

Abstract
A molecular dynamics-based protocol is proposed for finding and scoring protein-ligand binding poses. This protocol uses the recently developed reconnaissance metadynamics method, which employs a self-learning algorithm to construct a bias that pushes the system away from the kinetic traps where it would otherwise remain. The exploration of phase space with this algorithm is shown to be roughly six to eight times faster than unbiased molecular dynamics and is only limited by the time taken to diffuse about the surface of the protein. We apply this method to the well-studied trypsin–benzamidine system and show that we are able to refind all the poses obtained from a reference EADock blind docking calculation. These poses can be scored based on the length of time the system remains trapped in the pose. Alternatively, one can perform dimensionality reduction on the output trajectory and obtain a map of phase space that can be used in more expensive free-energy calculations.
Understanding how proteins interact with other molecules (ligands) is crucial when examining enzymatic catalysis, protein signaling, and a variety of other biological processes. It is also the basis for rational drug design and is thus an important technological problem. Ligand binding is primarily examined using X-ray crystallography experiments together with measurements of the binding free energies. Additionally, numerous computational methods have been applied to this problem so as to extract more detailed information. The fastest of these approaches are based on an extensive configurational search of the protein surface (docking), in which the various candidate poses found are scored in accordance with some approximate function that treats solvation, protein flexibility, and entropic effects in some approximate manner.
Free-energy methods, based on either molecular dynamics (MD) or Monte Carlo simulations, can be used to calculate accurate binding free energies (1–3). However, it is far more difficult to use these methods to search for candidate poses as the timescales involved in ligand binding are typically much longer than those that are accessible in MD. Thus, one often finds that the ligand becomes trapped in a kinetic basin on the surface of the protein and does not escape during the remainder of the calculation.
We recently developed a method, reconnaissance metadynamics, for increasing the rate at which high-dimensional configurational spaces are explored in MD simulations (4). This enhanced sampling is obtained by using a Gaussian mixture model to identify clusters in the stored trajectory. The positions of these clusters correspond to the kinetic basins in which the system would otherwise be trapped, which means that a history-dependent bias function that uses the information obtained from the clustering can be used to force the system away from the traps and into unexplored portions of phase space. In what follows we demonstrate how this algorithm can be used to examine the binding of benzamidine to trypsin, and perform a blind docking simulation based entirely on enhanced sampling.
Background
Extensive conformational search procedures combined with fast and simple scoring functions give a surprisingly good description of protein-ligand docking in a variety of systems. In fact, for a number of systems so-called blind docking calculations can be performed in which the binding pose is found without using any experimental insight (5). The two greatest, unsolved problems for this field are to find universal scoring functions and to develop protocols for incorporating protein flexibility (6). These two problems are interlinked as an accurate scoring function must take the energetic cost of the conformational changes into account. Standard biomolecular force fields, together with implicit solvent models, provide the best approach for balancing these contributions. However, empirical and knowledge-based scoring functions often perform better for certain classes of problems and are thus frequently employed (7–9).
Simulations based on MD force fields provide an alternative to simple docking calculations and both MD and Monte Carlo simulations have been used to locate sites with favorable interaction energy (10, 11). Furthermore, recent studies have exploited the power of modern computers to examine the process of ligand binding directly (12–14). In these calculations the ligand is initially placed outside the protein and MD is used to find favorable binding sites. In the limit of long simulation time, the sites are visited according to the Boltzmann distribution and thus can be scored based on the amount of time the ligand spends at each site. This approach allows one to incorporate the protein flexibility, to treat the water explicitly, and to use established techniques for improving the force fields. In addition, one can obtain dynamical information on the binding process as well as structural information. However, these calculations still use an enormous amount of computational time and produce so much data that specialist tools are required for analysis. For example, the recent paper on the binding of benzamidine to trypsin by Buch et al. (12) used 500 unbiased simulations at a length of 100 ns.
Using plain MD simulations for locating binding poses is expensive because kinetic traps prevent the ligand from diffusing freely over the whole protein surface during short simulations. This problem is encountered frequently in MD and can be resolved by using enhanced sampling methods. A number of such methods have been applied to ligand binding (15–30). Typically these methods accelerate sampling by either increasing the temperature or by introducing a bias that prevents the system from becoming trapped in a basin. The bias is often constructed in terms of a small number of collective variables (CVs) that are selected by the user based on what is known about the location of the binding site, the binding pathway, and the conformational changes in the protein that occur during binding (31, 32). Using these methods one can calculate binding free energies for a small number of putative poses (33). Alternatively, one can find new, poorly characterized binding sites by using them in tandem with docking calculations (34).
The reconnaissance metadynamics method (RMD) (4) inserts the rich data that can be obtained from short MD simulations into a self-learning algorithm and thereby generates local collective coordinates that can push the system away from the kinetic traps it encounters. This procedure saves one from selecting a small number of appropriate CVs at the outset and thus provides a way to perform simulations when the reaction mechanism is uncertain. Thus far we have applied this method to model systems for polypeptide folding (4) and to small clusters of water and argon (35). These studies have demonstrated that RMD performs an extensive exploration of the energetically accessible portions of phase space and that this method can be used to locate global minima in energy landscapes. However, in the problems that we have examined the free-energy landscape is dominated by energetic contributions so these systems could alternatively be studied through a combination of optimization and transition state searches (36). Applying the RMD algorithm to the blind docking problem, as we do in this paper, represents a far greater challenge to the methodology because ligand binding involves a delicate balance between enthalpic and entropic contributions.
Results and Discussion
We chose to examine the well-studied trypsin–benzamidine system, which has been extensively examined using free-energy perturbation (37). Both the benzamidine ligand and the trypsin protein are relatively rigid (38), so the binding site can be found using conventional blind docking (39).
One can use a large set of CVs in a reconnaissance metadynamics calculation and thus avoid many of the problems associated with choosing a small number of CVs for conventional metadynamics or umbrella sampling. However, it is important to realize that the CVs selected will influence the scope of the sampling. Thus for trypsin–benzamidine, where we know that the binding is not accompanied by large configurational changes in the protein, we selected CVs that describe only the position and orientation of the ligand relative to the protein and assume that MD alone will account for any protein flexibility. The CVs we chose are based on the distances between the C4, N1, and N2 atoms of the ligand (Fig. 1) and 16 uniformly spaced points on the protein surface (Materials and Methods). These distances are then transformed by a switching function so that whenever the ligand is far from the protein the collective variables have essentially the same values. The switching function is given by
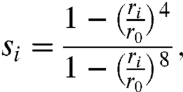 |
[1] |
where ri is the i th distance and r0 = 13 Å. This set of 48 coordinates contains redundancy. However, because the self-learning algorithm at the heart of the RMD algorithm selects the most appropriate linear combinations of these to push, this redundancy does not present a particular problem. What is important to stress is that the parameters in this function and the points on the surface are chosen without using any information on the location of the binding site. As such this approach is general enough that it could be used for any globular protein. In addition, this description of the ligand’s position, orientation, and conformation can be systematically refined by either increasing the number of points on the surface of the protein or by increasing the number of points in the ligand. However, the cost of the calculations will increase as the number of CVs is increased (Materials and Methods).
Fig. 1.
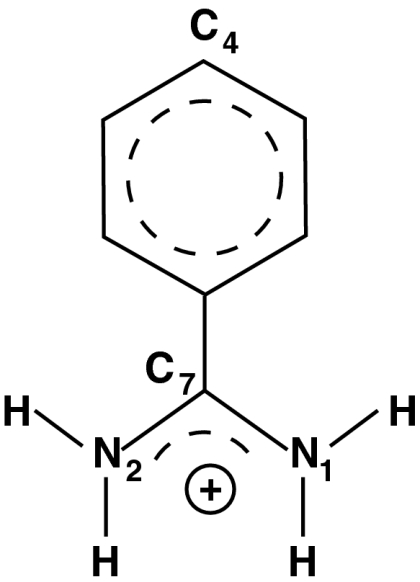
The benzamidine ligand with the atom labels used in the text.
Extent of Exploration.
To test whether or not RMD is doing a good job of exploring phase space we generated a set of putative binding poses using conventional blind docking. These calculations were done using EADock (40), which is known to reproduce the correct binding pose for a range of systems (41) and which generated a large number of structurally diverse poses (Table S1). We then ran 10, 200-ns reconnaissance metadynamics simulations and calculated the rmsd distance between snapshots taken every 10 ps from our trajectory and the 27 poses found in our EADock calculations. Fig. 2 shows that during our simulations we come close to every single one of these putative poses. More importantly, in five out of the ten simulations we were able to find the binding site. These results are in stark contrast to the results we obtain from MD simulations of similar length. During the course of these calculations we were only able to find a subset of the poses and the binding site was never visited. This observation appears, at first glance, to be at variance with the results of Buch et al. (12) who found that in 37% of their 100-ns, unbiased MD simulations on this system the experimental binding pose was found. However, in their simulations some information on the location of the binding site was employed, as constraints were applied on the relative position of the protein and ligand to ensure that the ligand only explored one side of the protein.
Fig. 2.

The extent to which the space is explored in the RMD and MD simulations. The results from 10 RMD and 10 MD simulations are shown and each column corresponds to one of the reference poses from the EADock calculation. The squares are colored according to the minimum rmsd between the trajectory frames and the reference pose. If the minimum rmsd from any pose is greater than 2.5 Å then we assume that it was not found during the simulation and color it white.
Fig. 3A provides an alternative representation of the data on the extent to which phase space is explored during the RMD and MD simulations. This figure shows the fraction of reference poses found as a function of time and suggests that RMD is on average six to eight times faster at finding poses than MD [fitting the curves in Fig. 3A to the function 1 - exp(-t/t0) we find that the ratio of t0 values for MD to RMD are 5.6, 7.5, and 8.3 for the three rmsd cutoffs we tested]. This increased speed does not appear particularly dramatic, but it is important to remember that if in any of the MD simulations the ligand had found the binding site it would have almost certainly remained there for the remainder of the simulation time, which is not what happens in the RMD simulations that find the binding site. In other words, the exploration in RMD is only slowed down because diffusion of the ligand about the protein is relatively slow—a fact of life that will be present in any method based on molecular dynamics.
Fig. 3.

Comparison of the exploration speed, cluster sizes, and interaction energies for the MD and RMD trajectories. (A) The fraction of the reference (EADock) poses found as a function of simulation time. A pose is found if the rmsd distance between it and the instantaneous position of the ligand drops to less than 2.5 Å. The average over all the RMD or MD runs (solid lines) is shown together with the standard deviation between runs. The averages obtained using cutoffs of 2 Å (dashed lines) and 3 Å (dotted lines) are also shown. Here, RMD consistently explores space more quickly than MD. (B and C ) The results from the clustering calculations. B shows the sizes of the top 100 clusters averaged over the 10 MD and 10 RMD simulations together with the standard deviations calculated for every 10th cluster. The clustering was done using a fixed rmsd cutoff of 1 Å. Hence, more diffuse clusters have fewer members. In reporting this data, we have multiplied the number of trajectory frames in each cluster by the time interval between the frames (10 ps) to get a residence time for each cluster. This figure clearly demonstrates that RMD is spending much less time in each pose, allowing a more efficient exploration of the configurational space. C shows a histogram of the single point protein-ligand interaction energy for the central frame in the top 10 (shaded area) and top 50 (unshaded areas) clusters from each of our RMD and MD simulations. Also shown is the histogram (dashed line) calculated from all the frames in the trajectory. Here, the results demonstrate that MD fails to find the low interaction energy poses that are found during the RMD simulations.
Generating Candidate Poses.
To generate meaningful output from any ligand-binding trajectory, it is necessary to predict which poses have high binding affinities, much as one scores poses in traditional docking calculations. Making such predictions from MD simulations is in principle straightforward, because the time spent in a given configuration is connected to its free energy. The only caveat is that one must see multiple transitions between states. If one appropriately accounts for the bias, similar strategies can be used in methods involving a bias potential. The problem with RMD is that multiple transitions between states are seldom observed because of the high-dimensionality space of collective variables. In contrast, when using methods like metadynamics, the small number of collective coordinates forces these transitions to occur.
In an RMD simulation it will take some time to generate sufficient bias to push the system out of a basin. The specific amount of time will depend on the basin’s depth and hence its kinetic stability. Low free-energy poses are usually narrow minima in the potential energy surface. These states will be both thermodynamically and kinetically stable. It may, therefore, be possible to find low free-energy poses by extracting the most populated clusters from an RMD trajectory. To further explore this idea, we analyzed the RMD trajectory frames using the method of Daura et al. (42) that is implemented in the GROMACS g_cluster utility. This procedure ranks each trajectory frame based on the number of neighboring frames that are within 1 Å rmsd. The top-ranked frame, together with all its neighbors, is then removed and the ranking process is repeated.
Fig. 3B shows that the clusters generated from the analysis of the RMD trajectories are much smaller than those generated from an analysis of the MD trajectories. This result confirms that the MD simulations are spending a great deal of time (up to 30 ns) trapped at a small number of sites on the protein surface. In contrast RMD spends at most 0.4 ns in any given pose and is thus able to explore more of the protein surface. In addition, this analysis of the RMD simulations identifies the binding pose as important. In three of the five RMD simulations that found the binding site, the cluster corresponding to the binding site is the most populated, whereas in the remaining two the binding site is ranked second and third.
Fig. 3C provides further evidence that clustering of the RMD trajectory gives reasonable binding poses. In this figure we show the vacuum interaction energy between the protein and the ligand for the top 50 clusters (i.e., the most populated ones) from each simulation. This interaction energy neglects solvent and entropic effects but is still often correlated with the binding free energy (43). Hence, the fact that the clusters found in RMD have consistently lower energies than those found in MD suggests that they correspond to more strongly bound conformations. Furthermore, if we examine all the frames in the trajectory we find that, in contrast with MD, the top clusters in RMD correspond to the structures with the lowest energies. There is no such shift in MD, which suggests that in these simulations the ligand becomes trapped in many basins that do not have particularly low interaction energies. As such, the MD simulations are too short to express the relationship between the residence time in a given structure and its free energy.
The clustering procedure does not take into account the bias, and thus some of the well-populated clusters might not correspond to minima on the unbiased free-energy surface. Hence, to probe the kinetic stabilities of the poses from one of the RMD simulations, we ran unbiased MD trajectories starting from the 136 most populated clusters. During these simulations we took the time spent within 2.5 Å rmsd of the initial configuration as a measure of the stability of the pose and found that 89 poses were stable for more than 100 ps, 25 were stable for more than 1 ns, and 7 of them were stable for more than 5 ns. Out of these seven poses, one was the crystallographic pose and one was a similar pose in which the ligand was separated from the Asp-189 residue by a water molecule. In addition, this set of poses contained the S2 and S3 states that were identified as stable in the MD studies of Buch et al. (12) (Table S2). Intriguingly, a stable pose (Fig. S1) was found in a part of the protein surface that was deliberately not explored in the MD investigation in reference (12). This configuration remains unchanged for 60 ns of unbiased MD and we predict that it is one of most stable interaction sites outside of the binding pocket. It is possible that, like the S2 site, it acts as a secondary binding site (44). For the EADock calculations a similar analysis showed that only eight of the poses generated were stable for more than 100 ps (Table S1) and that this set included the binding site, the S2 site, and a similar pose to that shown in Fig. S1.
Dimensionality Reduction.
Clustering is one way of examining the data from an extensive sampling of a high-dimensional phase space, such as that obtained from docking, MD, or an enhanced sampling calculation. An alternative is to perform dimensionality reduction (45, 46). This way of examining ligand binding is appealing, because the largest changes in the position of the ligand are those corresponding to motion across the two-dimensional protein surface so the data should lie on a low-dimensionality manifold. Furthermore, a low-dimensional representation of the protein surface is a useful tool for visualizing the kinetic information that can be extracted from MD-based approaches.
Many dimensionality reduction algorithms work by endeavoring to reproduce the rmsd distances between the trajectory frames in a lower-dimensionality space (47). Clearly the rmsd distances between the ligand in the various trajectory frames can be approximately reproduced in a three-dimensional space as they will be dominated by differences in position of the center of mass of the ligand. To further lower the dimensionality of the projection requires one either to incorporate periodicity in the low-dimensionality projection or to make less of the global features in phase space and, instead, focus on the local connectivity between basins. We recently developed the sketch-map algorithm (48) as a tool for analyzing trajectory data. This algorithm uses the second of these approaches to the problem as it endeavors to reproduce the immediate connectivity between states rather than the full set of distances between frames. The algorithm’s focus is controlled by transforming the distances in the high-dimensionality and low-dimensionality spaces using a sigmoid function. This procedure also ensures that close-together points are projected close together, whereas far apart points are projected far apart but not necessarily at the same distance.
We used the RMD trajectories to produce the sketch-map projections because, unlike our MD simulations which didn’t visit the binding site, we have sufficient sampling in the RMD to build a reliable map. To record the high-dimensional positions, we used the coordinates of the ligand’s C4, N1, and N2 atoms in a protein-centered frame of reference. Fig. 4A shows that the resulting two-dimensional map clearly separates the poses around the binding site from other low energy poses on the protein surface and that there are specific pathways and channels that connect the various clusters. Moreover, Fig. 4 B and C and Fig. S2 show that, in the area around the binding site, we are able to separate the metastable sites described by Buch et al. (12), in spite of the fact that some of them are rather close in space (center of mass separation of ∼4 Å). This result suggests that sketch-map is also able to describe the orientation of the ligand and that using multiple atoms to define the ligand’s position is worthwhile. The resolution can be further improved by constructing a map using only points that are close to the binding site (Fig. S3).
Fig. 4.

Results of the dimensionality reduction. (A) The two-dimensional, sketch-map representation of the configurations visited during the RMD simulations. The interval between the projected frames is 100 ps so there are approximately 20,000 points in this figure. The points are colored based on the minimum rmsd distance to the experimental binding site (exp) and the three other docking poses that are displayed in the Inset. The color scale only extends out to 10 Å, so if a point is further away from all of the sites than this distance it is colored gray. The docked pose P13 is the S2 metastable state reported in ref. 12, and P26 shares its pocket with the metastable state shown in Fig. S1. (B) A magnification of the area around the binding site highlighted by the red rectangle in A. Points in this figure are colored if they are within 2.5 Å rmsd of a specified pose. The poses indicated are the binding site (exp), two of the most stable EADock poses (P2 and P12), and the metastable poses described in ref. 12 (S3, TS1, TS2, and TS3; see Table S2). This last set of poses are the points along the pathway that Buch et al. (12) found most frequently connected the fully solvated ligand to the experimental binding pose. (C) The location of these poses on the protein surface. Each ligand molecule is colored, using the scheme from B, to indicate which pose is being shown. The same poses are shown in Fig. S2 together with a larger part of the protein surface.
We can use the projection shown in Fig. 4 to do a qualitative comparison between the results of our RMD simulation and the results of the extensive MD simulations by Buch et al. (12). In agreement with the previous study there is a significant population in the S3 state and a pathway from this state to the binding pose that passes through the TS1, TS2, and TS3 transition states. There are also other pathways between the bulk solvent and the binding site that pass through TS2 and TS3. In particular, during six of the ten binding or unbinding events that we observed, the ligand passed through the TS2 state on its way to or from the binding site, which suggests that this state is on the main binding pathway.
Conclusions
Molecular dynamics with explicit solvent has enormous potential for predicting protein-ligand interactions because it is based on a physically motivated and systematically improvable potential energy surface and because it incorporates conformational, solvent, and entropic effects in a physically consistent manner. Its one major drawback is that it is considerably more computationally expensive than using docking calculations based on a configurational search with approximate scoring functions. One reason for this expense is that there are many energetic basins on the surface of the protein which can kinetically trap the ligand and slow down diffusion. This problem can be resolved by using a simulation bias to force the system away from kinetic traps and to flatten the energy surface. However, the requirement to find a small set of CVs that describes all the potential traps makes it difficult to apply a suitable bias using many established methods. In contrast, in reconnaissance metadynamics we can use large numbers of collective variables and let the algorithm work out which linear combination best describes each trap. The procedure outlined in this paper can thus be used to tackle problems where conformational and solvent effects play a large role, which would be difficult to examine using standard docking. Furthermore, the method is considerably cheaper than unbiased MD.
Reconnaissance metadynamics simulations provide an extensive exploration of the low-energy portions of phase space. One can use this data to find the approximate locations for the various basins in the free-energy surface or alternatively use dimensionality reduction techniques to create low-dimensionality maps of phase space. The fact that these maps are low-dimensional allows one to reexplore the interesting parts of phase space using other, more quantitative, enhanced sampling algorithms. In future, we will use this idea to extract accurate free energies for the various binding poses found during the RMD simulations.
Materials and Methods
System Setup and Computational Details.
The simulations were performed using GROMACS 4.5 (49) and the PLUMED plug-in (50). We used the Amber ff99 force field (51) for the protein and TIP3P for the water molecules. For the ligand, van der Waals parameters were taken from the corresponding amino acids (phenylalanine and arginine), and appropriate charges were calculated using a RESP fit (52) to a Hartree–Fock calculation with the 6-31G* basis set—a procedure identical to that described in ref. 27. Long-range electrostatics was treated using the particle mesh Ewald approach with a grid spacing of 1.2 Å. A cutoff of 10 Å was used for all van der Waals and the direct electrostatic interactions and the neighbor list was updated every 10 steps. All production simulations were performed in the canonical ensemble at 300 K and this temperature was maintained using the stochastic velocity rescaling thermostat (53). To prevent the system from sampling fully solvated configurations we used a restraining wall that limited the exploration to configurations where the sum of all the switching functions between the C7 carbon and the points on the surface was greater than 1. This wall only has any effect when the minimum distance between the protein and the ligand is greater than 12 Å and represents a relatively small perturbation of the underlying energy surface.
The trypsin–benzamidine complex [Protein Data Bank (PDB) ID code 1J8A] (54) was used as the starting structure in this study. All histidines were protonated on the Nϵ site other than the catalytic H57, which was doubly protonated. This protein was then placed in a truncated octahedral simulation box that extended at least 7 Å from any protein atom. Prior to production a 10 ns constant pressure simulation, in which the protein atoms were initially restrained, was performed to equilibrate the system. Ten RMD production simulations were performed together with 10 MD simulations. These calculations were started from ten statistically inequivalent configurations, where the ligand was outside the protein. For each calculation we ran one RMD and one MD simulation. The initial starting configuration was generated by displacing the ligand from the binding site by 20 Å and running a short equilibration run. The remaining nine starting points were selected from the MD trajectory launched from the first point. In all these initial configurations the protein-ligand distance was greater than 10 Å. Furthermore, we visually inspected the starting configurations to ensure the widest possible spread of initial configurations.
RMD Setup.
Relevant points on the surface of the protein were selected by constructing a graph which had all the Cα atoms at its vertices and connections between any pair of vertices closer than 14 Å. A heuristic algorithm was then used to find the maximum independent set of this graph (55). This procedure produces a uniformly distributed set of Cα atoms on the surface. For trypsin these were the Cα atoms of residues 23, 47, 60, 74, 92, 97, 109, 127, 147, 159, 164, 173, 186, 193, 229, and 244. The switching function was set up so that its value for a test point moving along the protein surface (5 Å above it) changed smoothly from approximately 1 when it was immediately above one of the surface points to approximately 0.4 once it was above the neighboring surface point 14 Å away. For the reconnaissance metadynamics, data was collected every 0.5 ps, which was then clustered every 100 ps. The bias was constructed from the clusters that had a weight greater than 0.2 in these fits and by endeavoring to add hills of width 1.5 and height 1 kJ mol-1 every 2 ps. Hills were only added when the distance from one of the cluster centers (in the metric of that particular cluster) was less than 8.356—a distance that, at variance with previous applications of RMD, was kept constant for the entirety of the simulation.
As discussed in the main text we can easily create a more fine grained representation of the space by increasing the number of CVs and thus increasing the cost of the calculation. It is not straightforward to quantify the scaling with the number of CVs because it is unclear how much longer it will take to sample these higher dimensionality spaces. What we can say with certainty is that calculating the distance between a basin center and the instantaneous position scales with the square of the number of CVs. However, the cost of calculating the force because of the bias is for the most part small when compared to the cost of a single MD step.
Docking Calculations.
The docking calculations presented in this paper were used to provide a set of interesting poses that we could refind using our RMD simulations. We thus chose not to dwell on these calculations and just used the default (fast) protocol for EADock, which is provided on the Swissdock web server (56). The crystallographic structure of the protein (with the ligand removed) was used directly and 256 binding poses were obtained. These poses were then clustered using an rmsd cutoff of 2 Å and only clusters with at least five members were used. More details on these structures can be found in Table S1, which also shows that the crystallographic pose has an energy that is considerably lower than that of the other poses.
Sketch-Map Calculations.
The distances, d, between frames in the nine-dimensional space were transformed using 1 - [1 + (2a/b - 1)(d/σ)a]-b/a with σ, a, and b taking values of 20 Å, 1, and 3, respectively. The projection was then generated by minimizing the discrepancies between these transformed distances and the set of distances between the frames’ projections. These distances in the low-dimensionality space were once again transformed by the sigmoid function above, but in this case the a and b parameters were set to 2 and 3, respectively. The data from the 10 RMD trajectories was fitted by first projecting a set of 500 landmark points, 100 of which were selected at random and 400 of which were selected using farthest point sampling. Each point in this fit was weighted based on the number of unselected frames that fell within its voronoi polyhedra. Once this fitting was completed the unselected trajectory frames were mapped using the out-of-sample projection technique detailed in ref. 48.
Supplementary Material
Acknowledgments.
We thank Dr. M. Ceriotti for help with the sketch-map calculations and Dr. V. Limongelli for fruitful discussions. We also acknowledge computational resources from the central high-performance cluster of Eidgenössische Technische Hochschule Zurich (Brutus). Financial support for this work was obtained from European Union Grant ERC-2009-AdG-247075 and from The Swedish Research Council Grant 623-2009-821.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1201940109/-/DCSupplemental.
References
- 1.Gilson MK, Zhou HX. Calculation of protein-ligand binding affinities. Annu Rev Biophys Biomol Struct. 2007;36:21–42. doi: 10.1146/annurev.biophys.36.040306.132550. [DOI] [PubMed] [Google Scholar]
- 2.Singh N, Warshel A. Absolute binding free energy calculations: On the accuracy of computational scoring of protein-ligand interactions. Proteins: Struct Funct Bioinf. 2010;78:1705–1723. doi: 10.1002/prot.22687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Essex JW, Severance DL, Tirado-Rives J, Jorgensen WL. Monte Carlo simulations for proteins: Binding affinities for trypsin–benzamidine complexes via free-energy perturbations. J Phys Chem B. 1997;101:9663–9669. [Google Scholar]
- 4.Tribello G, Ceriotti M, Parrinello M. A self-learning algorithm for biased molecular dynamics. Proc Natl Acad Sci USA. 2010;107:17509–17514. doi: 10.1073/pnas.1011511107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hetenyi C, van der Spoel D. Toward prediction of functional protein pockets using blind docking and pocket search algorithms. Protein Science. 2011;20:880–893. doi: 10.1002/pro.618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang S, Zou X. Advances and challenges in protein-ligand docking. Int J Mol Sci. 2010;11:3016–3034. doi: 10.3390/ijms11083016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferrara P, Gohlke H, Price D, Klebe G, Brooks C. Assessing scoring functions for protein-ligand interactions. J Med Chem. 2004;47:3032–3047. doi: 10.1021/jm030489h. [DOI] [PubMed] [Google Scholar]
- 8.Warren G, et al. A critical assessment of docking programs and scoring functions. J Med Chem. 2006;49:5912–5931. doi: 10.1021/jm050362n. [DOI] [PubMed] [Google Scholar]
- 9.Huang S, Grinter S, Zou X. Scoring functions and their evaluation methods for protein-ligand docking: Recent advances and future directions. Phys Chem Chem Phys. 2010;12:12899–12908. doi: 10.1039/c0cp00151a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miranker A, Karplus M. An automated method for dynamic ligand design. Proteins: Struct Funct Bioinf. 1995;23:472–490. doi: 10.1002/prot.340230403. [DOI] [PubMed] [Google Scholar]
- 11.Carlson H, et al. Developing a dynamic pharmacophore model for HIV-1 integrase. J Med Chem. 2000;43:2100–2114. doi: 10.1021/jm990322h. [DOI] [PubMed] [Google Scholar]
- 12.Buch I, Giorgino T, De Fabritiis G. Complete reconstruction of an enzyme-inhibitor binding process by molecular dynamics simulations. Proc Natl Acad Sci USA. 2011;108:10184–10189. doi: 10.1073/pnas.1103547108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dror R, et al. Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc Natl Acad Sci USA. 2011;108:13118–13123. doi: 10.1073/pnas.1104614108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shan Y, et al. How does a drug molecule find its target binding site? J Am Chem Soc. 2011;133:9181–9183. doi: 10.1021/ja202726y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gallicchio E, Levy R. Advances in all atom sampling methods for modeling protein-ligand binding affinities. Curr Opin Struct Biol. 2011;21:161–166. doi: 10.1016/j.sbi.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Woods C, Jonathan W, King M. Enhanced configurational sampling in binding free-energy calculations. J Phys Chem B. 2003;107:13711–13718. [Google Scholar]
- 17.Knight J, Brooks C., III λ-dynamics free energy simulation methods. J Comput Chem. 2009;30:1692–1700. doi: 10.1002/jcc.21295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakajima N, Higo J, Kidera A, Nakamura H. Flexible docking of a ligand peptide to a receptor protein by multicanonical molecular dynamics simulation. Chem Phys Lett. 1997;278:297–301. [Google Scholar]
- 19.Higo J, Nishimura Y, Nakamura H. A free-energy landscape for coupled folding and binding of an intrinsically disordered protein in explicit solvent from detailed all-atom computations. J Am Chem Soc. 2011;133:10448–10458. doi: 10.1021/ja110338e. [DOI] [PubMed] [Google Scholar]
- 20.Torrie GM, Valleau JP. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J Chem Phys. 1977;23:187–199. [Google Scholar]
- 21.Hendrix D, Jarzynski C. A “fast growth” method of computing free energy differences. J Chem Phys. 2001;114:5974–5981. [Google Scholar]
- 22.Darve E, Pohorille A. Calculating free energies using average force. J Chem Phys. 2001;115:9169–9183. [Google Scholar]
- 23.Laio A, Parrinello M. Escaping free-energy minima. Proc Natl Acad Sci USA. 2002;99:12562–12566. doi: 10.1073/pnas.202427399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Woo HJ, Roux B. Calculation of absolute protein-ligand binding free energy from computer simulations. Proc Natl Acad Sci USA. 2005;102:6825–6830. doi: 10.1073/pnas.0409005102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jensen M, Park S, Tajkhorshid E, Schulten K. Energetics of glycerol conduction through aquaglyceroporin GlpF. Proc Natl Acad Sci USA. 2002;99:6731–6736. doi: 10.1073/pnas.102649299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cai W, Sun T, Liu P, Chipot C, Shao X. Inclusion mechanism of steroid drugs into β-cyclodextrins. Insights from free energy calculations. J Phys Chem B. 2009;113:7836–7843. doi: 10.1021/jp901825w. [DOI] [PubMed] [Google Scholar]
- 27.Gervasio F, Laio A, Parrinello M. Flexible docking in solution using metadynamics. J Am Chem Soc. 2005;127:2600–2607. doi: 10.1021/ja0445950. [DOI] [PubMed] [Google Scholar]
- 28.Kokubo H, Tanaka T, Okamoto Y. Ab initio prediction of protein-ligand binding structures by replica-exchange umbrella sampling simulations. J Comput Chem. 2011;32:2810–2821. doi: 10.1002/jcc.21860. [DOI] [PubMed] [Google Scholar]
- 29.Gallicchio E, Lapelosa M, Levy R. Binding energy distribution analysis method (BEDAM) for estimation of protein-ligand binding affinities. J Chem Theory Comput. 2010;6:2961–2977. doi: 10.1021/ct1002913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park I, Li C. Dynamic ligand-induced-fit simulation via enhanced conformational samplings and ensemble dockings: A survivin example. J Phys Chem B. 2010;114:5144–5153. doi: 10.1021/jp911085d. [DOI] [PubMed] [Google Scholar]
- 31.Provasi D, Bortolato A, Filizola M. Exploring molecular mechanisms of ligand recognition by opioid receptors with metadynamics. Biochemistry. 2009;48:10020–10029. doi: 10.1021/bi901494n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Limongelli V, et al. Molecular basis of cyclooxygenase enzymes (COXs) selective inhibition. Proc Natl Acad Sci USA. 2010;107:5411–5416. doi: 10.1073/pnas.0913377107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fidelak J, Juraszek J, Branduardi D, Bianciotto M, Gervasio F. Free-energy-based methods for binding profile determination in a congeneric series of CDK2 inhibitors. J Phys Chem B. 2010;114:9516–9524. doi: 10.1021/jp911689r. [DOI] [PubMed] [Google Scholar]
- 34.Masetti M, Cavalli A, Recanatini M, Gervasio F. Exploring complex protein-ligand recognition mechanisms with coarse metadynamics. J Phys Chem B. 2009;113:4807–4816. doi: 10.1021/jp803936q. [DOI] [PubMed] [Google Scholar]
- 35.Tribello G, Cuny J, Eshet H, Parrinello M. Exploring the free energy surfaces of clusters using reconnaissance metadynamics. J Chem Phys. 2011;135:114109. doi: 10.1063/1.3628676. [DOI] [PubMed] [Google Scholar]
- 36.Wales DJ. Energy Landscapes. Cambridge, UK: Cambridge Univ Press; 2003. [Google Scholar]
- 37.Wong C, McCammon J. Dynamics and design of enzymes and inhibitors. J Am Chem Soc. 1986;108:3830–3832. [Google Scholar]
- 38.Guvench O, Price D, Brooks C., III Receptor rigidity and ligand mobility in trypsin-ligand complexes. Proteins: Struct Funct Bioinf. 2005;58:407–417. doi: 10.1002/prot.20326. [DOI] [PubMed] [Google Scholar]
- 39.Hetényi C, van der Spoel D. Efficient docking of peptides to proteins without prior knowledge of the binding site. Protein Sci. 2002;11:1729–1737. doi: 10.1110/ps.0202302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grosdidier A, Zoete V, Michielin O. Fast docking using the CHARMM force field with EADock DSS. J Comput Chem. 2011;32:2149–2159. doi: 10.1002/jcc.21797. [DOI] [PubMed] [Google Scholar]
- 41.Grosdidier A, Zoete V, Michielin O. Blind docking of 260 protein-ligand complexes with EADock 2.0. J Comput Chem. 2009;30:2021–2030. doi: 10.1002/jcc.21202. [DOI] [PubMed] [Google Scholar]
- 42.Daura X, et al. Peptide folding: When simulation meets experiment. Angew Chem Int Ed. 1999;38:236–240. [Google Scholar]
- 43.He G, et al. Rank-ordering the binding affinity for FKBP12 and HLN1 neuraminidase inhibitors in the combination of a protein model with density functional theory. J Theor Comput Chem. 2011;10:541–565. [Google Scholar]
- 44.Oliveira M, et al. Tyrosine 151 is part of the substrate activation binding site of bovine trypsin identification by covalent labeling with p-diazoniumbenzamidine and kinetic characterization of tyr-151-(p-benzamidino)-azo-beta-trypsin. J Biol Chem. 1993;268:26893–26903. [PubMed] [Google Scholar]
- 45.Das P, Moll M, Stamati H, Kavraki L, Clementi C. Low-dimensional, free-energy landscapes of protein-folding reactions by nonlinear dimensionality reduction. Proc Natl Acad Sci USA. 2006;103:9885–9890. doi: 10.1073/pnas.0603553103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ferguson A, Panagiotopoulos A, Debenedetti P, Kevrekidis I. Systematic determination of order parameters for chain dynamics using diffusion maps. Proc Natl Acad Sci USA. 2010;107:13597–13602. doi: 10.1073/pnas.1003293107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cox T, Cox M. Multidimensional Scaling. London: Chapman & Hall; 1994. [Google Scholar]
- 48.Ceriotti M, Tribello G, Parrinello M. Simplifying the representation of complex free-energy landscapes using sketch-map. Proc Natl Acad Sci USA. 2011;108:13023–13028. doi: 10.1073/pnas.1108486108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hess B, Kutzner C, van der Spoel D, Lindahl E. Gromacs 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 50.Bonomi M, et al. Plumed: A portable plug in for free-energy calculations with molecular dynamics. Comput Phys Commun. 2009;180:1961–1972. [Google Scholar]
- 51.Wang J, Cieplak P, Kollman P. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J Comput Chem. 2000;21:1049–1074. [Google Scholar]
- 52.Bayly CI, Cieplak P, Cornell WD, Kollman PA. A well-behaved electrostatic potential based method using charge restraints for determining atom-centered charges: The RESP model. J Phys Chem. 1993;97:10269–10280. [Google Scholar]
- 53.Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys. 2007;126:014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 54.Cuesta-Seijo JA, García-Granda S. La tripsina como modelo de difracción de rayos x a alta resolución en proteínas. Bol R Soc Esp Hist Nat Secc Geol. 2002;97:123–129. [Google Scholar]
- 55.Balaji S, Swaminathan V, Kannan K. A simple algorithm to optimize maximum independent set. Advanced Modeling and Optimization. 2010;12:107–118. [Google Scholar]
- 56.Grosdidier A, Zoete V, Michielin O. Swissdock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011;39:W270–W277. doi: 10.1093/nar/gkr366. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.