspended in PBS, serially diluted and plated onto sorbitol MacConkey agar containing nalidixic acid (100 μg ml−1). After 18–24 h of incubation at 37°C, light pink colonies were distinguishable as EPEC. Random colonies were screened by PCR using nleH-specific primers for confirmation of EPEC. Each bacterial strain was tested in five independent experiments by using groups of at least three mice per strain. Data were analysed using the non-parametric, Mann–Whitney, two-tailed t-test. Differences were considered significant if P ≤ 0.05.
Serum cytokine and histological analyses
Serum KC levels were determined by ELISA using the Quantikine Mouse CXCL1/KC kit (R&D Systems). Each bacterial strain was tested in three independent experiments by using groups of at least three mice per strain. Data were analysed using the non-parametric, Mann–Whitney, two-tailed t-test. Differences were considered significant if P ≤ 0.05. For histological analysis, distal colonic tissues of control, EPEC- or ΔnleH1H2-infected mice were washed with PBS, fixed in formaldehyde and stained with H&E solutions.
Acknowledgments
This work was supported by the Crohn’s & Colitis Foundation of America (to K.J.R.), Department of Veterans Affairs Merit Award (to G.H.), and NIDDK grants R01 DK050694 (to G.H.), K01 DK081481 (to R.J.) and P01 DK067887 (to G.H.).
Footnotes
Supporting information
Additional supporting information may be found in the online version of this article.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Angot A, Vergunst A, Genin S, Peeters N. Exploitation of eukaryotic ubiquitin signaling pathways by effectors translocated by bacterial type III and type IV secretion systems. PLoS Pathog. 2007;3:e3. doi: 10.1371/journal.ppat.0030003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashida H, Kim M, Schmidt-Supprian M, Ma A, Ogawa M, Sasakawa C. A bacterial E3 ubiquitin ligase IpaH9.8 targets NEMO/IKKgamma to dampen the host NF-kappaB-mediated inflammatory response. Nat Cell Biol. 2010;12:66–73. doi: 10.1038/ncb2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HD, Frankel G. Enteropathogenic Escherichia coli: unravelling pathogenesis. FEMS Microbiol Rev. 2005;29:83–98. doi: 10.1016/j.femsre.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Collier-Hyams LS, Zeng H, Sun J, Tomlinson AD, Bao ZQ, Chen H, et al. Cutting edge: Salmonella AvrA effector inhibits the key proinflammatory, anti-apoptotic NF-kappa B pathway. J Immunol. 2002;169:2846–2850. doi: 10.4049/jimmunol.169.6.2846. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean P, Kenny B. The effector repertoire of enteropathogenic E. coli: ganging up on the host cell. Curr Opin Microbiol. 2009;12:101–109. doi: 10.1016/j.mib.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Li Y, Hardwidge PR, Frey EA, Pfuetzner RA, Lee S, et al. Regulation of type III secretion hierarchy of translocators and effectors in attaching and effacing bacterial pathogens. Infect Immun. 2005;73:2135–2146. doi: 10.1128/IAI.73.4.2135-2146.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis A, Kudo T, Kruidenier L, Girard F, Crepin VF, MacDonald TT, et al. The p50 subunit of NF-kappaB is critical for in vivo clearance of the noninvasive enteric pathogen Citrobacter rodentium. Infect Immun. 2008;76:4978–4988. doi: 10.1128/IAI.00736-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott SJ, Sperandio V, Giron JA, Shin S, Mellies JL, Wainwright L, et al. The locus of enterocyte effacement (LEE)-encoded regulator controls expression of both LEE- and non-LEE-encoded virulence factors in entero-pathogenic and enterohemorrhagic Escherichia coli. Infect Immun. 2000;68:6115–6126. doi: 10.1128/iai.68.11.6115-6126.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Wan F, Mateo K, Callegari E, Wang D, Deng W, et al. Bacterial effector binding to ribosomal protein s3 subverts NF-kappaB function. PLoS Pathog. 2009;5:e1000708. doi: 10.1371/journal.ppat.1000708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Angulo VA, Deng W, Thomas NA, Finlay BB, Puente JL. Regulation of expression and secretion of NleH, a new non-locus of enterocyte effacement-encoded effector in Citrobacter rodentium. J Bacteriol. 2008;190:2388–2399. doi: 10.1128/JB.01602-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraga A, Miller SI. A Salmonella enterica serovar typhimurium translocated leucine-rich repeat effector protein inhibits NF-kappa B-dependent gene expression. Infect Immun. 2003;71:4052–4058. doi: 10.1128/IAI.71.7.4052-4058.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauf N, Chakraborty T. Suppression of NF-kappa B activation and proinflammatory cytokine expression by Shiga toxin-producing Escherichia coli. J Immunol. 2003;170:2074–2082. doi: 10.4049/jimmunol.170.4.2074. [DOI] [PubMed] [Google Scholar]
- Hemrajani C, Marches O, Wiles S, Girard F, Dennis A, Dziva F, et al. Role of NleH, a type III secreted effector from attaching and effacing pathogens, in colonization of the bovine, ovine, and murine gut. Infect Immun. 2008;76:4804–4813. doi: 10.1128/IAI.00742-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemrajani C, Berger CN, Robinson KS, Marches O, Mousnier A, Frankel G. NleH effectors interact with Bax inhibitor-1 to block apoptosis during enteropathogenic Escherichia coli infection. Proc Natl Acad Sci USA. 2010;107:3129–3134. doi: 10.1073/pnas.0911609106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iguchi A, Thomson NR, Ogura Y, Saunders D, Ooka T, Henderson IR, et al. Complete genome sequence and comparative genome analysis of enteropathogenic Escherichia coli O127:H6 strain E2348/69. J Bacteriol. 2009;191:347–354. doi: 10.1128/JB.01238-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isberg RR, Normark S. Host microbe interactions: bacteria innate immune response: attack and counter-attack. Curr Opin Microbiol. 2000;3:3–13. [Google Scholar]
- Jarvis KG, Giron JA, Jerse AE, McDaniel TK, Donnenberg MS, Kaper JB. Enteropathogenic Escherichia coli contains a putative type III secretion system necessary for the export of proteins involved in attaching and effacing lesion formation. Proc Natl Acad Sci USA. 1995;92:7996–8000. doi: 10.1073/pnas.92.17.7996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RM, Wu H, Wentworth C, Luo L, Collier-Hyams L, Neish AS. Salmonella AvrA coordinates suppression of host immune and apoptotic defenses via JNK pathway blockade. Cell Host Microbe. 2008;3:233–244. doi: 10.1016/j.chom.2008.02.016. [DOI] [PubMed] [Google Scholar]
- Khan MA, Bouzari S, Ma C, Rosenberger CM, Bergstrom KS, Gibson DL, et al. Flagellin-dependent and -independent inflammatory responses following infection by enteropathogenic Escherichia coli and Citrobacter rodentium. Infect Immun. 2008;76:1410–1422. doi: 10.1128/IAI.01141-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DW, Lenzen G, Page AL, Legrain P, Sansonetti PJ, Parsot C. The Shigella flexneri effector OspG interferes with innate immune responses by targeting ubiquitin-conjugating enzymes. Proc Natl Acad Sci USA. 2005;102:14046–14051. doi: 10.1073/pnas.0504466102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Negrate G, Faustin B, Welsh K, Loeffler M, Krajewska M, Hasegawa P, et al. Salmonella secreted factor L deubiquitinase of Salmonella typhimurium inhibits NF-kappaB, suppresses IkappaBalpha ubiquitination and modulates innate immune responses. J Immunol. 2008;180:5045–5056. doi: 10.4049/jimmunol.180.7.5045. [DOI] [PubMed] [Google Scholar]
- Li Q, Verma IM. NF-kappaB regulation in the immune system. Nat Rev Immunol. 2002;2:725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- Lira SA, Zalamea P, Heinrich JN, Fuentes ME, Carrasco D, Lewin AC, et al. Expression of the chemokine N51/KC in the thymus and epidermis of transgenic mice results in marked infiltration of a single class of inflammatory cells. J Exp Med. 1994;180:2039–2048. doi: 10.1084/jem.180.6.2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loukiadis E, Nobe R, Herold S, Tramuta C, Ogura Y, Ooka T, et al. Distribution, functional expression, and genetic organization of Cif, a phage-encoded type III-secreted effector from enteropathogenic and enterohemorrhagic Escherichia coli. J Bacteriol. 2008;190:275–285. doi: 10.1128/JB.00844-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maresca M, Miller D, Quitard S, Dean P, Kenny B. Enteropathogenic Escherichia coli (EPEC) effector-mediated suppression of antimicrobial nitric oxide production in a small intestinal epithelial model system. Cell Microbiol. 2005;7:1749–1762. doi: 10.1111/j.1462-5822.2005.00587.x. [DOI] [PubMed] [Google Scholar]
- Merlin C, McAteer S, Masters M. Tools for characterization of Escherichia coli genes of unknown function. J Bacteriol. 2002;184:4573–4581. doi: 10.1128/JB.184.16.4573-4581.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal R, Peak-Chew SY, McMahon HT. Acetylation of MEK2 and I kappa B kinase (IKK) activation loop residues by YopJ inhibits signaling. Proc Natl Acad Sci USA. 2006;103:18574–18579. doi: 10.1073/pnas.0608995103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Orth K. In vitro signaling by MAPK and NFkappaB pathways inhibited by Yersinia YopJ. Methods Enzymol. 2008;438:343–353. doi: 10.1016/S0076-6879(07)38024-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro P, Flatau G, Lemichez E. Bacteria and the ubiquitin pathway. Curr Opin Microbiol. 2007;10:39–46. doi: 10.1016/j.mib.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Murphy KC, Campellone KG. Lambda Red-mediated recombinogenic engineering of enterohemorrhagic and enteropathogenic E. coli. BMC Mol Biol. 2003;4:11. doi: 10.1186/1471-2199-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadler C, Baruch K, Kobi S, Mills E, Haviv G, Farago M, et al. The type III secretion effector NleE inhibits NF-kappaB activation. PLoS Pathog. 2010;6:e1000743. doi: 10.1371/journal.ppat.1000743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neish AS. Bacterial inhibition of eukaryotic pro-inflammatory pathways. Immunol Res. 2004;29:175–186. doi: 10.1385/IR:29:1-3:175. [DOI] [PubMed] [Google Scholar]
- Neurath MF, Becker C, Barbulescu K. Role of NF-kappaB in immune and inflammatory responses in the gut. Gut. 1998;43:856–860. doi: 10.1136/gut.43.6.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson KS, Mousnier A, Hemrajani C, Fairweather N, Berger CN, Frankel G. The enteropathogenic Escherichia coli effector NleH inhibits apoptosis induced by Clostridium difficile toxin B. Microbiology. 2010;156:1815–1823. doi: 10.1099/mic.0.037259-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruchaud-Sparagano MH, Maresca M, Kenny B. Enteropathogenic Escherichia coli (EPEC) inactivate innate immune responses prior to compromising epithelial barrier function. Cell Microbiol. 2007;9:1909–1921. doi: 10.1111/j.1462-5822.2007.00923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savkovic SD, Koutsouris A, Hecht G. Activation of NF-kappaB in intestinal epithelial cells by enteropathogenic Escherichia coli. Am J Physiol. 1997;273:C1160–C1167. doi: 10.1152/ajpcell.1997.273.4.C1160. [DOI] [PubMed] [Google Scholar]
- Savkovic SD, Villanueva J, Turner JR, Matkowskyj KA, Hecht G. Mouse model of enteropathogenic Escherichia coli infection. Infect Immun. 2005;73:1161–1170. doi: 10.1128/IAI.73.2.1161-1170.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha WC, Liou HC, Tuomanen EI, Baltimore D. Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Cell. 1995;80:321–330. doi: 10.1016/0092-8674(95)90415-8. [DOI] [PubMed] [Google Scholar]
- Sharma R, Tesfay S, Tomson FL, Kanteti RP, Viswanathan VK, Hecht G. Balance of bacterial pro- and anti-inflammatory mediators dictates net effect of enteropathogenic Escherichia coli on intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2006;290:G685–G694. doi: 10.1152/ajpgi.00404.2005. [DOI] [PubMed] [Google Scholar]
- Shifflett DE, Clayburgh DR, Koutsouris A, Turner JR, Hecht GA. Enteropathogenic E. coli disrupts tight junction barrier function and structure in vivo. Lab Invest. 2005;85:1308–1324. doi: 10.1038/labinvest.3700330. [DOI] [PubMed] [Google Scholar]
- Simossis VA, Heringa J. PRALINE: a multiple sequence alignment toolbox that integrates homology-extended and secondary structure information. Nucleic Acids Res. 2005;33:W289–W294. doi: 10.1093/nar/gki390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tani M, Fuentes ME, Peterson JW, Trapp BD, Durham SK, Loy JK, et al. Neutrophil infiltration, glial reaction, and neurological disease in transgenic mice expressing the chemokine N51/KC in oligodendrocytes. J Clin Invest. 1996;98:529–539. doi: 10.1172/JCI118821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tato CM, Hunter CA. Host-pathogen interactions: subversion and utilization of the NF-kappa B pathway during infection. Infect Immun. 2002;70:3311–3317. doi: 10.1128/IAI.70.7.3311-3317.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor J. Infectious infantile enterittis, yesterday and today. Proc R Soc Med. 1970;63:1297–1301. [PMC free article] [PubMed] [Google Scholar]
- Tobe T, Beatson SA, Taniguchi H, Abe H, Bailey CM, Fivian A, et al. An extensive repertoire of type III secretion effectors in Escherichia coli O157 and the role of lambdoid phages in their dissemination. Proc Natl Acad Sci USA. 2006;103:14941–14946. doi: 10.1073/pnas.0604891103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan VK, Koutsouris A, Lukic S, Pilkinton M, Simonovic I, Simonovic M, Hecht G. Comparative analysis of EspF from enteropathogenic and enterohemorrhagic Escherichia coli in alteration of epithelial barrier function. Infect Immun. 2004;72:3218–3227. doi: 10.1128/IAI.72.6.3218-3227.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadolkowski EA, Burris JA, O’Brien AD. Mouse model for colonization and disease caused by enterohemorrhagic Escherichia coli O157:H7. Infect Immun. 1990;58:2438–2445. doi: 10.1128/iai.58.8.2438-2445.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Z, Petrof EO, Boone D, Claud EC, Sun J. Salmonella effector AvrA regulation of colonic epithelial cell inflammation by deubiquitination. Am J Pathol. 2007;171:882–892. doi: 10.2353/ajpath.2007.070220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Giron JA, Torres AG, Crawford JA, Negrete E, Vogel SN, Kaper JB. Flagellin of enteropathogenic Escherichia coli stimulates interleukin-8 production in T84 cells. Infect Immun. 2003;71:2120–2129. doi: 10.1128/IAI.71.4.2120-2129.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]