

Background: We have examined the role of the polybasic domain of Cdc42 in its membrane association and transforming capability.
Results: We show that a di-arginine motif within Cdc42 is essential for binding to PIP2-containing membranes and cellular transformation.
Conclusion: These findings demonstrate that Cdc42 binds to specific membrane sites to trigger oncogenic transformation.
Significance: These findings shed new light on how Cdc42 initiates transforming signals.
Keywords: Cdc42, Cell Biology, Cell Signaling, GTPase, Membrane Lipids, Rho, GDI, PIP2, Rho GTPases, Transformation
Abstract
Rho GTPases regulate a diverse range of processes that are dependent on their proper cellular localization. The membrane localization of these GTPases is due in large part to their carboxyl-terminal geranylgeranyl moiety. In addition, most of the Rho family members contain a cluster of positively charged residues (i.e. a “polybasic domain”), directly preceding their geranylgeranyl moiety, and it has been suggested that this domain serves to fine-tune their localization among different cellular membrane sites. Here, we have taken a closer look at the role of the polybasic domain of Cdc42 in its ability to bind to membranes and induce the transformation of fibroblasts. A FRET assay for the binding of Cdc42 to liposomes of defined composition showed that Cdc42 associates more strongly with liposomes containing phosphatidylinositol 4,5-bisphosphate (PIP2) when compared either with uncharged control membranes or with liposomes containing a charge-equivalent amount of phosphatidylserine. The carboxyl-terminal di-arginine motif (Arg-186 and Arg-187) was shown to play an essential role in the binding of Cdc42 to PIP2-containing membranes. We further showed that substitutions for the di-arginine motif, when introduced within a constitutively active (“fast cycling”) Cdc42(F28L) background, had little effect on the ability of the activated Cdc42 mutant to induce microspikes/filopodia in NIH 3T3 cells, whereas they eliminated its ability to transform fibroblasts. Taken together, these findings suggest that the di-arginine motif within the carboxyl terminus of Cdc42 is necessary for this GTPase to bind at membrane sites containing PIP2, where it can initiate signaling activities that are essential for the oncogenic transformation of cells.
Introduction
Members of the family of Rho GTPases regulate a variety of cellular processes that are dependent on the proper spatial orientation of proteins including cell polarity, vesicle trafficking, and migration (1–4). Despite the importance of the cellular localization of Rho GTPases for their signaling functions, a complete understanding of how they are targeted to the membrane sites that contain their specific biological effectors is still lacking. The ability of Rho GTPases to bind to membranes is largely mediated through their isoprenylation, which in most cases involves the geranylgeranylation of a carboxyl-terminal cysteine residue (5–7). This modification enables them to interact with either lipid membranes or RhoGDI,2 the latter being a key regulatory protein that influences the membrane versus cytosolic distribution of Rho GTPases including Cdc42, Rac1, and RhoA (8–13). RhoGDI stabilizes the soluble (cytosolic) form of these GTPases, such that its overexpression in mammalian cells has been shown to result in a dramatic shift in the population of Cdc42 from membranes to the cytosol (11).
Recently, we examined how RhoGDI influences the membrane association of Cdc42 and gained new insights into the mechanism by which this regulatory protein increases the soluble pool of the GTPase (14). In particular, the association of Cdc42 with lipid membranes was shown to be a dynamic process, such that it has an intrinsic capability to dissociate from membranes with a time scale of seconds. RhoGDI initially engages Cdc42 while it is bound to membranes and is subsequently released from membranes in a complex with Cdc42. The ability of RhoGDI to bind to the geranylgeranyl tail of Cdc42 helps to maintain the GTPase in the cytosol by slowing its reassociation with the membrane surface. We have proposed that this may have important biological consequences as it prevents Cdc42 from binding indiscriminately to membrane surfaces within cells, ensuring that Cdc42 binds to membrane sites that contain its specific signaling partners.
Although this model for RhoGDI function provides some intriguing clues regarding how Cdc42, as well as perhaps other Rho GTPases, is spatially regulated, it is not the complete picture. The fact that clear differences are observed in the cellular localization patterns of different Rho GTPases (i.e. RhoA, Rac1, Cdc42), even when considering individual isoforms of a particular protein (e.g. Rac1 versus Rac2 (15, 16)), would suggest that the distinct carboxyl-terminal ends of these proteins have important roles in determining the membrane locations from which they initiate signaling activities. Most Rho GTPases contain a cluster of positively charged residues directly preceding their geranylgeranyl moiety, suggesting that the carboxyl-terminal “polybasic regions” of Rho GTPases might contribute to their localization and positioning at the appropriate cellular membrane sites for signal propagation. Rac1 and Rac2, which differ by only 12 residues (with 5 of these residues being located within the polybasic region), show significantly different subcellular localizations (15, 16). Moreover, these two isoforms of Rac have been reported to interact with a different set of effectors in hematopoietic stem cells, and it was shown that their polybasic domains are sufficient to determine their relative ability to regulate superoxide production and chemotaxis in neutrophils, as well as to activate PAK (for p21-activated kinase) (17–19). A similar situation might also be true for the two splice variants of Cdc42 (i.e. the ubiquitous form of the protein, from here on referred to as Cdc42 (NP_001782), and the brain-specific isoform (Cdc42b (NP_426359)) that differ only in 8 of their 10 carboxyl-terminal residues (20). Cdc42b was shown to be capable of inducing striking filopodia at the axonal ends of cultured neurons, with this phenotype being significantly enhanced relative to what was observed with Cdc42.3
The polybasic domain of Cdc42 contains a pair of lysine residues and arginine residues (with these two sets of charged residues being separated by a serine). The exact positioning of positively charged residues in this region is conserved from yeast to humans. Our laboratory has previously demonstrated a role for the carboxyl-terminal di-lysine motif of Cdc42, via its interaction with the γCOP subunit of the COPI complex, in regulating intracellular trafficking as well as cell growth and transformation (21). However, thus far, the role of the conserved carboxyl-terminal di-arginine pair in Cdc42 signaling has not been determined.
Here, we have examined the importance of the di-arginine motif in the membrane association of Cdc42 and its role in the ability of hyperactivated Cdc42 to propagate signals and induce the transformation of fibroblasts. We show that the carboxyl-terminal di-arginine motif is necessary for the association of Cdc42 with PIP2-containing membranes, whereas the pair of lysine residues located just upstream from the di-arginine motif does not significantly influence binding to PIP2. Conversely, only the di-lysine motif is essential for the binding of γCOP. Substituting glutamine residues for the pair of carboxyl-terminal arginine residues has no effect on the ability of the constitutively active, fast cycling Cdc42(F28L) mutant to induce the generation of microspikes from the cell surface, whereas this substitution completely blocks the transforming capability of Cdc42(F28L). These findings demonstrate the importance of the carboxyl-terminal di-arginine residues of Cdc42 and their ability to associate with PIP2-containing membrane sites for the transforming activity of this GTPase.
EXPERIMENTAL PROCEDURES
Preparation of Insect Cell-expressed Cdc42
Cdc42 and its carboxyl-terminal mutants were purified as His6-tagged proteins following baculovirus-mediated expression in Spodoptera frugiperda (Sf21) insect cells. The sequence of the amino-terminal affinity tag and linker is MSYYHHHHHHDYDIPTTENLYFQGA. The amino acid residues for Cdc42 are numbered in the text with the start methionine for the untagged protein being position 1. All purification steps were performed at 4 °C. Stirred cultures of Sf21 cells (1 liter) underwent baculoviral infection for 48 h as carried out by Kinnakeet Biotechnology (Midlothian, VA). The cell pellets were resuspended in 40 ml of hypotonic buffer (20 mm sodium borate, pH 10.2, 5 mm MgCl2, 200 μm PMSF, 1 μg/ml aprotinin, and 1 μg/ml leupeptin) and disrupted by Dounce homogenization. The membrane-containing components of the lysate were centrifuged at 150,000 × g in a Ti70 rotor (Beckman Coulter) for 30 min. The supernatant containing non-prenylated Cdc42 was discarded, and the pellet was resuspended in 50 ml of TBS-containing magnesium (TBSM; 50 mm Tris, pH 7.5, 150 mm NaCl, and 5 mm MgCl2). These steps were repeated twice, and then the pellets were resuspended in TBSM that contained 1% Triton X-100. The lysate was further homogenized and mixed for 30 min on a rotisserie, resulting in the solubilization of the geranylgeranylated Cdc42. The remaining insoluble fraction was separated by centrifugation at 9,000 × g for 20 min at 4 °C, and the pellet was discarded. The supernatant, which contained the detergent-solubilized, isoprenylated His6-tagged Cdc42, was incubated for 30 min with chelating Sepharose beads (Qiagen) charged with Ni2+. The beads were washed with 400 ml of a high salt buffer (50 mm Tris, pH 7.5, 500 mm NaCl, 5 mm MgCl2, 0.1% CHAPS, and 20 mm imidazole), and the protein was then eluted with 5 ml of elution buffer (50 mm Tris, pH 7.5, 150 mm NaCl, 5 mm MgCl2, 0.1% CHAPS, and 500 mm imidazole). The fractions containing Cdc42 were pooled and concentrated to a volume of 1.5 ml.
Preparation of Escherichia coli-expressed RhoGDI
Bacterial cells expressing GST-RhoGDI were grown at 37 °C until an A600 of 0.8 was achieved. Induction was initiated by the addition of isopropyl 1-thio-β-d-galactopyranoside (1 mm), and the cells were allowed to incubate for an additional 3 h before pelleting at 6,000 × g for 10 min. The cell pellets were homogenized in TBSM and lysed by sonication. Cell debris was centrifuged at 20,000 × g for 30 min. Supernatants containing GST-tagged RhoGDI were incubated with glutathione beads (Amersham Biosciences) and equilibrated with TEDA buffer (20 mm Tris, pH 8.0, 1 mm EDTA, 1 mm DTT, and 1 mm sodium azide) for 30 min at 4 °C. The beads were washed with several column volumes of TEDA-containing 500 mm NaCl, and then after a final rinse with TBSM, the protein was eluted with 10 mm glutathione in TBSM. The eluents were concentrated in a 10-kDa molecular mass cutoff Amicon Ultra concentrator (Fisher). Protein concentrations were determined using the Bio-Rad protein assay kit with bovine serum albumin as a standard.
Preparation of Liposomes
Lipid vesicles that were used for fluorescence spectroscopy experiments were prepared by extrusion (Avanti mini-extruder). For larger liposomes (i.e. several microns in diameter) that were to be pelleted by low speed centrifugation, the technique of rapid solvent exchange was utilized (22). The control lipid composition in molar percentages was 35% PE, 25% PS, 5% PI, and 35% cholesterol (Nu Chek Preps). All lipids used in these studies were obtained from Avanti Polar Lipids, unless specified otherwise. In liposomes containing up to 5% PIP2, the molar percentage of PIP2 was achieved by replacement of an equivalent molar percentage of PI. Liposomes containing 10% PIP2 were prepared by replacing equivalent molar percentages of PI and PE (i.e. 5% each). In liposomes containing a higher percentage of PS, the additional PS replaced an equivalent molar percentage of PE.
Liposome Centrifugation Assays
To assay the binding of Cdc42 to liposomes, Cdc42 (20 pmol) was preloaded with [35S]GTPγS (1400 cpm/pmol) by EDTA-stimulated nucleotide exchange and incubated in 200 μl of 1 mg/ml lipids, prepared by rapid solvent exchange, for 10 min at room temperature. The mixture was centrifuged at 16,000 × g in a microcentrifuge for 10 min, resuspended in fresh buffer, and centrifuged again. Radioactivity was measured in the resulting supernatant and lipid pellet.
Fluorescence Assays for Interaction of Cdc42 with Liposomes
Fluorescence measurements were made using a Varian Cary Eclipse fluorometer in the counting mode. Excitation and emission wavelengths were 365 and 440 nm, respectively. One-ml samples were stirred continuously at 25 °C in TBSM.
To prepare hexadecanoyl aminofluorescein (HAF)-labeled lipids for FRET assays, 1.25 nmol of HAF (Molecular Probes) was vortexed in 50 μl of lipids (1 mg/ml). When monitoring the release of Cdc42 from liposomes, the GTPase was first loaded with Mant-GMPPNP and incubated with 30 μl of HAF-containing liposomes at room temperature for 5 min. The mixture was added to the cuvette, and at the designated time point, RhoGDIα (from here on designated RhoGDI) was added with stirring, generating traces that monitored the changes in Mant fluorescence due to changes in FRET between Mant-nucleotide-bound Cdc42 and HAF-labeled liposomes.
When monitoring the exchange of Cdc42 between different populations of liposomes, the GTPase was loaded with Mant-GMPPNP and incubated with 20 μl of unlabeled liposomes at room temperature for 5 min. The mixture was added to the cuvette, and at the designated time point, HAF-containing liposomes were added with stirring, generating traces that monitored the decrease in Mant fluorescence as Cdc42 associated with the HAF-labeled liposomes.
Cell Culture and Transfection
NIH 3T3 cells were cultured in DMEM plus 10% calf serum at 37 °C with 5% CO2. COS-7 cells were cultured in DMEM plus 10% fetal bovine serum (FBS) at 37 °C with 5% CO2. The cells were plated at 2 × 105 in a 60-mm dish, 18 h before transfection. For the production of stable cell lines, the selection of G418-resistant NIH 3T3 colonies was carried out 48 h after transfection by adding 400 μg/ml Geneticin to the culture medium. The cell colonies resistant to G418 were selected and subcultured in DMEM plus 10% calf serum and 200 μg/ml Geneticin.
For effector binding assays, COS-7 cells were transiently transfected with constructs expressing Myc-tagged PAK3 or HA-tagged γCOP using Lipofectamine (Invitrogen) and harvested 48 h later. Purified mutants of polyhistidine (His6)-tagged Cdc42 were immobilized on nickel-agarose and exposed to COS-7 cell lysates expressing either Myc-PAK3 or HA-γCOP. The beads were resuspended and pelleted (3×) in 1 ml of cold TBSM containing 0.1% CHAPS and then mixed with 100 μl of 1× SDS-loading buffer, prior to performing SDS-PAGE and Western blot analysis using primary antibodies against the epitope tags of the effector proteins.
Immunofluorescent Staining
NIH 3T3 cells transiently expressing HA-tagged wild-type Cdc42 and different HA-tagged Cdc42 mutants were plated on glass coverslips (Corning) 24 h following transfection. After subculture for 24 h, the cells were fixed and then processed for immunocytochemistry. Immunofluorescence labeling was performed on NIH 3T3 cells as described previously (23).
Transformation Assays
The transforming activities of the HA-Cdc42(F28L) and HA-Cdc42(F28L,R186Q,R187Q) mutants were assessed by growth in 1% serum and colony formation in soft agar. For growth in low serum, stable cell lines expressing the different Cdc42 mutants were plated, and cells were trypsinized and counted at 2-day intervals. For colony formation in soft agar, the stable cell lines were suspended in 0.3% agarose in DMEM, and colonies >50 μm were counted 14 days later.
RESULTS
Cdc42 Shows Enhanced Binding to Liposomes Containing PIP2
We were interested in examining how certain lipid compositions influence the membrane association of prenylated Cdc42 to gain insight into the underlying factors that might be important in determining its cellular membrane localization. Of particular interest were the physiologically relevant lipid PIP2 and membrane domains commonly referred to as lipid rafts as both have been suggested to be important in Rho GTPase function (24). As an initial step toward examining these questions, [35S]GTPγS-bound Cdc42 was mixed with large unilamellar liposomes, prepared by rapid solvent exchange. These liposomes are large enough to be pelleted by centrifugation and thus provide a rapid and convenient screen for the relative ability of Cdc42 to partition into vesicles of varying defined lipid compositions by monitoring its presence in the pellet fraction. These assays indicated that the association of Cdc42 with liposomes mimicking the composition of lipid rafts was much less effective when compared with lipid compositions modeled after the inner leaflet of the plasma membrane, which contained 35% PE, 25% PS, 5% PI, and 35% cholesterol, as well as liposomes in which PI was replaced with an equivalent molar percentage of PIP2 (i.e. 35% PE, 25% PS, 5% PIP2, and 35% cholesterol) (supplemental Fig. S1). Based on these initial screens, we then set out to take a closer look at the potential electrostatic contribution between the positively charged and membrane-proximal polybasic region of Cdc42 and the highly negatively charged headgroup of PIP2. This was done by utilizing assays that take advantage of FRET approaches, which enable us to monitor the binding of Cdc42 to liposomes in real time and with greater sensitivity.
We first examined the association of Cdc42 with liposomes of different lipid compositions in the presence of RhoGDI. Recently, we showed that this regulatory protein exerts an important influence on the distribution of Cdc42 between membrane and soluble fractions by affecting the rate at which Cdc42 is able to rebind to liposomes following its dissociation from the lipid bilayer, rather than by directly accelerating its rate of dissociation from the membrane, as assumed previously (14). Thus, the presence of RhoGDI, by sequestering Cdc42 in the soluble fraction following its release from membranes, helped us to specifically compare the rates at which this GTPase dissociated from liposomes of different lipid compositions, and in particular, to see how negatively charged lipids such as PIP2 might affect the rate at which Cdc42 dissociates from membranes.
Fig. 1A depicts the FRET assay that we used to examine the dissociation of Cdc42 from liposomes of different defined lipid compositions, prepared by extrusion, in the presence of RhoGDI. In this assay, the fluorescence of Mant-labeled guanine nucleotides bound to Cdc42 provides a spectroscopic read-out for the association of Cdc42 with fluorescein-labeled lipids, i.e. as a result of the quenching of Mant fluorescence due to FRET. To start the experiment, recombinant GDP-bound Cdc42, expressed in Sf21 cells and purified as a His6-tagged protein, was exchanged with Mant-GMPPNP by treatment with EDTA. The Mant-GMPPNP-bound Cdc42 was incubated with HAF-labeled liposomes for 5 min. This resulted in an ∼50% quenching of the Mant fluorescence as an outcome of the binding of Mant-GMPPNP-Cdc42 to the HAF-containing liposomes. RhoGDI was added, and the dissociation of Mant-GMPPNP-Cdc42 from the liposomes was followed over time, as monitored by the increased Mant fluorescence due to the loss of FRET (Fig. 1B). A standard lipid composition for the liposomes was 35% PE, 25% PS, 5% PI, and 35% cholesterol (from here on referred to as control liposomes). We found that doubling the percentage of PS in the liposomes (i.e. 10% PE, 50% PS, 5% PI, and 35% cholesterol) had no effect on the rate or extent of Cdc42 dissociation from the lipid vesicles. In contrast, when using liposomes that contained 5% PIP2 (i.e. 35% PE, 25% PS, 5% PIP2, and 35% cholesterol), the dissociation of Cdc42 from the vesicles in the presence of RhoGDI was delayed, with the effect of PIP2 being dose-dependent as further increases in its molar percentage up to 10% (i.e. 30% PE, 25% PS, 10% PIP2, and 35% cholesterol) showed an even greater reduction in the dissociation rate of Cdc42 from liposomes.
FIGURE 1.

Cdc42 exhibits dose-dependent increase in affinity for PIP2-containing membranes. A, schematic of GDI-mediated release of Mant-guanine nucleotide-bound Cdc42 from liposomes containing HAF. B, insect cell recombinant Cdc42 (50 nm) was preloaded with Mant-GMPPNP and mixed with liposomes (30 μm bulk lipid concentration) containing HAF, with the indicated molar percentages of PIP2 and PS. The lipid composition for control liposomes was 35% cholesterol, 35% PE, 25% PS, and 5% PI. In some cases, PIP2 replaced an equal molar percentage of PI, and additional PS or PIP2, beyond 5%, replaced an equal molar percentage of PE. At the 1-min time point, RhoGDI (1 μm) was added, and the release of Cdc42 from the HAF-labeled membranes was monitored by the increase in Mant-nucleotide fluorescence. C, schematic of the intervesicle transfer of geranylgeranylated Cdc42 between the surfaces of unlabeled liposomes and liposomes containing HAF. D, Mant-GMPPNP-bound Cdc42 (30 nm) was incubated with unlabeled liposomes (20 μm bulk lipid concentration). At the zero time point, liposomes (20 μm bulk lipid concentration) containing HAF-labeled lipids, ± 5% PIP2, were added, and the exchange of Cdc42 between the different populations of liposomes was monitored through the quenching of Mant fluorescence.
Given these results suggesting that Cdc42 might associate with a higher affinity to liposomes containing PIP2, we set up a FRET assay to monitor the exchange of Cdc42 between two different populations of liposomes (Fig. 1C). Specifically, Mant-GMPPNP-loaded Cdc42 was initially bound to control liposomes lacking PIP2, and then because of its intrinsic capability to dissociate from these vesicles, it was free to reassociate with either control liposomes labeled with HAF (Fig. 1D, designated −PIP2) or HAF-labeled liposomes of varying lipid composition relative to the control vesicles. Using this read-out, we found that Mant-GMPPNP-Cdc42 was exchanged between the two populations of liposomes at identical rates when comparing control liposomes with liposomes containing a 2-fold greater amount of PS (i.e. 50% PS instead of 25% PS) (not shown). However, when we examined the exchange of Mant-GMPPNP-Cdc42 between control liposomes and HAF-labeled liposomes that contained 5% PIP2, there was a clear increase in the amount of Cdc42 that partitioned into the PIP2-containing vesicles (Fig. 1D). The faster rate of quenching of Mant fluorescence depicts a case where the Mant-GMPPNP-loaded Cdc42 binds to the PIP2-containing, HAF-labeled liposomes and appears to partition more favorably to these vesicles.
We then set out to further establish that PIP2 enhances the binding of Cdc42 to lipid vesicles through titration experiments in which we monitored the interaction of Mant-GMPPNP-bound Cdc42 with HAF-labeled liposomes containing 50% PS (which possess a net negative charge equivalent to vesicles containing 5% PIP2) versus HAF liposomes containing 5% PIP2. The fractional change in Mant-GMPPNP fluorescence after each addition of vesicles was plotted as a function of the bulk concentration of lipids (Fig. 2A) and fit to Equation 1,
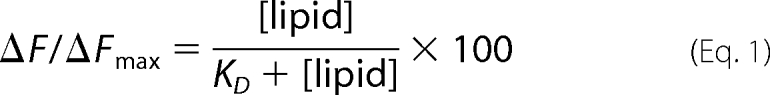 |
where KD represents the apparent dissociation constant for the interaction of Mant-GMPPNP-bound Cdc42 with the liposomes. We obtained KD values of ∼3 and ∼25 μm, respectively, for the binding of Cdc42 to liposomes containing 5% PIP2 versus 50% PS (Fig. 2A). Thus, Cdc42 loaded with Mant-GMPPNP binds with a higher affinity to PIP2-containing lipid vesicles when compared with liposomes containing PS, even when higher concentrations of PS relative to PIP2 are used. Thus, PIP2 appears to provide a binding advantage for Cdc42 to the lipid bilayer that cannot simply be mimicked by other negatively charged phospholipids.
FIGURE 2.

PIP2 influences membrane binding of geranylgeranylated Cdc42. A, insect (Sf21) cell recombinant Cdc42 (50 nm), loaded with Mant-GMPPNP, was mixed with increasing concentrations of HAF-labeled liposomes prepared by extrusion, either containing 50% PS (together with 35% cholesterol, 10% PE, and 5% PI (closed circles)) or 5% PIP2 (together with 35% cholesterol, 25% PS, and 35% PE (open circles)). The relative percentage of Mant-nucleotide quenching was plotted with respect to the bulk lipid concentration and fit to Equation 1 (see “Results”), where Fmax represents the maximal level of Mant-nucleotide quenching by fluorescein, ΔF/ΔFmax represents the proportional quenching with respect to the maximum, and KD represents the apparent dissociation constant for the interaction of Cdc42 with liposomes. B, insect cell recombinant Cdc42 (50 nm) was preloaded with Mant-GMPPNP and mixed with liposomes (30 μm bulk lipid concentration) containing HAF, with the indicated molar percentages of PI(4,5)P2, PI(3,5)P2, and phosphatidylinositol 4-phosphate (PI4P). The lipid composition for control liposomes was 35% cholesterol, 35% PE, 25% PS, and 5% PI, where the phosphoinositide isomers replaced an equal molar percentage of PI. At the zero time point, RhoGDI (1 μm) was added, and the release of Cdc42 from the membranes was monitored by the increase in Mant fluorescence.
We then compared the binding of Cdc42 with liposomes containing 5% PIP2 versus liposomes containing the physiologically less common isomer phosphatidylinositol 3,5-bisphosphate (PI(3,5)P2), which differs only in the positioning of one of its headgroup phosphate molecules. Fig. 2B shows that liposomes containing 5% PI(3,5)P2 had a similar influence on the kinetics of vesicle exchange as those containing 5% PI(4,5)P2 as both isomers imparted a similar decrease in the membrane release of Cdc42, relative to control liposomes, when assayed in the presence of RhoGDI. We find that no fewer than two phosphates per inositol headgroup are necessary to maintain this contact with Cdc42 as replacing PIP2 with an equal molar percentage of phosphatidylinositol 4-phosphate gave results that were indistinguishable from control liposomes. This suggests that the membrane binding of Cdc42 favors PIP2 as a result of its density of negatively charged moieties.
The Di-arginine Pair Located at Carboxyl Terminus of Cdc42 Is Required for Effects of PIP2 on Membrane Binding
Given the likely importance of the polybasic region of Cdc42 for its ability to bind to membranes, we next examined whether it might contribute to the regulatory effects of PIP2. The polybasic region of Cdc42 consists of a pair of lysine residues in tandem with a pair of arginine residues that lie just upstream from the C terminus and the site of attachment of the geranylgeranyl moiety. We had previously shown that the di-lysine motif was required for the binding of Cdc42 to the γCOP subunit of the COPI complex and that this interaction was important both for intracellular trafficking and for the ability of constitutively active (fast cycling) Cdc42 to transform cells (21). As depicted in Fig. 3A, the carboxyl-terminal di-lysine motif of Cdc42 may point away from the membrane surface (to be accessible for binding to γCOP). Assuming that the proximal di-arginine motif is not necessary for binding to γCOP, it then would be free to be positioned toward the membrane surface, so that it could aid in the membrane binding of Cdc42. We tested this idea by examining the ability of wild-type, GTPγS-bound Cdc42 and different Cdc42 mutants containing substitutions within the di-arginine and di-lysine motifs to bind to γCOP. HA-tagged γCOP was expressed in COS-7 cells, and then the lysates were incubated with insect cell-expressed, recombinant His6-tagged wild-type Cdc42 or with either the His6-Cdc42(R186Q,R187Q) double mutant (i.e. designated the di-glutamine or QQ mutant) or His6-Cdc42(K183S,K184S) (designated the di-serine or SS mutant), bound to nickel beads. The beads were then washed and pelleted, and the samples were examined by Western blotting. Fig. 3B shows that γCOP associates with wild-type Cdc42 and the Cdc42 di-glutamine mutant, but is incapable of binding to the Cdc42 di-serine mutant, consistent with our previous findings that it is the di-lysine motif that is required for binding to the COPI complex. Importantly, the Cdc42 di-glutamine mutant is not only able to bind to γCOP, but also interacts with other Cdc42-target/effector proteins such as PAK3 that bind in the classical manner through the “effector loop” (Switch I domain) (Fig. 3B).
FIGURE 3.

Carboxyl-terminal di-arginine motif of Cdc42 is necessary for its binding to PIP2-containing membranes. A, depiction of the membrane-binding interface of Cdc42 and the proposed orientation of its carboxyl terminus. B, insect cell recombinant His6-tagged, wild-type (WT) Cdc42 (1 μg) and the His6-tagged Cdc42 (K183S,K184S) and Cdc42(R186Q,R187Q) mutants, designated as SS and QQ, respectively, were loaded with GTPγS, prebound to nickel affinity beads, and incubated with lysates from COS7 cells transiently expressing either the HA-tagged γCOP subunit or the Myc-tagged PAK3. Eluents were analyzed by Western blotting with anti-HA or anti-Myc antibodies to assess the binding of γCOP and PAK to Cdc42, respectively. C, insect cell recombinant wild-type His6-tagged Cdc42 (50 nm), the His6-tagged Cdc42(R186Q,R187Q) (50 nm), or the His6-tagged Cdc42(K183S,K184S) mutant (50 nm) was preloaded with Mant-GMPPNP and mixed with liposomes (30 μm lipid), ± 10% PIP2. At the 1-min time point, RhoGDI (1 μm) was added, and the release of Cdc42 from the membranes was monitored by the increase in Mant-nucleotide fluorescence. D, intervesicle transfer of Mant-GMPPNP-bound wild-type Cdc42 (30 nm) or the Cdc42 QQ mutant (30 nm) from unlabeled liposomes (20 μm bulk lipid concentration) to liposomes containing HAF (20 μm bulk lipid concentration), ± 5% PIP2.
We then examined how the Cdc42 di-serine and di-glutamine mutants interacted with PIP2-containing liposomes, using two different approaches. One approach involved examining the ability of Mant-GMPPNP-loaded wild-type Cdc42 and the different Cdc42 mutants to dissociate either from HAF-labeled control liposomes or from HAF-labeled liposomes containing 10% PIP2, in the presence of RhoGDI, as read out by an increase in Mant fluorescence (Fig. 3C). Similar to the results shown in Fig. 1B, the presence of PIP2 in the liposomes significantly slowed the rate of dissociation of Cdc42-Mant-GMPPNP from the HAF-labeled vesicles. The Cdc42 di-serine mutant exhibited a similar reduced rate of dissociation from the PIP2-containing liposomes, whereas the Mant-GMPPNP-loaded Cdc42 di-glutamine mutant exhibited a rate of dissociation from PIP2-containing vesicles (Fig. 3C), as well as control vesicles lacking PIP2 (not shown), approaching that for the dissociation of wild-type Cdc42 from control liposomes.
In a second set of experiments, we examined the ability of wild-type Cdc42, versus the Cdc42 di-serine and di-glutamine mutants, to exchange between control liposomes and liposomes containing 5% PIP2, as monitored in real time by the quenching of Mant fluorescence that accompanies the dissociation of Cdc42 from unlabeled liposomes and its subsequent binding to HAF-labeled vesicles. As shown in Fig. 3D, the Cdc42 di-glutamine mutant exhibited a rate of exchange between control liposomes and liposomes containing PIP2 that was significantly slower than wild-type Cdc42 (compare blue and red traces, respectively) and instead was identical to the rate of exchange of wild-type Cdc42 between control liposomes lacking PIP2 (black trace). These results further demonstrate that it is the di-arginine pair within the carboxyl-terminal region of Cdc42 that is essential for the high affinity membrane binding conferred by PIP2.
Disrupting the Interaction between Cdc42 and PIP2 Selectively Blocks Its Ability to Induce Transformation of Fibroblasts while Preserving Its Effects on Cell Morphology
In light of the role played by the di-arginine pair within the carboxyl-terminal domain of Cdc42 in binding to PIP2-containing membranes, we were interested in examining the cellular consequences of mutating these residues. First, we set out to see whether there might be gross changes in the overall cellular localization of Cdc42 containing substitutions at this site. Based on immunofluorescence experiments, we thus far have not detected significant differences in the overall cellular localization for Cdc42 that can be attributed to substitutions for the carboxyl-terminal di-arginine pair. Some examples are shown in Fig. 4, where we have compared the localization of the HA-tagged wild-type Cdc42, the constitutively active HA-Cdc42(F28L) mutant, and the HA-Cdc42(F28L,R186Q,R187Q) triple mutant. Each of these Cdc42 constructs showed plasma membrane staining and Golgi staining as observed previously for this GTPase (25).
FIGURE 4.

Subcellular localization of Cdc42 and its microspike formation were not detectably altered by disrupting its interaction with PIP2. NIH 3T3 cells were transiently transfected with HA epitope-tagged Cdc42, Cdc42(F28L), or Cdc42(F28L,R186Q,R187Q). The subcellular localization was examined by anti-HA staining (left panels). Actin cytoskeletal morphology was examined by rhodamine-conjugated phalloidin staining (right panels). Arrows point to microspikes.
We then set out to probe for the functional consequences of substituting for the di-arginine pair on Cdc42. We first examined whether these changes influenced the ability of the constitutively active Cdc42(F28L) molecule to stimulate microspike formation, i.e. one of the classical read-outs for Cdc42 cellular function (26). However, as shown in Fig. 4, we did not detect significant differences in the ability of the activated Cdc42(F28L) mutant, versus the Cdc42(F28L,R186Q,R187Q) triple mutant, to induce the microspike phenotype in NIH 3T3 cells (see arrows).
We next generated NIH 3T3 cell lines stably expressing the HA-Cdc42(F28L) mutant and a HA-Cdc42(F28L,R186Q,R187Q) triple mutant (Fig. 5A shows their relative expression) to see whether substitutions for the carboxyl-terminal di-arginine pair might impact the ability of activated Cdc42(F28L) to induce cellular transformation. Here, we obtained some very interesting and striking results. A number of previous studies have shown that the Cdc42(F28L) mutant is able to induce the transformation of fibroblasts as read out by growth in low serum or by colony formation in soft agar. This is further demonstrated in Fig. 5, B–D. However, the HA-Cdc42(F28L,R186Q,R187Q) triple mutant, when stably expressed in NIH 3T3 cells at levels comparable with HA-Cdc42(F28L) (Fig. 5A), showed little ability to stimulate growth in low serum (Fig. 5B) or to exhibit anchorage independent growth (Fig. 5, C and D). Collectively, these results highlight two important points. First, they show that it is possible to uncouple the ability of an activated Cdc42 mutant to induce actin cytoskeletal changes and microspike formation from its ability to drive cellular transformation. Secondly, they suggest that the ability of constitutively active Cdc42 to associate with PIP2 is essential for its transforming activity.
FIGURE 5.

Binding to PIP2 is required for Cdc42-stimulated cell growth and transformation. A, NIH 3T3 cell lines that stably expressed HA-tagged Cdc42(F28L) or Cdc42(F28L,R186Q,R187Q) were generated. Cdc42 protein was detected in insect cell lysates by immunoblotting with an anti-HA antibody. B, NIH 3T3 cells that stably expressed HA-tagged Cdc42(F28L) or Cdc42(F28L,R186Q,R187Q) were cultured at a low concentration (1%) of calf serum, harvested, and counted at 2-day intervals over a 1-week period. Error bars indicate S.D. C, anchorage-independent growth of HA-Cdc42(F28L) and HA-Cdc42(F28L,R186Q,R187Q)-expressing NIH 3T3 cells was determined by colony formation in soft agar. After ∼2 weeks, plates were examined and photographed. D, the colonies from the soft agar plates shown in C were scored. In each experiment, four duplicate 35-mm plates were counted at four randomly chosen areas. Error bars in indicate S.D.
To obtain additional evidence for the importance of PIP2 in Cdc42(F28L)-induced cellular transformation, we performed two sets of experiments. One involved examining the effects of phenylarsine oxide on Cdc42-mediated transformation, given the reports that it inhibits PIP2 production in cells by blocking the actions of phosphatidylinositol 4-phosphate kinase (27, 28). Supplemental Fig. S2A shows that phenylarsine oxide strongly inhibited the ability of Cdc42(F28L) to confer NIH 3T3 cells with the transformed phenotype of being capable of growing in low serum, whereas HeLa cells were significantly less sensitive to this treatment under identical experimental conditions. The second approach involved transiently expressing a mutated RFP-labeled, lipidated MARCKS protein that is effective in binding PIP2 in a sustained manner (i.e. without being susceptible to the PKC-mediated reversal of this interaction (29)) into Cdc42(F28L)-expressing cells (supplemental Fig. S2B). The expression of the MARCKS mutant, but not the RFP vector control, gave rise to a strong inhibition of Cdc42(F28L)-induced transformation. Taken together, the results of these experiments were consistent with the idea that the interaction of Cdc42(F28L) with PIP2-containing membranes was necessary for this oncogenic GTPase to confer its full transforming capability.
DISCUSSION
Carboxyl-terminal Di-arginine Motif of Cdc42 Targets It to Membranes Containing PIP2
The Rho family GTPase, Cdc42, helps to regulate a broad array of cellular events ranging from actin cytoskeletal remodeling and polarity-dependent processes to cell growth and differentiation. It has been commonly assumed that most of these activities are dependent upon the interactions of activated Cdc42 with its signaling targets and effector proteins along the surfaces of cellular membranes. Thus, in this study, we set out to better understand how the membrane binding of Cdc42 is mediated, and in particular, the role played by a stretch of basic amino acid residues located within its carboxyl-terminal end. As a first step toward addressing these questions, we used synthetic liposomes as model membranes with compositions consistent with the constituent lipids of the inner leaflet of the plasma membrane. The negatively charged lipids PS and PIP2, which are both major components of the inner leaflet of the plasma membrane (16, 30–32), have been shown to be involved in targeting polybasic domain-containing proteins to the plasma membrane. We examined the contributions from each of these anionic lipids by selectively including one or the other in liposomes. For these experiments, we chose levels of PIP2 that were typically 5–10% of the total lipid in the liposomes. The estimated concentration range in cells has been suggested to lie between 0.2 and 3%, assuming that the lipids are randomly distributed (33, 34). However, this does not take into account the existence of membrane domain heterogeneity, and in particular, the presence of PIP2-enriched domains, where the levels of PIP2 can exceed 5–10% of the total lipid (35–39). We found that when Cdc42 was bound to liposomes containing PIP2, its rate of dissociation from the membrane surface in the presence of RhoGDI was significantly reduced when compared with its dissociation from control lipid vesicles. No such effect was seen for Cdc42 in liposomes containing excess concentrations of PS. Furthermore, Cdc42 was more efficient at partitioning into liposomes containing PIP2 when compared with control vesicles. Collectively, these results indicate that PIP2 plays a specific role by enhancing the binding of Cdc42 to lipid bilayers that cannot be simply accounted for by a bulk negative charge of anionic liposomes, but rather is influenced by the charge density of the individual lipid.
Cdc42 contains a polybasic region within its carboxyl-terminal end, located just upstream from a covalently attached geranylgeranyl moiety. The polybasic region consists of di-lysine and di-arginine motifs, separated by a single serine residue. Because the di-arginine motif directly precedes the geranylgeranyl moiety, we speculated that it might be in a better position to contact the membrane when compared with the di-lysine motif. Furthermore, the di-lysine motif has been shown to make important contacts with the γCOP subunit of the membrane-associated COPI complex, an interaction that is directed away from the membrane surface (21). In fact, we show here that the di-arginine motif did not contribute to the interaction between Cdc42 and the γCOP subunit. However, it is necessary for the ability of Cdc42 to bind with higher affinity to liposomes containing PIP2. These findings then raised the question of what might be the cellular consequences of the binding of Cdc42 to PIP2.
Binding of Cdc42 to PIP2 Has Important Roles in Its Ability to Impact Cell Growth
The anionic lipid PI(4,5)P2 has been shown to accumulate at locations where Cdc42 is known to function, in particular, at the site of bud formation in Saccharomyces cerevisiae and at sites of cytokinesis and phagocytosis in metazoan cells (34, 39–42).4 Moreover, PIP2 has been shown to bind to guanine nucleotide exchange factors (GEFs), scaffolding partners, and effectors of Cdc42 (43–47), as well as to bind to and inhibit Rho GTPase-activating proteins (GAPs) (48). Therefore, the recruitment of Cdc42 to PIP2-enriched regions on the plasma membrane could facilitate its ability to become activated and/or to signal through its effector proteins. To see whether this might indeed be the case, we substituted 2 glutamine residues for the di-arginine motif within an activated Cdc42(F28L) background. Indeed, we found that these substitutions had a profound impact on the ability of this oncogenic mutant to induce cellular transformation, as read out by the growth of cells under serum-deprived conditions or the formation of colonies in soft agar. Consistent with the importance of PIP2 for Cdc42(F28L)-induced cellular transformation, both the use of the inhibitor phenylarsine oxide that blocks PIP2 production and the introduction of a MARCKS mutant that sequesters PIP2 markedly inhibited the ability of this oncogenic GTPase to transform fibroblasts. Although these experiments have their limitations, as increasing concentrations of phenylarsine oxide and the MARCKS mutant can have deleterious effects on normal cells, in both cases, we were able to achieve effects in concentration ranges that were specific for Cdc42-transformed cells. Moreover, substitutions of the 2 arginine residues on Cdc42(F28L) essential for binding PIP2 did not directly impact its ability to interact directly with downstream signaling effector proteins. This was even the case for the γCOP subunit whose binding to Cdc42 requires a di-lysine motif that is immediately proximal to the 2 critical arginine residues. Interestingly, we also found that mutating the di-arginine motif did not significantly affect the ability of activated Cdc42 to stimulate the formation of microspikes, which represents one of the best known cellular responses to Cdc42 (26). Thus, substitutions for the di-arginine motif uncoupled the ability of Cdc42 to trigger actin cytoskeletal rearrangements necessary for generating microspikes/filopodia from the stimulation of those signaling events that underlie the ability of Cdc42 to induce transformed phenotypes.
What do these findings imply for the role of PIP2 in the cellular actions of Cdc42? An examination of the epitope-tagged forms of Cdc42(F28L) versus the Cdc42(F28L,R186Q, R187Q) triple mutant by immunofluorescence indicates that substituting for the carboxyl-terminal di-arginine motif does not lead to marked changes in the cellular localization of this GTPase. Apparently, Cdc42 is able to interact with the specific effector protein(s) necessary for mediating the actin cytoskeletal changes required for microspike formation from plasma membrane sites, independent of whether or not they contain PIP2. On the other hand, the ability of Cdc42 to engage the essential effector(s) for cellular transformation requires that Cdc42 binds to specific membrane locations that are rich in PIP2. This could be the result of one or more Cdc42 effector proteins, which are necessary for sending transforming signals, having the capability to bind PIP2 so as to be recruited to these specific membrane locations.
The ability of PIP2 to increase the overall affinity of Cdc42 for the membrane could provide an added advantage of enabling these membrane sites to better compete with RhoGDI for binding the geranylgeranyl moiety of Cdc42. This also could have interesting implications with regard to how the different classes of regulatory proteins for Cdc42 influence its ability to cycle on and off membranes. Recently, we have found that Rho GTPases, when complexed to RhoGDI in solution, are still susceptible to the actions of RhoGEFs, enabling them to undergo GDP-GTP exchange.5 Once GTP-bound Cdc42 encounters a membrane, its affinity for RhoGDI is significantly weakened (14). This should result in the dissociation of RhoGDI from Cdc42, allowing it to bind effector proteins and stimulate signaling activities. These signals would then be terminated by the actions of RhoGAPs that catalyze the hydrolysis of GTP, thus yielding GDP-bound Cdc42 that binds to RhoGDI with higher affinity at the membrane (14). However, because the rate at which Cdc42 dissociates from membranes, even when bound to RhoGDI, is limited by its intrinsic ability to dissociate from lipid bilayers (14), the presence of PIP2 might significantly slow its membrane release and thereby provide an opportunity for a GEF to catalyze another round of GDP-GTP exchange at the membrane signaling site, resulting in a net accumulation of active Cdc42 at the membrane.
Still, another intriguing possibility is that the binding of PIP2 to the carboxyl-terminal end of Cdc42 might help the GTPase to assume the proper activated conformational state to engage a specific effector protein that is essential for transformation and/or to induce a specific change in the activity of the effector. Previous work from our laboratory showed that the ability of RhoGDI to distinguish between the GDP- and GTP-bound forms of Cdc42 was dependent upon Cdc42 being associated with membranes (14). Specifically, although the binding of RhoGDI to the GDP- versus GTP-bound forms of Cdc42 in solution was essentially indistinguishable, clear differences were observed when monitoring the interactions of these nucleotide-bound forms of Cdc42 with liposomes. This then implies that the lipid bilayer interacts with Cdc42 in a manner that significantly influences its activated conformational state, which in turn engages specific target/effector proteins. Moreover, the presence of PIP2 at specific membrane sites might further tune the conformation of activated Cdc42, enabling it to engage specific targets, through the interactions of this lipid with the carboxyl-terminal di-arginine motif of the GTPase. Future studies will be further directed toward examining how the lipid bilayer might help to influence GTPase structure or the capacity of activated Cdc42 to form signaling complexes that are important for its transforming capability and whether a specific Cdc42 effector protein(s) is recruited to membrane sites where PIP2 has accumulated.
Supplementary Material
Acknowledgments
We thank Cindy Westmiller for expert secretarial assistance. We also thank Dr. Ronald P. Magnusson at Kinnakeet Biotechnology for production of baculovirus-infected insect cell pellets and the Baird Laboratory at Cornell University for providing us with the RFP-MARCKS-ED SA4 plasmid.

This article contains supplemental Figs. S1 and S2.
M. Endo and R. A. Cerione, unpublished results.
U. Golebiewska, J. G. Kay, T. Masters, S. Grinstein, W. Im, R. W. Pastor, S. Scarlata, and S. McLaughlin, personal communication.
T. Freisinger, B. Klunder, J. L. Johnson, A. Neves, T. Schmidt, M. Costanzo, C. Boone, E. Frey, and R. Welcher-Soldner, in preparation.
- RhoGDI
- Rho guanine nucleotide dissociation inhibitor
- PIP2
- phosphatidylinositol 4,5-bisphosphate
- PI(3,5)P2
- phosphatidylinositol 3,5-bisphosphate
- PI(4,5)P2
- phosphatidylinositol 4,5-bisphosphate
- PE
- phosphatidylethanolamine
- PI
- phosphatidylinositol
- PS
- phosphatidylserine
- COPI
- coat protein complex I
- HAF
- hexadecanoyl aminofluorescein
- PAK
- p21-activated kinase
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate
- GMPPNP
- 5′-guanylyl imidodiphosphate
- Mant
- methylanthraniloyl
- TBSM
- TBS-containing magnesium
- MARCKS
- myristoylated alanine-rich C kinase substrate
- GEF
- guanine nucleotide exchange factor.
REFERENCES
- 1. Jaffe A. B., Hall A. (2005) Rho GTPases: biochemistry and biology. Annu. Rev. Cell Dev. Biol. 21, 247–269 [DOI] [PubMed] [Google Scholar]
- 2. Harris K. P., Tepass U. (2010) Cdc42 and vesicle trafficking in polarized cells. Traffic 11, 1272–1279 [DOI] [PubMed] [Google Scholar]
- 3. Heasman S. J., Ridley A. J. (2008) Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 9, 690–701 [DOI] [PubMed] [Google Scholar]
- 4. Erickson J. W., Cerione R. A. (2001) Multiple roles for Cdc42 in cell regulation. Curr. Opin. Cell Biol. 13, 153–157 [DOI] [PubMed] [Google Scholar]
- 5. Struckhoff A. P., Rana M. K., Worthylake R. A. (2011) RhoA can lead the way in tumor cell invasion and metastasis. Front. Biosci. 16, 1915–1926 [DOI] [PubMed] [Google Scholar]
- 6. Silvius J. R., l'Heureux F. (1994) Fluorometric evaluation of the affinities of isoprenylated peptides for lipid bilayers. Biochemistry 33, 3014–3022 [DOI] [PubMed] [Google Scholar]
- 7. Ziman M., Preuss D., Mulholland J., O'Brien J. M., Botstein D., Johnson D. I. (1993) Subcellular localization of Cdc42p, a Saccharomyces cerevisiae GTP-binding protein involved in the control of cell polarity. Mol. Biol. Cell 4, 1307–1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fukumoto Y., Kaibuchi K., Hori Y., Fujioka H., Araki S., Ueda T., Kikuchi A., Takai Y. (1990) Molecular cloning and characterization of a novel type of regulatory protein (GDI) for the rho proteins, ras p21-like small GTP-binding proteins. Oncogene 5, 1321–1328 [PubMed] [Google Scholar]
- 9. Leonard D., Hart M. J., Platko J. V., Eva A., Henzel W., Evans T., Cerione R. A. (1992) The identification and characterization of a GDP-dissociation inhibitor (GDI) for the CDC42Hs protein. J. Biol. Chem. 267, 22860–22868 [PubMed] [Google Scholar]
- 10. Nomanbhoy T. K., Cerione R. (1996) Characterization of the interaction between RhoGDI and Cdc42Hs using fluorescence spectroscopy. J. Biol. Chem. 271, 10004–10009 [DOI] [PubMed] [Google Scholar]
- 11. Michaelson D., Silletti J., Murphy G., D'Eustachio P., Rush M., Philips M. R. (2001) Differential localization of Rho GTPases in live cells: regulation by hypervariable regions and RhoGDI binding. J. Cell Biol. 152, 111–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Roberts P. J., Mitin N., Keller P. J., Chenette E. J., Madigan J. P., Currin R. O., Cox A. D., Wilson O., Kirschmeier P., Der C. J. (2008) Rho family GTPase modification and dependence on CAAX motif-signaled posttranslational modification. J. Biol. Chem. 283, 25150–25163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moissoglu K., Slepchenko B. M., Meller N., Horwitz A. F., Schwartz M. A. (2006) In vivo dynamics of Rac-membrane interactions. Mol. Biol. Cell 17, 2770–2779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Johnson J. L., Erickson J. W., Cerione R. A. (2009) New insights into how the Rho guanine nucleotide dissociation inhibitor regulates the interaction of Cdc42 with membranes. J. Biol. Chem. 284, 23860–23871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Magalhaes M. A., Glogauer M. (2010) Pivotal advance: phospholipids determine net membrane surface charge resulting in differential localization of active Rac1 and Rac2. J. Leukoc. Biol. 87, 545–555 [DOI] [PubMed] [Google Scholar]
- 16. Yeung T., Gilbert G. E., Shi J., Silvius J., Kapus A., Grinstein S. (2008) Membrane phosphatidylserine regulates surface charge and protein localization. Science 319, 210–213 [DOI] [PubMed] [Google Scholar]
- 17. Gu Y., Filippi M. D., Cancelas J. A., Siefring J. E., Williams E. P., Jasti A. C., Harris C. E., Lee A. W., Prabhakar R., Atkinson S. J., Kwiatkowski D. J., Williams D. A. (2003) Hematopoietic cell regulation by Rac1 and Rac2 guanosine triphosphatases. Science 302, 445–449 [DOI] [PubMed] [Google Scholar]
- 18. Yamauchi A., Marchal C. C., Molitoris J., Pech N., Knaus U., Towe J., Atkinson S. J., Dinauer M. C. (2005) Rac GTPase isoform-specific regulation of NADPH oxidase and chemotaxis in murine neutrophils in vivo: role of the C-terminal polybasic domain. J. Biol. Chem. 280, 953–964 [DOI] [PubMed] [Google Scholar]
- 19. Knaus U. G., Wang Y., Reilly A. M., Warnock D., Jackson J. H. (1998) Structural requirements for PAK activation by Rac GTPases. J. Biol. Chem. 273, 21512–21518 [DOI] [PubMed] [Google Scholar]
- 20. Kang R., Wan J., Arstikaitis P., Takahashi H., Huang K., Bailey A. O., Thompson J. X., Roth A. F., Drisdel R. C., Mastro R., Green W. N., Yates J. R., 3rd, Davis N. G., El-Husseini A. (2008) Neural palmitoyl-proteomics reveals dynamic synaptic palmitoylation. Nature 456, 904–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wu W. J., Erickson J. W., Lin R., Cerione R. A. (2000) The γ-subunit of the coatomer complex binds Cdc42 to mediate transformation. Nature 405, 800–804 [DOI] [PubMed] [Google Scholar]
- 22. Buboltz J. T., Feigenson G. W. (1999) A novel strategy for the preparation of liposomes: rapid solvent exchange. Biochim. Biophys. Acta 1417, 232–245 [DOI] [PubMed] [Google Scholar]
- 23. Antonyak M. A., Li B., Boroughs L. K., Johnson J. L., Druso J. E., Bryant K. L., Holowka D. A., Cerione R. A. (2011) Cancer cell-derived microvesicles induce transformation by transferring tissue transglutaminase and fibronectin to recipient cells. Proc. Natl. Acad. Sci. 108, 4852–4857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. del Pozo M. A., Alderson N. B., Kiosses W. B., Chiang H. H., Anderson R. G., Schwartz M. A. (2004) Integrins regulate Rac targeting by internalization of membrane domains. Science 303, 839–842 [DOI] [PubMed] [Google Scholar]
- 25. Erickson J. W., Zhang C., Kahn R. A., Evans T., Cerione R. A. (1996) Mammalian Cdc42 is a brefeldin A-sensitive component of the Golgi apparatus. J. Biol. Chem. 271, 26850–26854 [DOI] [PubMed] [Google Scholar]
- 26. Nobes C. D., Hall A. (1995) Rho, Rac, and Cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 81, 53–62 [DOI] [PubMed] [Google Scholar]
- 27. Micheva K. D., Holz R. W., Smith S. J. (2001) Regulation of presynaptic phosphatidylinositol 4,5-biphosphate by neuronal activity. J. Cell Biol. 154, 355–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gwanyanya A., Sipido K. R., Vereecke J., Mubagwa K. (2006) ATP and PIP2 dependence of the magnesium-inhibited, TRPM7-like cation channel in cardiac myocytes. Am. J. Physiol. Cell Physiol. 291, C627–CC635 [DOI] [PubMed] [Google Scholar]
- 29. Gadi D., Wagenknecht-Wiesner A., Holowka D., Baird B. (2011) Sequestration of phosphoinositides by mutated MARCKS effector domain inhibits stimulated Ca2+ mobilization and degranulation in mast cells. Mol. Biol. Cell 22, 4908–4917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Di Paolo G., De Camilli P. (2006) Phosphoinositides in cell regulation and membrane dynamics. Nature 443, 651–657 [DOI] [PubMed] [Google Scholar]
- 31. Golebiewska U., Nyako M., Woturski W., Zaitseva I., McLaughlin S. (2008) Diffusion coefficient of fluorescent phosphatidylinositol 4,5-bisphosphate in the plasma membrane of cells. Mol. Biol. Cell 19, 1663–1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Heo W. D., Inoue T., Park W. S., Kim M. L., Park B. O., Wandless T. J., Meyer T. (2006) PI(3,4,5)P3 and PI(4,5)P2 lipids target proteins with polybasic clusters to the plasma membrane. Science 314, 1458–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Raucher D., Stauffer T., Chen W., Shen K., Guo S., York J. D., Sheetz M. P., Meyer T. (2000) Phosphatidylinositol 4,5-bisphosphate functions as a second messenger that regulates cytoskeleton-plasma membrane adhesion. Cell 100, 221–228 [DOI] [PubMed] [Google Scholar]
- 34. McLaughlin S., Wang J., Gambhir A., Murray D. (2002) PIP2 and proteins: interactions, organization, and information flow. Annu. Rev. Biophys. Biomol. Struct. 31, 151–175 [DOI] [PubMed] [Google Scholar]
- 35. Redfern D. A., Gericke A. (2005) pH-dependent domain formation in phosphatidylinositol polyphosphate/phosphatidylcholine mixed vesicles. J. Lipid Res. 46, 504–515 [DOI] [PubMed] [Google Scholar]
- 36. Cho H., Kim Y. A., Yoon J. Y., Lee D., Kim J. H., Lee S. H., Ho W. K. (2005) Low mobility of phosphatidylinositol 4,5-bisphosphate underlies receptor specificity of Gq-mediated ion channel regulation in atrial myocytes. Proc. Natl. Acad. Sci. 102, 15241–15246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fooksman D. R., Shaikh S. R., Boyle S., Edidin M. (2009) Cutting edge: phosphatidylinositol 4,5-bisphosphate concentration at the APC side of the immunological synapse is required for effector T cell function. J. Immunol. 182, 5179–5182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. van den Bogaart G., Meyenberg K., Risselada H. J., Amin H., Willig K. I., Hubrich B. E., Dier M., Hell S. W., Grubmüller H., Diederichsen U., Jahn R. (2011) Membrane protein sequestering by ionic protein-lipid interactions. Nature 479, 552–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Botelho R. J., Teruel M., Dierckman R., Anderson R., Wells A., York J. D., Meyer T., Grinstein S. (2000) Localized biphasic changes in phosphatidylinositol 4,5-bisphosphate at sites of phagocytosis. J. Cell Biol. 151, 1353–1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Field S. J., Madson N., Kerr M. L., Galbraith K. A., Kennedy C. E., Tahiliani M., Wilkins A., Cantley L. C. (2005) PtdIns(4,5)P2 functions at the cleavage furrow during cytokinesis. Curr. Biol. 15, 1407–1412 [DOI] [PubMed] [Google Scholar]
- 41. Janetopoulos C., Devreotes P. (2006) Phosphoinositide signaling plays a key role in cytokinesis. J. Cell Biol. 174, 485–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bertin A., McMurray M. A., Thai L., Garcia G., 3rd, Votin V., Grob P., Allyn T., Thorner J., Nogales E. (2010) Phosphatidylinositol 4,5-bisphosphate promotes budding yeast septin filament assembly and organization. J. Mol. Biol. 404, 711–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Papayannopoulos V., Co C., Prehoda K. E., Snapper S., Taunton J., Lim W. A. (2005) A polybasic motif allows N-WASP to act as a sensor of PIP2 density. Mol. Cell 17, 181–191 [DOI] [PubMed] [Google Scholar]
- 44. Das B., Shu X., Day G. J., Han J., Krishna U. M., Falck J. R., Broek D. (2000) Control of intramolecular interactions between the pleckstrin homology and Dbl homology domains of Vav and Sos1 regulates Rac binding. J. Biol. Chem. 275, 15074–15081 [DOI] [PubMed] [Google Scholar]
- 45. Crompton A. M., Foley L. H., Wood A., Roscoe W., Stokoe D., McCormick F., Symons M., Bollag G. (2000) Regulation of Tiam1 nucleotide exchange activity by pleckstrin domain binding ligands. J. Biol. Chem. 275, 25751–25759 [DOI] [PubMed] [Google Scholar]
- 46. Martin-Belmonte F., Gassama A., Datta A., Yu W., Rescher U., Gerke V., Mostov K. (2007) PTEN-mediated apical segregation of phosphoinositides controls epithelial morphogenesis through Cdc42. Cell 128, 383–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Baumeister M. A., Martinu L., Rossman K. L., Sondek J., Lemmon M. A., Chou M. M. (2003) Loss of phosphatidylinositol 3-phosphate binding by the C-terminal Tiam-1 pleckstrin homology domain prevents in vivo Rac1 activation without affecting membrane targeting. J. Biol. Chem. 278, 11457–11464 [DOI] [PubMed] [Google Scholar]
- 48. Ligeti E., Dagher M. C., Hernandez S. E., Koleske A. J., Settleman J. (2004) Phospholipids can switch the GTPase substrate preference of a GTPase-activating protein. J. Biol. Chem. 279, 5055–5058 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.