

Abstract
Heparin is a polysaccharide-based natural product that is used clinically as an anticoagulant drug. Heparan sulfate 3-O-sulfotransferase (3-OST) is an enzyme that transfers a sulfo group to the 3-OH position of a glucosamine unit. 3-OST is present in multiple isoforms, and the polysaccharides modified by these different isoforms perform distinct biological functions. 3-OST isoform 1 (3-OST-1) is the key enzyme for the biosynthesis of anticoagulant heparin. Here, we report the crystal structure of the ternary complex of 3-OST-1, 3′-phosphoadenosine 5′-phosphate, and a heptasaccharide substrate. Comparisons to previously determined structures of 3-OST-3 reveal unique binding modes used by the different isoforms of 3-OST for distinguishing the fine structures of saccharide substrates. Our data demonstrate that the saccharide substrates display distinct conformations when interacting with the different 3-OST isoforms. Site-directed mutagenesis data suggest that several key amino residues, including Lys259, Thr256, and Trp283 in 3-OST-3 and Arg268 in 3-OST-1, play important roles in substrate binding and specificity between isoforms. These results deepen our understanding of the biosynthetic mechanism of heparan sulfate and provide structural information for engineering enzymes for an enhanced biosynthetic approach to heparin production.
Keywords: oligosaccharides, substrate specificity, ternary complex, heparan sulfate
Heparan sulfate (HS) is a highly sulfated polysaccharide that is commonly found on the mammalian cell surface and in the extracellular matrix. HS participates in a wide range of physiological and pathophysiological functions, including embryonic development, inflammatory responses, blood coagulation, and assisting viral/bacterial infections (1). HS consists of a disaccharide repeating unit with glucuronic acid (GlcA) or iduronic acid (IdoA) and glucosamine, each of which is capable of carrying sulfo groups. The position of the sulfo groups as well as the location of the IdoA unit dictates the functions of HS (1). The placement of sulfo groups and IdoA units involves a series of HS biosynthetic enzymes, including an epimerase and various sulfotransferases. The heparan sulfate 3-O-sulfotransferases (3-OSTs) are a key sulfotransferase family in the HS biosynthetic pathway.
The 3-OSTs transfer a sulfo group to the 3-OH position of an N-sulfo glucosamine or an N-unsubstituted glucosamine unit to form 3-O-sulfo glucosamine (GlcNS3S ± 6S or GlcNH23S ± 6S). The 3-OST family consists of seven different isoforms that enable introduction of a GlcNS3S unit within distinct carbohydrate contexts (2). For example, 3-OST-1 sulfates the glucosamine unit that is flanked by a GlcA unit on its nonreducing side, and 3-OST-3 sulfates the glucosamine unit that is flanked by an IdoA2S unit on its nonreducing side (Fig. S1). Consequently, the HS modified by different 3-OST isoforms displays different affinities to physiological and pathophysiological targets. The HS modified by 3-OST-1 has high affinity to antithrombin, bestowing this polysaccharide with anticoagulant activity, whereas the HS modified by 3-OST-3 has high affinity to the glycoprotein D of herpes simplex virus type 1, which serves as an entry receptor for the virus (3). The distinct roles of 3-O-sulfation in directing HS function offer a model to understand the regulation of HS biosynthesis, especially the mechanism used by the isoforms to specifically recognize the saccharide structures around the modification site.
Understanding the biosynthesis of HS is not only critical for dissecting its structure–function relationships in vivo, but also plays a significant role in aiding HS-based drug discovery. Heparin, a clinically used anticoagulant drug, is a specialized form of HS that contains a higher sulfation level and is more densely populated with IdoA units. Heparin is currently isolated from porcine intestine or bovine lung through a poorly regulated supply chain. The worldwide distribution of contaminated heparin in 2007 raised the concerns over the safety and reliability of animal-sourced heparins (4). A cost-effective method for preparing synthetic heparin and securing the safety of the heparin supply chain is, therefore, highly desirable. Furthermore, a facile method for preparing heparin enables engineering of HS/heparin with improved anticoagulant efficacy and reduced side effects, as well as exploiting heparin or heparin-like molecules for the development of anticancer and antiviral drugs (5, 6). Although heparin fragments can be synthesized by a purely chemical method (7), the synthesis is extremely challenging, especially for products larger than a hexsaccharide. A new chemoenzymatic approach provides a promising alternative method to prepare heparin and HS fragments with high efficiency and purity (8). Improved knowledge of the mechanism of action of HS biosynthetic enzymes will further enhance the chemoenzymatic method for preparing therapeutic heparins.
In this article, we report a crystal structure of a ternary complex of 3-OST-1/3′-phosphoadenosine 5′-phosphate (PAP) with an acceptor saccharide substrate. Detailed comparison with previously reported crystal structures of 3-OST-1, 3-OST-3 (9, 10), and the ternary 3-OST-3/PAP/tetrasaccharide complex, the crystal structure of 3-OST-1/PAP/heptasaccharide reveals different saccharide binding modes among the 3-OST isoforms (9, 10). Comparisons to the 3-OST-3/PAP/tetrasaccharide structure suggest that Lys259 in 3-OST-3 plays a key role in binding the 2-O-sulfo group of IdoA2S on the nonreducing side of the acceptor site. Despite substrates with the same sequence at the reducing end of the substrate, the 3-OSTs bind the substrate in different orientations. These results suggest that substrate binding by these enzymes is accomplished through a complex mechanism—not determined solely by a single saccharide unit, but by an intricate interplay of substrate sequence and conformation with protein side chains within the binding cleft.
Results and Discussion
Overall Structure of 3-OST-1/PAP/Heptasaccharide Ternary Complex.
The catalytic domain of 3-OST-1 (Gly48-His311; Fig. 1A) was cocrystallized with PAP and a synthesized heptasaccharide substrate (Figs. 1B and 2 and Table S1), and crystals of the ternary complex diffracted at 1.85 Å. This crystal form contained two molecules per asymmetric unit (rmsd of 0.53 Å over 232 Cα atoms). The final model contains residues Ser54-His311 in molecule A, and Thr55-His311 in molecule B. The PAP—the cofactor product of sulfo transfer from 3′-phosphoadenosine 5′-phosphosulfate (PAPS)—is bound and well-ordered in both molecules. The protein displays the expected α/β motif with a phosphosulfate binding loop interacting with PAP (Fig. 1). The heptasaccharide is present in the same conformation in both molecules in the asymmetric unit. The substrate bound in molecule A is very well defined, but appears to exhibit partial occupancy in molecule B.
Fig. 1.

Sequence and structure of 3-O-sulfotransferase isoforms. (A) Structure-based sequence alignment of 3-OST-1 and 3-OST-3. Secondary structural elements are shown, numbered, and labeled, with α-helices as rectangles and β-sheets as arrows. Disordered residues Asp185–Gly186 (from molecules A and B) are shadowed by a cyan box. Residues disordered only in molecule B are shadowed by an orange box. Catalytic residues are colored in red, disulfides in yellow, and residues mutated in this study in magenta. (B) Ribbon diagram of m3-OST-1 with PAP (pink) and bound heptasaccharide substrate (purple). The substrate bound in molecule A is the most well ordered and will therefore be used in all figures. All secondary structural elements are numbered as in A. Disulfide bond between Cys260 and Cys269 is drawn in yellow. Structural figure was generated using PyMOL (18).
Fig. 2.

Heptasaccharide substrate binding to 3-OST-1. (A) 3-OST-1 secondary structural elements shown as a ribbon diagram (green), with the bound heptasaccharide substrate drawn in stick in purple. Simulated annealing Fo–Fc omit electron density map (gray) contoured at 2σ. (B) Stereo diagram of the protein–substrate interactions. The Cα backbone of 3-OST-1 is traced in tan. Side chains involved in substrate (purple) binding interactions are shown in green. Putative hydrogen bonding interactions are shown as dashed lines. PAP is drawn in pink. Stereo diagram was generated using MOLSCRIPT (19).
Compared with the previously reported 3-OST-1 binary complex (10), the ternary complex revealed no large-scale structural changes (rmsd of 0.43 Å over 242 Cα atoms) upon acceptor substrate binding (Fig. S2). The only notable difference between the binary and ternary complexes is a disordering of the loop containing residues Asp185–Gly186 in both molecules in the asymmetric unit in the ternary complex (Fig. 1B). In the binary complex crystal form, this loop participates in multiple lattice contacts, likely maintaining its structure. This loop is remotely located from the substrate binding cleft of 3-OST-1 and would, therefore, be unlikely to influence binding. The long coil enclosing the PAP binding site (residues Ala277–Val281) in the ternary complex is also partially disordered in molecule B; however, this coil is packed against a neighboring symmetry-related molecule in molecule A, which helps order this region. The binding of the heptasaccharide is easily accommodated within the binding cleft, requiring only slight rearrangements of three side chains—Arg67, Lys173, and His271.
Interaction Between Heptasaccharide Substrate and 3-OST-1.
Of the two 3-OST-1 molecules in the asymmetric unit, the substrate binding cleft of molecule A shows significantly clearer density for the bound substrate than in molecule B (Fig. 2A). The final refined model for the substrate in molecule B yields an occupancy of 0.7. In addition, the substrate bound in molecule A is slightly better ordered on the reducing end of the sugar, which allows for modeling of the GlcA residue H-6 (Fig. 2A). The electron density for residue H-6 was not sufficient for modeling in molecule B. Residue H-7, however, is too disordered to model in either molecule.
The heptasaccharide appears to interact in the substrate binding cleft through a multitude of hydrogen bonding interactions (Fig. 2B and Fig. S3A). All glucosamine residues in the substrate, including the nonreducing end terminal sugar (H-1, GlcNAc6S; Fig. 3A), exhibit a 4C1 chair conformation. The 6-O-sulfate of H-1 interacts only with the backbone amide nitrogen of His271, placing residue H-1 within the “steric gate,” a unique structural motif that reportedly influences the substrate specificity (11). The side chain of His271 assumes a different rotamer to accommodate the substrate binding within the gate.
Fig. 3.

Structures of the substrates for 3-O-sulfotransferases. (A) The heptasaccharide substrate used for 3-OST-1 crystallization and mutational analysis. (B) The tetrasaccharide used for the crystallization of the ternary complex of 3-OST-3/PAP/tetrasaccharide (9). (C) The octasaccharide substrate used for analyzing 3-OST-3 wild-type and mutant proteins described in this study. The tetrasaccharide and the octasaccharide were purified from heparin lyase-degraded heparin (15), whereas the heptasaccharide was enzymatically synthesized (8). The 3-O-sulfation site in each substrate is indicated by an arrow. The identical trisaccharide regions between the heptasaccharide and tetrasaccharide are depicted in a dashed box. All substrates are shown with the nonreducing end on the left and the reducing end on the right.
H-2, the GlcA residue that confers binding specificity for 3-OST-1, also adopts the 4C1 chair conformation. There are multiple putative hydrogen bonding interactions between the carboxylate group of GlcA residue H-2 and the positively charged side chains of Arg72, Lys123, and Lys274 lining the substrate binding cleft. A possible hydrogen bonding interaction between the 3-OH of H-2 and Ser273 was also observed.
The acceptor site for sulfation lies in residue H-3, which is referred to as the acceptor sugar. The 3-OH group of H-3 is positioned 5.6 Å from the 5′-phosphate of the PAP, suggesting that the acceptor site would lie within close proximity to the sulfo donor, allowing transfer of the sulfo group from PAPS to the 3-OH position. Additionally, the 3-OH is also within hydrogen bonding distance (2.7 Å) of Glu90, which has been proposed to act as the general base for an SN2-like in-line displacement reaction mechanism (12). Binding of the acceptor sugar in this location is aided by an interaction between the N-sulfo group and the amide backbone nitrogen of Ala126. Thus, H-3 is held in a channel between the PAP binding site and a ridge formed by the loop between β-strand 3 and α-helix 4. The acceptor sugar is the most ordered unit of the heptasaccharide substrate with an average B-factor of 28.9 Å2 vs. 38.2 Å2 for the entire ligand. Away from the acceptor sugar along the oligosaccharide substrate, there is a concomitant increase in thermal motion.
Residue H-4 is a 2-O-sulfo-l-iduronic acid (IdoA2S). Although an IdoA2S can interconvert between the 1C4 chair and the 2S0 skew-boat conformation (13, 14), the IdoA2S bound to 3-OST-1 is found in the 1C4 chair conformation. The 2-O-sulfo group makes a single putative hydrogen bond with Arg197 of 3-OST-1. The carboxylate group is involved in multiple hydrogen bonding interactions with Arg67, Asn163, and Asn167. The head group of Arg67 rotates slightly upon binding of the substrate to optimize the interaction with H-4.
The penultimate ordered residue on the reducing end of the substrate is another GlcNS6S (H-5). The 6-O-sulfo group putatively hydrogen bonds with Arg197, whereas the N-sulfo group binds in a cavity within hydrogen bonding distance of His168, Lys171, and Lys173 (disordered in the binary complex). In addition, the 3-OH interacts with the side chain of Asn167. Residue H-6 makes no hydrogen bonding contacts with the protein in molecule A, and is correspondingly less ordered than the rest of the substrate.
Distinct Substrate Conformations for 3-OST-1 and 3-OST-3.
Comparison of the structure of the ternary complex of 3-OST-1/PAP/heptasaccharide with previously reported 3-OST-3/PAP/tetrasaccharide (structure of the tetrasaccharide shown in Fig. 3B) (9) reveals different substrate binding modes used by the different isoforms. The heptasaccharide bound to 3-OST-1 and the tetrasaccharide bound to 3-OST-3 occupy similar regions of the substrate binding cleft, proximal to the catalytic center (Fig. 4A). The conformations of the acceptor sugar (residues H-3 and T-2; Figs. 3 and 4A) are nearly identical, differing only in the torsion of the 6-O-sulfo group around the C5–C6 bond. In the ternary complexes of both 3-OST-1 and 3-OST-3, the position of this sulfo group is likely influenced by an interaction with symmetry-related molecules, consistent with the biochemical evidence that the 6-O-sulfo group on the acceptor sugar is nonessential for 3-OST-1 or 3-OST-3 modifications (2). Residues H-2 and T-1, on the nonreducing side of the acceptor sugar, also adopt nearly identical conformations, despite being different saccharide entities (GlcA vs. ΔUA2S, where ΔUA is 4-deoxy-α-l-threo-hex-4-eno-pyranosyluronic acid). The nonreducing saccharide of the tetrasaccharide substrate carries a ΔUA2S instead of an IdoA2S, because this tetrasaccharide was isolated from heparin lyase-degraded heparin. As a result, the ring conformation of ΔUA2S is slightly distorted in a 2H1 conformation, flattening one side of the ring to a planar conformation (Fig. 3B) (9). Based on previous studies, the 2-O-sulfo group is critical for substrate recognition by 3-OST-3 (3). The key residues in 3-OST-3 responsible for binding the 2-O-sulfo group are Arg370, Gln255, and Lys259 (Fig. 4B and Fig. S3B). Of these, the arginine and glutamine are conserved between 3-OST-1 and 3-OST-3 (Fig. 1A). Lys259 is well conserved among orthologs of 3-OST-3 (Fig. S4A), but is replaced by the structurally equivalent Asn167 in 3-OST-1 (Fig. 1A).
Fig. 4.

Comparison of HS binding to 3-OST-1 and 3-OST-3. (A) Comparison of substrate conformations of the 3-OST-1 bound heptasaccharide (purple) and the 3-OST-3 bound tetrasaccharide (gray, PDB ID code 1T8U) (9). The position of the 3-O-sulfation is labeled with a pink asterisk. The surface of 3-OST-1 is rendered in green. Residues of the heptasaccharide are labeled in white, as in Fig. 3A. (B) Superposition of 3-OST-1 (green) and 3-OST-3 (gray, PDB ID code 1T8U) (9) residues involved in binding their respective oligosaccharide substrates [heptasaccharide (purple) and tetrasaccharide (light gray)]. Putative hydrogen bonding interactions between 3-OST-3 and the tetrasaccharide are shown as dashed lines. The 3-O-sulfation site is labeled with a pink asterisk. (C) Superposition of the heptasaccharide substrate (purple) bound to 3-OST-1 with the tetrasaccharide substrate (gray) bound to 3-OST-3 (PDB ID code 1T8U) (9), highlighting the structural differences between the reducing ends of the chain. (D) Superposition of 3-OST-1 (green) and 3-OST-3 (gray, PDB ID code 1T8U) (9). PAP from the 3-OST-1 structure is drawn in pink. N and C termini are labeled from each respective protein. α-helix 6 helix jutting from the top of the substrate binding cleft is labeled with a red asterisk. (E) Sodium metal ion interactions in the 3-OST-3 substrate binding site. α-helix 6 from 3-OST-3 (9) is shown as a gray ribbon, and the sodium ion as a purple sphere. Interactions coordinating the sodium ion are shown as solid black lines. Potential hydrogen bond between Trp283 and the 6-O-sulfo group on residue T-4 is shown as a dashed line.
Examination of the interactions beyond the acceptor site, toward the reducing end of the substrate, reveals notable differences. The tetrasaccharide substrate has three reducing-end saccharide units in common with the heptasaccharide used in the complex with 3-OST-1 (depicted in a dashed box in Fig. 3 A and B). Despite this sequence identity, the bound substrates display surprisingly different conformations. The glycosidic bonds display significantly distinct angles in this common trisaccharide domain (Fig. 4C). The tetrasaccharide was found to be “kinked” around the glycosidic bond between residues T-2 and T-3 (9). The kink is also observed in the heptasaccharide between residues H-3 and H-4, but is rotated ∼22.3° with respect to residues T-2 and T-3 of the tetrasaccharide (via a dihedral defined by C1–O4–C4–C5; Fig. 4C). An obvious difference between the substrates is found in the conformations of the IdoA2S residues (H-4 and T-3). In 3-OST-1, this residue adopted the 1C4 chair, whereas the IdoA2S bound to 3-OST-3 was found in the 2S0 skew-boat conformation (Fig. 4 A and C). Conversion from the 2S0 skew boat to the 1C4 chair conformation moves the 2-O-sulfo group from the equatorial position to an axial position, which provides an additional hydrogen bonding interaction with the Arg197 side chain of 3-OST-1 (Fig. 2B and Fig. S3A). The difference in the IdoA2S conformations synergistically adds to the different torsions about the kink, sharply diverting the reducing-end saccharide units of the two substrates toward different orientations (Fig. 4C). These distinct oligosaccharide conformations may also play a role in substrate specificity between 3-OST-1 and 3-OST-3. The presence of the 2-O-sulfo group on residue T-1 is critical for binding to 3-OST-3, whereas this 2-O-sulfo group is absent on H-2 residue for 3-OST-1. However, if the same residue was bound to 3-OST-1, in the conformation exhibited in the ternary complex with the heptasaccharide, there would likely be charge repulsion between the 2-O-sulfo group and the carboxylate of the IdoA2S on H-4 (Fig. 4B). Altering the reducing side IdoA2S conformation from 1C4 chair (as in 3-OST-1) to 2S0 skew boat (as in the 3-OST-3) increases the distance to the carboxylate group from 2.8 Å (Fig. 4B, pink dashed line) to 3.2 Å (Fig. 4B, orange dashed line). Thus, the presence of Lys259 in 3-OST-3, combined with the 2S0 conformation of the T-3 residue, also alleviates charge repulsion within the substrate.
Another possible contributing factor to the differences in substrate binding modes between 3-OST-1 and 3-OST-3 may be found within α-helix 6 (Figs. 1 and 4D). Structural comparison of the reducing end of the substrate binding clefts of 3-OST-1 and 3-OST-3 shows a notable difference in the length of the α-helix that forms the top wall of the substrate binding cleft (Figs. 1B and 4D, red asterisk). This helix consists of residues Pro245–Lys259 of 3-OST-3 and Pro153–Lys171 of 3-OST-1. The additional amino acid residues of 3-OST-1 make this helix one entire helical turn longer than that of 3-OST-3, providing additional interactions between the protein and the reducing end of the substrate (Fig. 2B), such as the side chain of His168 of 3-OST-1 that interacts with the N-sulfo group of H-5.
An additional contributing factor to substrate orientation is the participation of a cation in the binding of enzyme and substrate. A likely sodium ion was observed in the binding cleft of 3-OST-3 (Fig. 4E), which mediates interactions between the protein and T-4 of the tetrasaccharide (9). This sodium ion is not present in the structure of the 3-OST-3 apoprotein (PDB ID code 1T8T), despite having identical sodium concentrations, suggesting a specific role in substrate binding for 3-OST-3. The presence of this ion may help orient the T-4 unit of the tetrasaccharide substrate, increasing its binding affinity.
Mutational Analysis.
To explore the possible substrate recognition mechanisms suggested by crystal studies, we prepared various 3-OST-1 and 3-OST-3 mutants. Two structurally defined oligosaccharide substrates were used for the activity and substrate specificity analysis: the heptasaccharide (3-OST-1 specific) and an octasaccharide (3-OST-3 specific; Fig. 3 A and C). An octasaccharide substrate, rather than the tetrasaccharide substrate, was used for the analysis of 3-OST-3 mutants because the octasaccharide substrate exhibited higher binding affinity and susceptibility to 3-OST-3 sulfation. The 3-O-sulfation sites within both substrates by the respective enzymes are well characterized (8, 15).
The reactivity of the oligosaccharide substrates toward 3-OST-1 and 3-OST-3 modification was measured by the amount of 35S-labeled products, whereas the substrate selectivity was confirmed by the product identification using a HPLC method (Table 1 and SI Materials and Methods). As expected, the susceptibility of the heptasaccharide to 3-OST-1 modification was 40-fold higher than modification by 3-OST-3 (Table 1). Conversely, the susceptibility of the octasaccharide substrate to 3-OST-3 modification was 5.6-fold higher than modification by 3-OST-1 (Table 1). These results illustrated basal substrate selectivity by the different 3-OST isoforms. We next determined the identities of the 3-O-sulfated oligosaccharide products. Because the oligosaccharide substrates contain several GlcNS6S units, it is necessary to ensure that only a single 3-O-sulfo group was introduced onto the acceptor site. We expected that wild-type 3-OST-1 generates a single 3-O-sulfated heptasaccharide when it is incubated with its substrate; likewise, that a single 3-O-sulfated octasaccharide is formed when 3-OST-3 is incubated with the octasaccharide substrate as analyzed by high-resolution DEAE-HPLC (Figs. S5 and S6, respectively).
Table 1.
Reactivity of 3-OST mutants toward heptasaccharide and octasaccharide substrates and product analysis
| Mutant proteins | Heptasaccharide* substrate, %† | Identity of 3-O-sulfated heptasaccharide product‡ | Octasaccharide§ substrate, %† | Identity of 3-O-sulfated octasaccharide product† |
| 3-OST-1 WT | 100 | The anticipated 3-O-sulfated heptasaccharide was found | 18.2 ± 0.9 | Multiple products |
| 3-OST-1 V164E | 20.3 ± 1.5 | Multiple products | 2.9 ± 0.9 | Not determined |
| 3-OST-1 N167A | 40.4 ± 4.2 | Similar to wild type | 3.9 ± 1.0 | Not determined |
| 3-OST-1 H168A | 51.1 ± 7.1 | Similar to wild type | 5.0 ± 3.0 | Not determined |
| 3-OST-1 H168E | 19.1 ± 1.4 | Similar to wild type | 2.3 ± 0.9 | Not determined |
| 3-OST-1 K171A | 83.9 ± 5.0 | Similar to wild type | 2.2 ± 0.0 | Not determined |
| 3-OST-1 R268A | 7.2 ± 1.4 | Similar to wild type | 7.6 ± 2.7 | Not determined |
| 3-OST-3 WT | 2.5 ± 1.2 | Multiple products | 100 | The anticipated 3-O-sulfated octasaccharide was found |
| 3-OST-3 T256A | 1.1 ± 0.3 | Multiple products | 101.5 ± 15.3 | Similar to wild type |
| 3-OST-3 T256V | 2.2 ± 0.3 | Multiple products | 59.4 ± 10.7 | Similar to wild type |
| 3-OST-3 T256E | 0.2 ± 0.1 | Multiple products | 13.1 ± 1.6 | Multiple products |
| 3-OST-3 K259A | 0.3 ± 0.1 | Multiple products | 13.1 ± 1.6 | Multiple products |
| 3-OST-3 W283A | 0.2 ± 0.1 | Multiple products | 7.6 ± 3.2 | Similar to wild type |
Contribution of a sodium ion to substrate specificity.
Based on the crystal structure, Thr256 and Asp252 of 3-OST-3 bind to the sodium ion (Fig. 4E) (9). Sequence alignments of 3-OST-1 and 3-OST-3 illustrated that Thr256 is unique to 3-OST-3, and appears to be evolutionarily well conserved (Fig. S4A). The structurally homologous residue in 3-OST-1 is a valine (Fig. 1A). To investigate the role of this residue in substrate binding, Thr256 of 3-OST-3 was replaced with three different residues—alanine, valine, or glutamate. The 3-OST-3 T256A mutant exhibited reactivity on the 3-OST-3–specific octasaccharide substrate (Fig. 3C), similar to that of wild-type 3-OST-3, whereas the 3-OST-3 T256V and 3-OST-3 T256E mutants showed increasingly diminished reactivity (Table 1). The orthogonal 3-OST-1 V164E mutant also exhibited severely diminished reactivity on its heptasaccharide substrate (Table 1). Interestingly, product-identity analysis using DEAE-HPLC revealed that both the 3-OST-1 V164E and the 3-OST-3 T256E mutants generated multiple sulfation products (Table 1 and Figs. S5 and S6), which would suggest that glutamate substitution near the reducing end of the chain may affect substrate binding to 3-OST-3 by prohibiting interaction via the sodium ion, ultimately increasing the promiscuity of 3-OST-3. It is tempting to speculate that the presence of the metal ion may help orient the substrate in a catalytically accessible conformation.
Contribution of reducing-end saccharides to substrate binding and specificity.
Examination of the 3-OST-3 binding pocket surrounding the N-sulfo group of T-4 reveals that one wall is formed by a tryptophan residue (Trp283). Trp283 is involved in a single hydrogen bonding interaction with the terminal sugar (Fig. 4E). This tryptophan is conserved across mammalian orthologs of 3-OST-3 (Fig. S4A), but not in 3-OST-1 (Fig. 1A and Fig. S4B). In 3-OST-1, this tryptophan is replaced by a tyrosine (Tyr192), likely resulting in the loss of a hydrogen bond with the reducing end of the substrate. The 3-OST-3 W283A exhibited greatly diminished reactivity (7.6 ± 3.2% compared with wild type; Table 1) and displayed a 41-fold reduction in catalytic efficiency (the values of kcat/Km; Table S2). It should be noted that the level of the expression of the 3-OST-3 W283A was severely decreased, and its behavior suggested concomitantly decreased stability and/or reduced solubility. Therefore, maintaining the structure of the sulfate binding pocket near the reducing end of the substrate binding cleft appears to be crucial for 3-OST-3 structural stability and reactivity.
Role of α-helix 6 on substrate binding.
A series of mutants along α-helix 6 were prepared. The Lys259 of 3-OST-3 was replaced with alanine, and the following substitution mutations were created in 3-OST-1: N167A, H168A, and K171A. The 3-OST-3 K259A mutant was markedly decreased in reactivity toward the octasaccharide substrate. The product identity analysis reveals that 3-OST-3 K259A generated multiple sulfation products (Table 1), suggesting that substitution of this lysine residue alters both reactivity and substrate specificity of the resulting protein. Because Lys259 interacts with the 2-O-sulfo group that is so critical for substrate binding by 3-OST-3, this interaction provides a molecular explanation for substrate specificity at that particular residue. Lys259 is juxtaposed between the 2-O-sulfo group on T-1, and the carboxylate group on residue T-3, in an optimal position to alleviate electrostatic repulsion between them. Unlike the profound effect by the mutation Lys259 in 3-OST-3, mutations in α-helix 6 had less of an effect on the enzyme activity and substrate specificity of 3-OST-1. The individual 3-OST-1 mutants K171A, N167A, and H168A exhibited only modest decreases in reactivity toward the heptasaccharide substrate compared with wild type (Table 1 and Table S2). The 3-OST-1 H168A mutant displayed a more substantial decrease in reactivity. These mutations in 3-OST-1 did not appear to affect substrate specificity.
Role of the steric gate in binding to the nonreducing end of the substrate.
It has previously been suggested that the residues interacting with the nonreducing end of the saccharide substrate could influence substrate specificity via a steric gate motif (11). The ternary complex of 3-OST-1 with the heptasaccharide substrate permitted us to view the interaction between the nonreducing-end saccharide unit and the amino acid residues situated at the steric gate. Indeed, the saccharide unit on the nonreducing end of the heptasaccharide (Fig. 5; residue H-1) is positioned directly between the gate residues His271 and Glu88. Immediately past the gate, the positively charged side chain of Arg268 may represent a continuation of the substrate binding cleft (Fig. 5). Consistent with this conclusion, mutation of Arg268 resulted in a severe loss of sulfotransferase activity (7.2 ± 1.4% of wild-type 3-OST-1) (Table 1). Decreased sulfotransferase activity correlates well with decreased binding affinity and catalytic efficiency (Table S2). Because the NH1 atom of Arg268 lies 3.6 Å from the 6-O-sulfo group of residue H-1, there is a favorable electrostatic interaction and the potential for a long-range hydrogen bonding interaction, bridged by a water molecule. Also, further extension of the substrate chain by one residue on the nonreducing end could place the chain in direct contact with Arg268. The importance of R268A yields insights into the putative behavior of 3-OSTs in the presence of a long HS polysaccharide substrate, demonstrating that such long-range interactions can affect enzyme reactivity distal from the catalytic center. Furthermore, our results confirm the role of the steric gate to influence substrate specificity between the 3-OST isoforms (11).
Fig. 5.
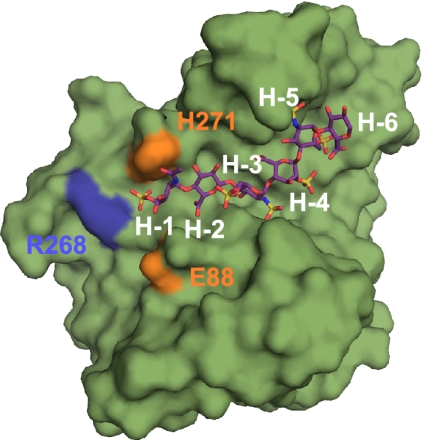
Long-range nonreducing-end interactions between 3-OST-1 and the bound heptasaccharide substrate. 3-OST-1 is shown as a surface rendering in green. The heptasaccharide is drawn in stick in purple. Gate residues His271 and Glu88 are shown in orange. The position of Arg268 is shown in blue. Residues from the heptasaccharide are labeled in white.
Comparison of Oligosaccharide Conformations When Bound to 3-OST-1 vs. Antithrombin.
It has been demonstrated that 3-O-sulfation of a pentasaccharide sequence identical to that contained in the 3-OST-1 substrate (residues H-1 through H-5) used in this study increases the binding affinity of heparin to antithrombin by 1,000-fold (16). However, the IdoA2S (H-4) bound to 3-OST-1 is present in a different conformation. For antithrombin binding, the equivalent IdoA2S adopts the 2S0 skew-boat conformation, which is critically important for high-affinity binding to antithrombin (17). When bound to 3-OST-1, however, this IdoA2S residue in the heptasaccharide—the precursor of the antithrombin-binding pentasaccharide domain—is clearly present in a 1C4 chair conformation. Although the conformation of an IdoA2S unit is interchangeable between the 1C4 and 2S0 conformations depending on the protein binding partner (9), a scenario that the 3-O-sulfonation drives the neighboring IdoA2S unit to favor the 2S0 skew-boat conformation is certainly possible.
Conclusions
Unlike that of nucleic acids and proteins, the biosynthesis of HS polysaccharide is not a template-driven process. HS sulfotransferases, including 3-OSTs, are primarily responsible for controlling the sulfation patterns in HS, which consequently determine the function of HS polysaccharides. The unique functions of 3-O-sulfated HS underscore the importance of investigating the mechanism for the control of HS biosynthesis. Although we have solved the ternary complex structure of 3-OST-3/PAP/tetrasaccharide (9), the substrate recognition mechanism remained unclear due to the lack of the ternary structure of other 3-OST isoforms, especially 3-OST-1. Despite that the binary structure of 3-OST-1/PAP was initially solved in 2003, our recent success in synthesizing structurally homogeneous HS oligosaccharides provides a vital reagent for completing this study (8). The substrate is synthesized to a length sufficient for displaying the interactions between the saccharides with the key amino acid residues of 3-OST-1. Our findings demonstrate that each 3-OST isoform provides a unique perspective on modes of substrate binding and transforms the polysaccharides with distinct sulfation patterns. The structural information lays a foundation for targeted protein engineering toward the development of an enzyme-based approach to synthesize heparin and HS. These efforts will enhance the ability of enzyme-based synthetic methods, leading to specifically tailored heparin with anticoagulation, anticancer, and other potentially therapeutic uses.
Materials and Methods
Crystals of the 3-OST-1/PAP/heptasaccharide complex were obtained using a microbatch technique, by mixing 1.5 μL of the complex [12.9 mg/mL 3-OST-1, 5 mM heptasaccharide, 4 mM PAP, 23.6 mM Tris (pH 7.5), 142 mM NaCl] with 2.5 μL of 0.1 M sodium citrate (pH 5.5) and 20% (wt/vol) PEG 3000. The heptasaccharide was synthesized through a chemoenzymatic approach as previously described (8), and the structure was confirmed by ESI-MS analysis (Fig. S7). Crystals grew to a usable size after 10-d incubation at room temperature. The crystals were transferred in two steps to a cryoprotectant solution containing 0.1 M sodium citrate (pH 5.5), 0.1 M NaCl, 4 mM PAP, 20 mM heptasaccharide, 30% (wt/vol) PEG3000, and 7.6% (vol/vol) ethylene glycol. Following cryoprotection, the crystals were flash frozen in liquid nitrogen and placed into a stream of liquid nitrogen gas cooled to −180 °C for data collection. The data were collected at 1.85 Å on the Southeast Regional Collaborative Access Team 22-ID beamline at the Advanced Proton Source, Argonne National Laboratory.
Additional experimental procedures are presented in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank G. Mueller and S. Garantziotis for critical reading of the manuscript, and Prof. Peng George Wang (Georgia State University) for providing the plasmid expressing NAHK. This research was supported by the Division of Intramural Research of the National Institute of Environmental Health Sciences, National Institutes of Health (NIH) Grant 1 ZIA ES102645-03; and NIH Grants AI050050, HL094463, and HL096972. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences Contract W-31-109-Eng-38.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: Coordinates for murine 3-OST-1 with the bound heptasaccharide substrate reported in this paper have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 3UAN).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1117923109/-/DCSupplemental.
References
- 1.Bishop JR, Schuksz M, Esko JD. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature. 2007;446:1030–1037. doi: 10.1038/nature05817. [DOI] [PubMed] [Google Scholar]
- 2.Shworak NW, et al. Multiple isoforms of heparan sulfate D-glucosaminyl 3-O-sulfotransferase. Isolation, characterization, and expression of human cdnas and identification of distinct genomic loci. J Biol Chem. 1999;274:5170–5184. doi: 10.1074/jbc.274.8.5170. [DOI] [PubMed] [Google Scholar]
- 3.Liu J, Pedersen LC. Anticoagulant heparan sulfate: Structural specificity and biosynthesis. Appl Microbiol Biotechnol. 2007;74:263–272. doi: 10.1007/s00253-006-0722-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu H, Zhang Z, Linhardt RJ. Lessons learned from the contamination of heparin. Nat Prod Rep. 2009;26:313–321. doi: 10.1039/b819896a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shriver Z, Raguram S, Sasisekharan R. Glycomics: A pathway to a class of new and improved therapeutics. Nat Rev Drug Discov. 2004;3:863–873. doi: 10.1038/nrd1521. [DOI] [PubMed] [Google Scholar]
- 6.Baleux F, et al. A synthetic CD4-heparan sulfate glycoconjugate inhibits CCR5 and CXCR4 HIV-1 attachment and entry. Nat Chem Biol. 2009;5:743–748. doi: 10.1038/nchembio.207. [DOI] [PubMed] [Google Scholar]
- 7.Arungundram S, et al. Modular synthesis of heparan sulfate oligosaccharides for structure-activity relationship studies. J Am Chem Soc. 2009;131:17394–17405. doi: 10.1021/ja907358k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu Y, et al. Chemoenzymatic synthesis of homogeneous ultralow molecular weight heparins. Science. 2011;334:498–501. doi: 10.1126/science.1207478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moon A, et al. Structural analysis of the sulfotransferase (3-OST-3) involved in the biosynthesis of an entry receptor of herpes simplex virus 1. J Biol Chem. 2004;279:45185–45193. doi: 10.1074/jbc.M405013200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Edavettal SC, et al. Crystal structure and mutational analysis of heparan sulfate 3-O-sulfotransferase isoform 1. J Biol Chem. 2004;279:25789–25797. doi: 10.1074/jbc.M401089200. [DOI] [PubMed] [Google Scholar]
- 11.Xu D, Moon AF, Song D, Pedersen LC, Liu J. Engineering sulfotransferases to modify heparan sulfate. Nat Chem Biol. 2008;4:200–202. doi: 10.1038/nchembio.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Edavettal SC, et al. A conformational change in heparan sulfate 3-O-sulfotransferase-1 is induced by binding to heparan sulfate. Biochemistry. 2004;43:4680–4688. doi: 10.1021/bi0499112. [DOI] [PubMed] [Google Scholar]
- 13.Ferro DR, et al. Conformer populations of L-iduronic acid residues in glycosaminoglycan sequences. Carbohydr Res. 1990;195:157–167. doi: 10.1016/0008-6215(90)84164-p. [DOI] [PubMed] [Google Scholar]
- 14.Mulloy B, Forster MJ. Conformation and dynamics of heparin and heparan sulfate. Glycobiology. 2000;10:1147–1156. doi: 10.1093/glycob/10.11.1147. [DOI] [PubMed] [Google Scholar]
- 15.Copeland RJ, et al. Using a 3-O-sulfated heparin octasaccharide to inhibit the entry of herpes simplex virus type 1. Biochemistry. 2008;47:5774–5783. doi: 10.1021/bi800205t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Atha DH, Lormeau J-C, Petitou M, Rosenberg RD, Choay J. Contribution of monosaccharide residues in heparin binding to antithrombin III. Biochemistry. 1985;24:6723–6729. doi: 10.1021/bi00344a063. [DOI] [PubMed] [Google Scholar]
- 17.Das SK, et al. Synthesis of conformationally locked L-iduronic acid derivatives: Direct evidence for a critical role of the skew-boat 2S0 conformer in the activation of antithrombin by heparin. Chemistry. 2001;7:4821–4834. doi: 10.1002/1521-3765(20011119)7:22<4821::aid-chem4821>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 18.DeLano WL. San Carlos, CA: DeLano Scientific; 2002. The PyMOL Molecular Graphics System. [Google Scholar]
- 19.Kraulis PJ. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J Appl Cryst. 1991;24:946–950. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.